

Abstract
Myxoid liposarcomas (MLSs) are genetically defined by the presence of DDIT3 gene fusions and most commonly arise in the extremities of young adults. Whether MLSs develop primarily in the retroperitoneum is controversial, and a recent retrospective study found no molecularly confirmed examples. Because MLSs tend to metastasize to deep soft tissues, purported examples of primary retroperitoneal lesions might represent distant metastasis, most commonly from extremities. In addition, well-differentiated or dedifferentiated liposarcomas, which are characterized by MDM2 amplifications, may exhibit prominent myxoid changes and mimic MLSs. Here, we document 5 cases of MLSs that originated in the retroperitoneum that were identified through critical clinicopathologic reevaluation. These cases accounted for 2.3% of 214 primary retroperitoneal liposarcomas and 3.2% of 156 MLSs in our database. They occurred in 3 men and 2 women with a median age of 32 years. All tumors were localized to the retroperitoneum at presentation, and no patient developed extra-abdominal recurrences during the clinical course (median, 50 mo). All 5 cases exhibited at least focal classic histologic findings. All harbored DDIT3 gene rearrangements, and none harbored MDM2 amplifications according to fluorescence in situ hybridization. This study demonstrates that primary MLSs can occur in the retroperitoneum, albeit rarely, and can be accurately diagnosed through combined clinicopathologic and molecular analysis.
Key Words: liposarcoma, retroperitoneum, diagnosis, fluorescence in situ hybridization
Myxoid liposarcomas (MLSs) account for 15% to 20% of all liposarcomas1 and tend to affect young adults, with the incidence peaking in the fourth to fifth decades of life. MLSs typically arise in deep soft tissues of the extremities; uncommon sites include the head and neck, subcutis, and thorax. Histologically, MLSs exhibit a mixture of uniform oval-shaped cells and signet-ring cell lipoblasts on a background comprising myxoid stroma and prominent arborizing capillary vasculature. The round cell component, defined by markedly increased cellularity, is predictive of aggressive behavior when comprising a significant proportion of the tumor volume. MLSs are genetically characterized by the presence of FUS-DDIT3 (>90%) or EWSR1-DDIT3 (<10%) fusion genes.2–4
Whether primary MLSs can develop in the retroperitoneum has recently become a matter of debate. Previously, the retroperitoneum was listed as a relatively common site of MLSs. In 1962, Enzinger and Winslow5 reported that 25% of MLSs occurred in the retroperitoneum, and more than a third of retroperitoneal liposarcomas were classified as MLSs. However, later published reports described the retroperitoneum as an uncommon site of MLSs,6,7 and, more recently, primary retroperitoneal MLSs have been considered rare1 or even nonexistent.8 This drastic shift in viewpoint stems from several factors. First, clinicopathologic studies established that MLSs have a unique proclivity to metastasize to deep soft tissues and bones,9–12 and the retroperitoneum represents one of the most common metastatic sites of these tumors.10 In addition, advances in clinical imaging have facilitated systemic surveys of tumor distribution. As a result, patients who present with metastatic retroperitoneal MLSs and would have previously been diagnosed with primary retroperitoneal MLSs can now be precisely staged by imaging.
Furthermore, molecular genetic evidence has refined the classifications of liposarcomas; as a result, some tumors that were previously classified as MLSs are currently diagnosed as well-differentiated liposarcomas (WDLSs) or dedifferentiated liposarcomas (DDLSs). WDLSs and DDLSs often affect the retroperitoneum and abdominal cavity,1 and some WDLSs or DDLSs may exhibit relatively uniform spindle cell proliferation on a background comprising abundant myxoid matrix and prominent plexiform capillaries, leading to a significant risk of misclassification as primary retroperitoneal MLSs.13 Whereas MLSs are genetically defined by DDIT3 gene rearrangement, WDLSs and DDLSs are characterized by MDM2 and CDK4 gene amplifications and overexpression of the respective protein products,14,15 and these specific genetic changes are diagnostically useful in histologically ambiguous cases.
To better understand the true incidence and characteristics of primary retroperitoneal MLSs, we retrospectively searched for potential cases of primary retroperitoneal MLS and critically reevaluated their clinical, radiologic, and histologic features. We hereby document 5 cases of primary retroperitoneal MLSs with confirmatory molecular genetic data.
MATERIALS AND METHODS
Patients
We electronically searched the pathology database of the National Cancer Center Hospital in Tokyo for potential cases of primary retroperitoneal MLS accessioned between 1998 and February 2015. Among a total of 219 cases (299 samples) recorded as retroperitoneal liposarcomas, we identified 11 candidate tumors (search terms: [“myxoid liposarcoma” OR “liposarcoma, myxoid type”] AND “retroperitoneum”). The remaining 208 cases were WDLSs/DDLSs (n=205) or pleomorphic liposarcomas (n=3), and all had originated from the retroperitoneum. From the 11 candidate tumors, a careful review of the clinical records and pathology materials excluded 6 cases from further analysis for the following reasons: (1) 3 cases had a previous history of MLS arising in the limbs (buttock in 2 cases and thigh in 1 case) and retroperitoneal tumors were considered metastases; (2) 1 patient presented with multiple soft tissue masses, including the retroperitoneal mass, and the primary site could not be confirmed; (3) 1 patient underwent resection of a “recurrent” MLS in the groin 1 year after resection of the retroperitoneal tumor, and the exact order of tumor development could not be verified because of the incomplete imaging studies; and (4) 1 case was excluded because the tumor contained multinucleated floret-like giant cells and spindle cell fascicles, and the diagnosis was revised as WDLS/DDLS with myxoid change. The remaining 5 cases exhibited histology compatible with MLS and met the strict clinical criteria of primary retroperitoneal origin and were therefore further analyzed to determine the DDIT3 and MDM2 gene status.
Fluorescence In Situ Hybridization
Fluorescence in situ hybridization (FISH) analysis was performed on formalin-fixed, paraffin-embedded, 4-µm-thick tumor sections. To examine DDIT3 rearrangement, we used the Vysis DDIT3 Break Apart FISH Probe Kit (Abbott Molecular, Abbott Park, IL). For MDM2 amplification status, we used the ZytoLight SPEC MDM2/CEN 12 Dual Color Probe (ZytoVision GmbH, Bremerhaven, Germany) and/or the Vysis LSI MDM2 SpectrumOrange Probe (Abbott Molecular) combined with Vysis CEP 12 (D12Z3) SpectrumGreen Probe (Abbott Molecular). FISH images were captured using the Metafer Slide Scanning Platform (MetaSystems, Altlussheim, Germany), and 100 nonoverlapping tumor cells were examined. For DDIT3, tumors in which >20% of the cells showed split signals were considered positive for gene rearrangement. An MDM2/control probe ratio of >2.0 in ≥10% of the nuclei was considered positive for MDM2 amplification.
RESULTS
Clinical Findings
Clinicopathologic data of the 5 patients with primary retroperitoneal MLSs are summarized in Table 1. The patients included 3 men and 2 women, with ages at diagnosis ranging from 30 to 73 years (median, 32 y). All primary tumors were localized to the retroperitoneum, and physical examination did not detect tumors elsewhere. Case 1 underwent computed tomography (CT; neck to thigh) and positron emission tomography (PET) scans (head to thigh), the latter of which indicated increased 18F-fluorodeoxyglucose uptake (maximum standard uptake value, 4.7) only in the retroperitoneum. Case 2 underwent CT and magnetic resonance imaging (MRI) scans from neck to toe and a whole-body PET scan (maximum standard uptake value, 1.3 in the recurrent retroperitoneal lesion). The other 3 cases underwent CT (chest to groin) and MRI scans (abdomen to groin). The maximum tumor diameters ranged from 10 to 36 cm (average, 20 cm). MRI revealed a mixed pattern of hypointensity and hyperintensity on T1-weighted images (WI) and hyperintensity on T2WI (Fig. 1). CT revealed slightly heterogenous, isodense masses. Shell-like mineralization was noted in case 5.
TABLE 1.
Summary of Clinicopathologic Data of MLSs That Originated in the Retroperitoneum
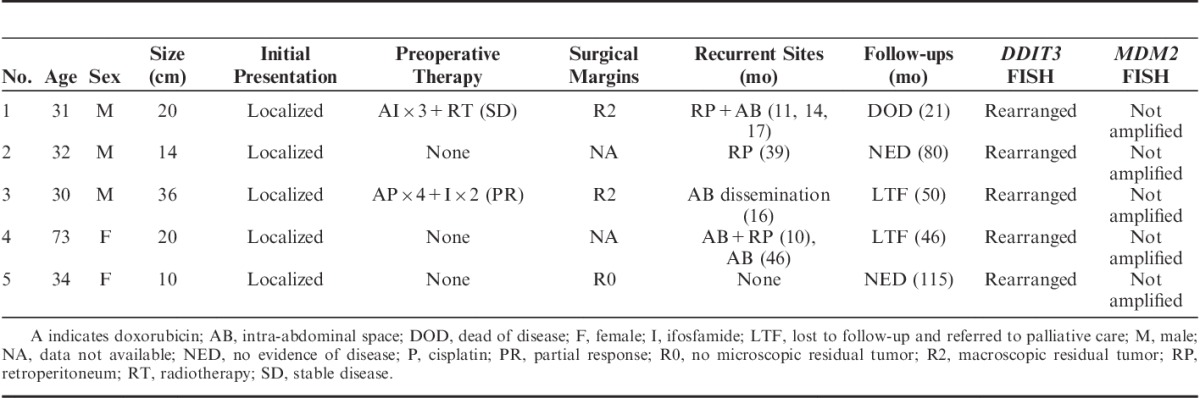
FIGURE 1.
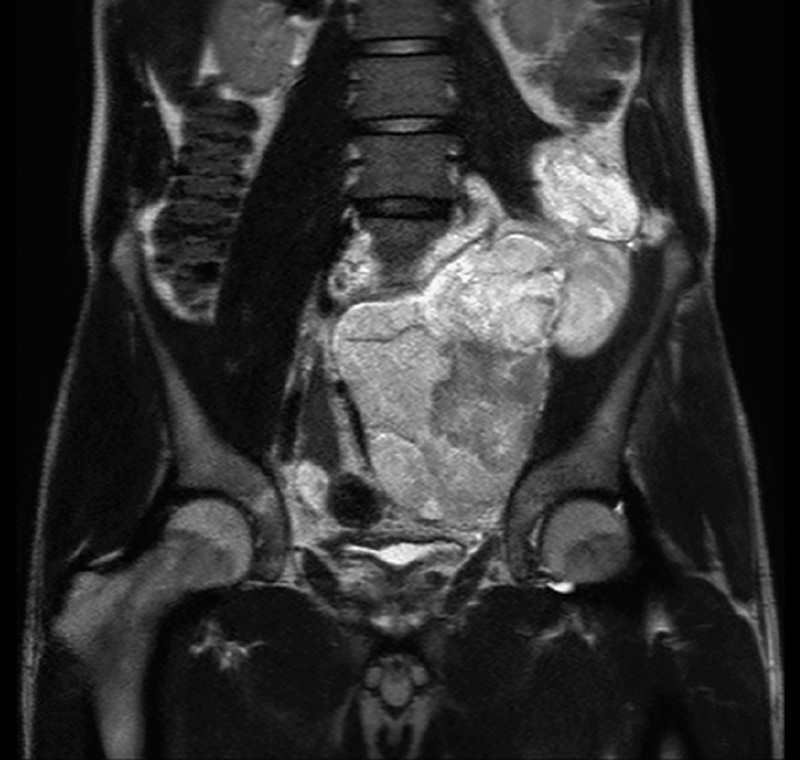
T2-weighted magnetic resonance image of a primary retroperitoneal MLS.
All primary tumors were surgically excised, after neoadjuvant therapies were administered in 2 cases. All but 1 patient developed recurrent disease, and the recurrent sites were anatomically confined to the retroperitoneal or intra-abdominal regions. No patients developed extra-abdominal soft tissue masses during their courses (range, 21 to 115 mo), as supported by physical examination and/or clinical interview. After a median follow-up of 50 months, 2 patients remained disease free for >5 years; the remaining patients either died of the disease or were referred to palliative care units because of advanced disease.
Histologic Findings
All 5 cases exhibited at least focal areas with classic histologic findings of MLS, including proliferating uniform, oval-shaped cells on a myxoid background with a rich plexiform capillary network (Fig. 2). Signet-ring cell lipoblasts were occasionally noted. In addition, prominent hyalinization and a focal area with reduced vascular density were each observed in 1 tumor. Histologically, all recurrent tumors exhibited similar features as the respective primary tumors except for the tumor in case 1, which contained an emergent round cell component at recurrence. Two cases were immunohistochemically examined during the original workups, and both tumors were found to be negative for MDM2 and CDK4.
FIGURE 2.
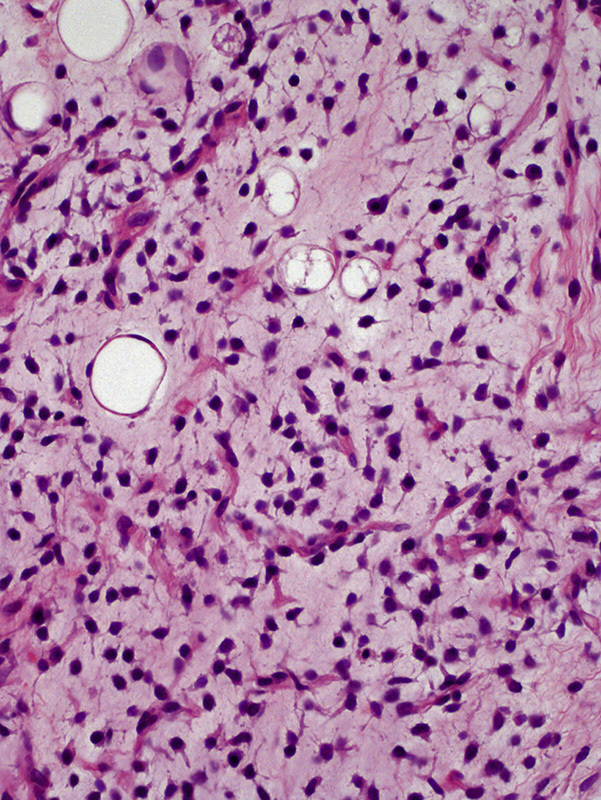
Primary retroperitoneal MLS showed a classic histologic appearance, characterized by a mixture of uniform oval cells and signet-ring cell lipoblasts on a background comprising myxoid stroma and plexiform capillary network (hematoxylin and eosin staining).
Molecular Cytogenetic Findings
All 5 cases harbored DDIT3 gene rearrangements (Fig. 3A). None of the cases harbored MDM2 amplifications (Fig. 3B).
FIGURE 3.
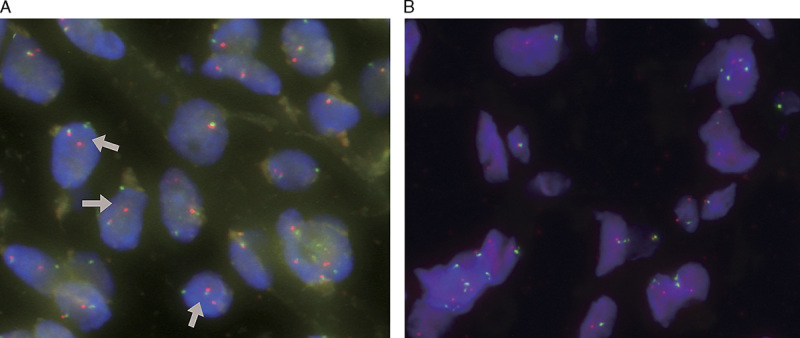
A, Primary retroperitoneal MLSs consistently harbored DDIT3 gene rearrangement (DDIT3 break-apart FISH assay; arrows indicate splits). B, All primary retroperitoneal MLSs lacked MDM2 gene amplifications (red signals indicate MDM2; green signals indicate CEP12).
DISCUSSION
In this report, we have documented 5 cases of genetically confirmed MLSs that originated from the retroperitoneum. These cases were identified from among 214 cases of primary retroperitoneal liposarcomas (2.3%) during the review period and represented only 3.2% of 156 myxoid/round cell liposarcoma cases diagnosed during this period. However, the calculated incidences are likely overestimated because of referral bias, as retroperitoneal WDLSs/DDLSs that are untreatable and MLSs in the limbs that were readily managed at local hospitals tend not to be referred to us for pathologic review. Overall, after a critical reevaluation of clinicopathologic parameters, we confirmed the rarity of primary retroperitoneal MLSs.
This rarity likely explains the results of a recent study by de Vreeze et al,8 who did not identify any genetically confirmed cases of primary retroperitoneal MLS in a smaller cohort of liposarcomas (n=68). When the authors analyzed 16 tumors originally diagnosed as retroperitoneal MLS, all exhibited MDM2 and CDK4 immunoreactivity or MDM2 amplification but lacked FUS-DDIT3 or EWSR1-DDIT3 fusion genes and were accordingly reclassified as WDLSs or DDLSs. We are aware of 2 potential cases of genetically proven primary retroperitoneal MLS16 that were described as “localized” to the “retroperitoneum to lower abdomen,” although detailed clinical information regarding these cases was not provided. In addition, some large series of MLS cases included those in the retroperitoneum17–19; however, those studies lacked either molecular genetic data or clinicoradiologic documentation to confirm a primary retroperitoneal origin. The present study unequivocally demonstrates that MLSs do primarily occur in the retroperitoneum and can be accurately diagnosed through combined clinicopathologic and molecular analysis.
Our study highlights the need for a careful clinical workup before the diagnosis of primary retroperitoneal MLS. Five of the initial 11 candidate cases were excluded because metastatic spread to the retroperitoneum could not be entirely ruled out. Careful clinical interview including an inquiry regarding the remote history is important. According to our review of 3 published series,9,11,12 12% (8/66) of MLS patients presented with the first metastases >5 years after the initial presentation, including 2 cases with a long interval between the initial presentation and metastatic disease (18 and 25 y).9,12 A thorough physical examination is also mandatory for accurate identification of the primary site. In addition, a variety of imaging modalities are available to rule out possible primary tumors in the extremities, particularly the lower extremities where most MLSs develop.9–12,16 These modalities include MRI, enhanced CT, and 18F-fluorodeoxyglucose PET/CT, with a caveat that the latter may show low tracer uptake.12,20 However, extensive imaging studies may not always be economically feasible, and clinical parameters often supplant such assessments in actual practice settings. Among our 5 cases, only case 2 involved a systemic imaging workup, whereas case 1 included MRI and CT scans of the thighs, the most common primary site for MLS. Although the radiologic studies did not cover extremities in the remaining 3 cases, we believe that the primary retroperitoneal origins in all 5 cases were confirmed by the absence of tumors elsewhere over a relatively long follow-up period (median, 50 mo).
As de Vreeze et al rightly noted,8 it can be difficult to distinguish MLSs from WDLSs/DDLSs with myxoid changes in the retroperitoneum. In our study, 1 case of WDLS/DDLS was initially misinterpreted as MLS. This distinction is of paramount importance for appropriate treatment. WDLSs/DDLSs are usually resistant to radiotherapy and chemotherapy, whereas MLSs are sensitive to these modalities.21,22 Furthermore, trabectedin, a recently developed agent that interferes with the binding of fusion genes and target promoters,23 has shown promise against MLS.24,25 Although the differential diagnosis is ultimately made possible by genetic means, it can be facilitated by the combined use of conventional modalities, including clinical, radiologic, and histologic findings.
Clinically, MLSs typically arise in younger patients, compared with WDLSs/DDLSs.1 MLSs may affect children, in whom WDLSs/DDLSs are distinctly rare.1 Radiologically, WDLSs/DDLSs present as multinodular masses that may contain a purely lipomatous component, whereas MLSs exhibit hypointense to isointense signals on T1WI and hyperintense signals on T2WI and occasionally exhibit intermixing with lipomatous areas in a marbled or nebulous textural manner.26 Intratumoral mineralization might suggest WDLSs/DDLSs, as it is more common in these tumor types.27 Nonetheless, decisions should not be made solely on these distinctive clinicoradiologic parameters, as exemplified in the present study by case 4, which involved an elderly patient, and case 5, which exhibited shell-like mineralization. Histologic examination of MLSs generally reveals uniform monomorphic cytomorphology, in contrast to at least focal nuclear pleomorphism observed in WDLSs/DDLSs with myxoid changes. The tumor cells in WDLSs/DDLSs with myxoid changes tend to be spindled, whereas those in MLSs are typically oval with less conspicuous cytoplasms. In addition, plexiform thin-walled vasculature is characteristic of MLSs, whereas the vasculature associated with WDLSs/DDLSs is commonly coarse and curvilinear; however, WDLSs/DDLSs may also show a plexiform and delicate form that is indistinguishable from the pattern noted in MLSs.8 MDM2 and CDK4 immunohistochemistry may be a practical surrogate for the molecular analysis of MDM2 amplification.28
Another rare liposarcoma variant that should be differentiated from MLS in young patients is the so-called pleomorphic MLS (also known as myxoid pleomorphic liposarcoma).29 This variant typically occurs in the mediastinum; however, cases involving the retroperitoneal/abdominal regions have been reported. Unlike MLSs, pleomorphic MLSs harbor pleomorphic liposarcoma-like components and lack the DDIT3 gene fusion.
In summary, we conclude that MLS rarely occurs in the retroperitoneum, and the primary site alone should not be used to rule out a diagnosis of MLS. However, the rarity of such cases demands considerable diagnostic caution in clinical practice settings. Particular attention should be paid to the distinction from WDLSs/DDLSs with myxoid changes and the possibility of retroperitoneal metastasis from extraneous sites.
Footnotes
Conflicts of Interest and Source of Funding: Supported in part by the National Cancer Center Research and Development Fund (26-A-9 and 26-A-21) and the Practical Research for Innovative Cancer Control from the Japan Agency for Medical Research and Development (AMED, 15ck0106089h0002). The authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article.
REFERENCES
- 1.Fletcher CDM, Bridge JA, Hogendoorn PCW, et al. World Health Organization Classification of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2013. [Google Scholar]
- 2.Aman P, Ron D, Mandahl N, et al. Rearrangement of the transcription factor gene CHOP in myxoid liposarcomas with t(12;16)(q13;p11). Genes Chromosomes Cancer. 1992;5:278–285. [DOI] [PubMed] [Google Scholar]
- 3.Rabbitts TH, Forster A, Larson R, et al. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet. 1993;4:175–180. [DOI] [PubMed] [Google Scholar]
- 4.Panagopoulos I, Hoglund M, Mertens F, et al. Fusion of the EWS and CHOP genes in myxoid liposarcoma. Oncogene. 1996;12:489–494. [PubMed] [Google Scholar]
- 5.Enzinger FM, Winslow DJ. Liposarcoma. A study of 103 cases. Virchows Arch Pathol Anat Physiol Klin Med. 1962;335:367–388. [PubMed] [Google Scholar]
- 6.Singer S, Antonescu CR, Riedel E, et al. Histologic subtype and margin of resection predict pattern of recurrence and survival for retroperitoneal liposarcoma. Ann Surg. 2003;238:358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldblum JR, Folpe AL, Weiss SW. Enzinger and Weiss’s soft tissue tumors, 6th ed Philadelphia, PA: Elsevier; 2014. [Google Scholar]
- 8.de Vreeze RS, de Jong D, Tielen IH, et al. Primary retroperitoneal myxoid/round cell liposarcoma is a nonexisting disease: an immunohistochemical and molecular biological analysis. Mod Pathol. 2009;22:223–231. [DOI] [PubMed] [Google Scholar]
- 9.Cheng EY, Springfield DS, Mankin HJ. Frequent incidence of extrapulmonary sites of initial metastasis in patients with liposarcoma. Cancer. 1995;75:1120–1127. [DOI] [PubMed] [Google Scholar]
- 10.Pearlstone DB, Pisters PW, Bold RJ, et al. Patterns of recurrence in extremity liposarcoma: implications for staging and follow-up. Cancer. 1999;85:85–92. [DOI] [PubMed] [Google Scholar]
- 11.Schwab JH, Boland PJ, Antonescu C, et al. Spinal metastases from myxoid liposarcoma warrant screening with magnetic resonance imaging. Cancer. 2007;110:1815–1822. [DOI] [PubMed] [Google Scholar]
- 12.Sheah K, Ouellette HA, Torriani M, et al. Metastatic myxoid liposarcomas: imaging and histopathologic findings. Skeletal Radiol. 2008;37:251–258. [DOI] [PubMed] [Google Scholar]
- 13.Sioletic S, Dal Cin P, Fletcher CD, et al. Well-differentiated and dedifferentiated liposarcomas with prominent myxoid stroma: analysis of 56 cases. Histopathology. 2013;62:287–293. [DOI] [PubMed] [Google Scholar]
- 14.Dei Tos AP, Doglioni C, Piccinin S, et al. Coordinated expression and amplification of the MDM2, CDK4, and HMGI-C genes in atypical lipomatous tumours. J Pathol. 2000;190:531–536. [DOI] [PubMed] [Google Scholar]
- 15.Sirvent N, Coindre JM, Maire G, et al. Detection of MDM2-CDK4 amplification by fluorescence in situ hybridization in 200 paraffin-embedded tumor samples: utility in diagnosing adipocytic lesions and comparison with immunohistochemistry and real-time PCR. Am J Surg Pathol. 2007;31:1476–1489. [DOI] [PubMed] [Google Scholar]
- 16.Antonescu CR, Tschernyavsky SJ, Decuseara R, et al. Prognostic impact of P53 status, TLS-CHOP fusion transcript structure, and histological grade in myxoid liposarcoma: a molecular and clinicopathologic study of 82 cases. Clin Cancer Res. 2001;7:3977–3987. [PubMed] [Google Scholar]
- 17.Kilpatrick SE, Doyon J, Choong PF, et al. The clinicopathologic spectrum of myxoid and round cell liposarcoma. A study of 95 cases. Cancer. 1996;77:1450–1458. [DOI] [PubMed] [Google Scholar]
- 18.Hoffman A, Ghadimi MP, Demicco EG, et al. Localized and metastatic myxoid/round cell liposarcoma: clinical and molecular observations. Cancer. 2013;119:1868–1877. [DOI] [PubMed] [Google Scholar]
- 19.Moreau LC, Turcotte R, Ferguson P, et al. Myxoid/round cell liposarcoma (MRCLS) revisited: an analysis of 418 primarily managed cases. Ann Surg Oncol. 2012;19:1081–1088. [DOI] [PubMed] [Google Scholar]
- 20.Schwab JH, Healey JH. FDG-PET lacks sufficient sensitivity to detect myxoid liposarcoma spinal metastases detected by MRI. Sarcoma. 2007;2007:36785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pitson G, Robinson P, Wilke D, et al. Radiation response: an additional unique signature of myxoid liposarcoma. Int J Radiat Oncol Biol Phys. 2004;60:522–526. [DOI] [PubMed] [Google Scholar]
- 22.Jones RL, Fisher C, Al-Muderis O, et al. Differential sensitivity of liposarcoma subtypes to chemotherapy. Eur J Cancer. 2005;41:2853–2860. [DOI] [PubMed] [Google Scholar]
- 23.Di Giandomenico S, Frapolli R, Bello E, et al. Mode of action of trabectedin in myxoid liposarcomas. Oncogene. 2014;33:5201–5210. [DOI] [PubMed] [Google Scholar]
- 24.Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol. 2007;8:595–602. [DOI] [PubMed] [Google Scholar]
- 25.Gronchi A, Bui BN, Bonvalot S, et al. Phase II clinical trial of neoadjuvant trabectedin in patients with advanced localized myxoid liposarcoma. Ann Oncol. 2012;23:771–776. [DOI] [PubMed] [Google Scholar]
- 26.El Ouni F, Jemni H, Trabelsi A, et al. Liposarcoma of the extremities: MR imaging features and their correlation with pathologic data. Orthop Traumatol Surg Res. 2010;96:876–883. [DOI] [PubMed] [Google Scholar]
- 27.Murphey MD, Arcara LK, Fanburg-Smith J. From the archives of the AFIP: imaging of musculoskeletal liposarcoma with radiologic-pathologic correlation. Radiographics. 2005;25:1371–1395. [DOI] [PubMed] [Google Scholar]
- 28.Binh MB, Sastre-Garau X, Guillou L, et al. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. Am J Surg Pathol. 2005;29:1340–1347. [DOI] [PubMed] [Google Scholar]
- 29.Alaggio R, Coffin CM, Weiss SW, et al. Liposarcomas in young patients: a study of 82 cases occurring in patients younger than 22 years of age. Am J Surg Pathol. 2009;33:645–658. [DOI] [PubMed] [Google Scholar]