

Abstract
Yeast tRNA ligase (Trl1) is an essential trifunctional enzyme that repairs RNA breaks with 2′,3′-cyclic-PO4 and 5′-OH ends. Trl1 is composed of C-terminal cyclic phosphodiesterase and central polynucleotide kinase domains that heal the broken ends to generate the 3′-OH, 2′-PO4, and 5′-PO4 termini required for sealing by an N-terminal ligase domain. Trl1 enzymes are found in all human fungal pathogens and they are promising targets for antifungal drug discovery because: (i) their domain structures and biochemical mechanisms are unique compared to the mammalian RtcB-type tRNA splicing enzyme; and (ii) there are no obvious homologs of the Trl1 ligase domain in mammalian proteomes. Here we characterize the tRNA ligases of two human fungal pathogens: Coccidioides immitis and Aspergillus fumigatus. The biological activity of CimTrl1 and AfuTrl1 was verified by showing that their expression complements a Saccharomyces cerevisiae trl1Δ mutant. Purified recombinant AfuTrl1 and CimTrl1 proteins were catalytically active in joining 2′,3′-cyclic-PO4 and 5′-OH ends in vitro, either as full-length proteins or as a mixture of separately produced healing and sealing domains. The biochemical properties of CimTrl1 and AfuTrl1 are similar to those of budding yeast Trl1, particularly with respect to their preferential use of GTP as the phosphate donor for the polynucleotide kinase reaction. Our findings provide genetic and biochemical tools to screen for inhibitors of tRNA ligases from pathogenic fungi.
Keywords: antifungal target, tRNA splicing
INTRODUCTION
Fungal tRNA ligases, exemplified by Saccharomyces cerevisiae Trl1 (SceTrl1), are essential agents in the repair of programmed tRNA and mRNA breaks with 2′,3′-cyclic phosphate and 5′-OH ends that are generated during tRNA splicing and the unfolded protein response. SceTrl1 performs three distinct RNA repair reactions: (i) The 2′,3′-cyclic phosphate (>p) end is hydrolyzed to a 3′-OH, 2′-PO4 by a cyclic phosphodiesterase (CPD); (ii) the 5′-OH end is phosphorylated by an NTP-dependent polynucleotide kinase; and (iii) the 3′-OH, 2′-PO4, and 5′-PO4 ends are sealed by an ATP-dependent RNA ligase to form an unconventional 2′-PO4, 3′–5′ phosphodiester at the splice junction (Greer et al. 1983). SceTrl1 and its homolog KlaTrl1 from Kluyveromyces lactis are composed of three separable catalytic domains: a C-terminal CPD module belonging to the 2H phosphoesterase superfamily; a central kinase module of the P-loop phosphotransferase superfamily; and an N-terminal ligase domain that belongs to the covalent nucleotidyltransferase superfamily (Apostol et al. 1991; Sawaya et al. 2003; Wang and Shuman 2005; Wang et al. 2006; Remus and Shuman 2014).
The CPD and kinase reactions are collectively referred to as end-healing (Schwer et al. 2004). Healing provides the proper termini for end sealing, which is dependent on the 2′-PO4 moiety. The sealing phase of tRNA splicing requires ATP and proceeds via three adenylate transfer steps. First, the ligase domain reacts with ATP to form a covalent ligase-(lysyl-Nζ)-AMP intermediate and displace pyrophosphate. Second, ligase transfers AMP to the 5′-PO4 RNA terminus to form an RNA–adenylate intermediate (AppRNA). Third, ligase directs the attack of the 3′-OH on AppRNA to form the splice junction and displace AMP.
Fungal Trl1 enzymes are potential therapeutic targets because their domain structures and biochemical mechanisms are unique compared to the RtcB-type tRNA repair systems elaborated by metazoans, archaea, and many bacteria (Popow et al. 2011; Tanaka and Shuman 2011; Tanaka et al. 2011a; Chakravarty and Shuman 2012; Chakravarty et al. 2012; Englert et al. 2012; Desai et al. 2013; Maughan and Shuman 2015). RtcB is a GTP-dependent RNA ligase that splices 3′-PO4 and 5′-OH ends via a novel chemical mechanism entailing the formation of covalent RtcB-(histidinyl)-GMP and RNA3′pp5′G intermediates. There is no 5′ kinase end-healing step in the RtcB pathway of RNA repair. RtcB is absent from the proteomes of most fungi. Conversely, mammalian proteomes have no discernible homologs of the sealing domain of fungal tRNA ligase. These features highlight the sealing components of tRNA ligases as untapped targets for the discovery of new antifungals. Even more attractive is the prospect of inhibiting multiple enzymatic steps in the fungal tRNA splicing pathway, which would reduce the risk of the emergence of resistance. For that scenario to be plausible, the CPD or kinase modules of fungal Trl1 enzymes must have “druggable” properties. The kinase domains of KlaTrl1 and SceTrl1 are unique among the polynucleotide kinase enzymes characterized to date in that they have a strong preference for GTP as the phosphate donor (Westaway et al. 1993; Sawaya et al. 2003; Remus and Shuman 2014). This raises the prospect that the fungal kinase donor site could be targeted by a small molecule inhibitor that interacts with the guanine specificity determinant(s) of Trl1, but does not bind to the donor sites of the many other cellular P-loop phosphotransferases, which either have no NTP donor preference or prefer ATP as substrates.
To fortify the case for fungal tRNA ligases as a drug target, it is imperative to understand the properties of Trl1 enzymes produced by fungi that cause human disease, i.e., it would be naïve to presume that all fungal tRNA ligases have the same properties as the enzymes from nonpathogenic yeast such as S. cerevisiae and K. lactis. To that end, we have initiated here a characterization of the tRNA ligases from two fungal pathogens: Aspergillus fumigatus and Coccidioides immitis. Coccidioides immitis is the causative agent of San Joaquin Valley Fever, a human infectious disease acquired by inhaling airborne fungal spores. Extrapulmonary dissemination of C. immitis infection, particularly meningitis, can be deadly (Stockamp and Thompson 2016). Valley Fever is prevalent in the US desert Southwest region (California, Arizona, New Mexico, Nevada, Texas), where it constitutes a major public health crisis for local residents (especially the elderly, immune-compromised, and ethnic minorities) and for the concentrated inmate population of the many prisons located in the area (Goodyear 2014). Aspergillus fumigatus is a saprophytic soil fungus that causes lung disease when airborn conidia are inhaled. Immunocompetent individuals may develop aspergilloma or allergic bronchopulmonary aspergillosis. Immunosuppressed individuals (especially those with hematological malignancies or who have undergone bone marrow or solid organ transplantation) may develop invasive aspergillosis, a severe and often fatal infection (Dagenais and Keller 2009).
RESULTS
Genetic complementation confirms biological activity of fungal pathogen tRNA ligases
We showed previously that expression of heterologous RNA repair systems can complement the lethality of a trl1Δ knockout in S. cerevisiae (Schwer et al. 2004; Wang et al. 2006; Nandakumar et al. 2008; Tanaka et al. 2011b). Here we cloned the ORFs encoding Candida albicans (Cal), A. fumigatus (Afu), and C. immitis (Cim) Trl1 enzymes into yeast CEN plasmids wherein their expression is driven by the S. cerevisiae TPI1 promoter. We tested complementation by plasmid shuffle in a S. cerevisiae trl1Δ p(CEN URA3 TRL1) strain. All three Trl1 orthologs from pathogenic fungi complemented trl1Δ. As shown in Figure 1, the AfuTRL1 and CalTRL1 strains grew as well as the SceTRL1 control when spot-tested on rich agar medium at 20°C–37°C. The CimTRL1 strain grew as well as SceTRL1 at 20°C–34°C, but formed smaller colonies at 37°C. Having affirmed that the pathogen Trl1s are biologically active as tRNA ligases, we aimed to produce and purify recombinant AfuTrl1 and CimTrl1 proteins and elucidate their properties.
FIGURE 1.

Genetic complementation of S. cerevisiae trl1Δ by fungal pathogen tRNA ligases. Serial dilutions of trl1Δ cells transformed with CEN plasmids bearing SceTRL1, CalTRL1, AfuTRL1, or CimTRL1 genes were spotted to YPD agar plates, which were incubated at 20°C, 25°C, 30°C, 34°C, and 37°C.
RNA end-joining activities of recombinant AfuTrl1 and CimTrl1
We produced the full-length 834-aa AfuTrl1 (calculated mass 94.2 kDa) and CimTrl1 (94.4 kDa) proteins in Escherichia coli as His10Smt3 fusions and purified them from soluble extracts by sequential Ni-affinity chromatography/imidazole elution, removal of the His10Smt3 tag by treatment with Ulp1 protease, recovery of the tag-free Trl1 proteins in the flow-through of a second Ni-affinity column, and a final Superose-200 gel-filtration step. AfuTrl1 and CimTrl1 were monomeric by gel filtration. SDS–PAGE analysis of the AfuTrl1 and CimTrl1 preparations is shown in Figure 2A.
FIGURE 2.

RNA end-joining activities of recombinant AfuTrl1 and CimTrl1. (A) Aliquots (5 µg) of the recombinant AfuTrl1 and CimTrl1 preparations were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (kDa) of marker proteins are indicated on the left. (B,C) AfuTrl1 and CimTrl1 were reacted with a 3′ 32P-labeled 20-mer HORNA>p substrate (depicted at the top of panel B with the 32P-label denoted by •) or a 3′ 32P-labeled 10-mer HORNA>p substrate (depicted at the top of panel C). Complete reaction mixtures (20 µL) containing 50 mM Tris–HCl (pH 7.5), 2 mM DTT, 10 mM MgCl2, 100 µM ATP, 100 µM GTP, 20 nM HORNA>p, and 50 nM Trl1 were incubated for 5 min at 22°C. Individual reaction components were included (+) or omitted (−) as specified below the lanes; mixtures lacking 10 mM MgCl2 were supplemented with 10 mM EDTA where indicated by (+). The reactions were quenched with formamide/EDTA and the products were analyzed by urea–PAGE. Autoradiographs of the gels are shown. The positions and identities of the HORNA>p substrate (*), 5′-phosphorylated kinase product (5′-P), and ligation products (circle and multimers) are indicated on the left in panel B. The positions and identities of the input substrate and healed intermediates (specified by their termini), and circular ligation product are indicated on the left and right in panel C.
To assay the composite healing and sealing pathway of Trl1 enzymes, we used a 20-mer RNA with 5′-OH and 2′,3′-cyclic phosphate (>p) ends and a single radiolabel between the 3′-terminal and penultimate nucleosides (Fig. 2B). The HORNA>p substrate (20 nM) was reacted for 5 min at 22°C with 50 nM AfuTrl1 or CimTrl1 in the presence of 10 mM Mg2+, 0.1 mM ATP, and 0.1 mM GTP, and the products were analyzed by urea–PAGE. AfuTrl1 and CimTrl1 converted all of the HORNA>p substrate into ligated products, these being a circular RNA formed by intramolecular ligation (that migrated ahead of the substrate strand) and RNA multimers formed via intermolecular end joining (that migrated well behind the substrate strand) (Fig. 2B).
The requirements for AfuTrl1 and CimTrl1 activity were queried by systematic omission of reaction components. Absent Mg2+, formation of the ligated circle and multimer RNAs was suppressed and the predominant product was shifted to a species that migrated between the substrate strand and the circle; this RNA (denoted by the arrowhead in Fig. 2B) corresponds to the 5′-phosphorylated product of the Trl1 polynucleotide kinase reaction. (Note that the PAGE system used does not separate a 20-mer RNA with a 2′,3′-cyclic phosphate terminus versus a 2′-monophosphate, so we cannot directly gauge the extent of the CDP reaction with the 20-mer substrate.) Omission of Mg2+ and inclusion of 10 mM EDTA in the reaction mixtures suppressed the 5′ kinase reaction, suggesting that residual divalent cation was carried along with the kinase during the purification procedure. When GTP was omitted, the ligation reaction proceeded to completion, signifying that 0.1 mM ATP sufficed as a phosphate donor for the AfuTrl1 and CimTrl1 kinases. Yet, when ATP was omitted, ligation was just as efficient, signifying that: (i) 0.1 mM GTP sufficed as a kinase donor substrate; and (ii) the AfuTrl1 and CimTrl1 preparations contained a significant fraction of preformed covalent ligase–AMP intermediate. When ATP and GTP were jointly omitted, no reaction was observed (i.e., no change in mobility of the RNA substrate), which signifies that there was no NTP prebound to the kinase.
In order to track all three Trl1 reactions in a one-pot format (Remus and Shuman 2014), we used a 10-mer HORNA>p substrate with a single radiolabel between the 3′-terminal and penultimate nucleosides (Fig. 2C) and performed the same series of reactions under complete and component-omitted conditions. (The PAGE system is capable of resolving a 10-mer RNA with a 2′,3′-cyclic phosphate terminus versus a 10-mer with a 2′-monophosphate.) In the presence of Mg2+, ATP, and GTP, AfuTrl1 and CimTrl1 converted all of the input 10-mer into a more rapidly migrating 10-mer circle (Fig. 2C); we saw no multimeric products of intermolecular end joining. (Previous studies showed that plant AtRNL and K. lactis Trl1 also form only 10-mer circles when reacted with a 10-mer HORNA>p substrate, whereas they, like AfuTrl1 and CimTrl1, generate a mixture of circle and multimer products when reacted with a 20-mer HORNA>p [Remus and Shuman 2013, 2014].) When Mg2+ was omitted, the 10-mer HORNA>p substrate was converted by AfuTrl1 and CimTrl1 to a 5′-phosphorylated kinase reaction product of intermediate mobility. When ATP and GTP were jointly omitted, the ligase and kinase reactions were suppressed, but we could now observe the conversion of all of the input 10-mer HORNA>p substrate to the CPDase reaction product HORNAp, which migrated just slightly ahead of HORNA>p (Fig. 2C). The CPDase product HORNAp was also predominant when EDTA was present in the reaction lacking magnesium. Thus, the CPDase reaction of AfuTrl1 and CimTrl1 requires neither divalent cation nor NTP. As noted above, and for the same reasons, we found that omission of ATP or GTP alone did not affect circularization of the 10-mer substrate.
The effect of Trl1 concentration on the efficiency and product distribution of the reaction with the 20-mer HORNA>p substrate is shown in Figure 3, where 20 nM HORNA>p was incubated for 30 min with 0.5, 1, 2, 5, 10, 20, 50, or 100 nM AfuTrl1 or CimTrl1. The salient points are that: (i) the 5′-PO4 species was prominent at substoichiometric Trl1 concentrations (comprising 20%–26% of the total labeled RNA at 0.5–1 nM Trl1) and declined steadily with increasing enzyme, consistent with its intermediacy in ligation; and (ii) ligated RNAs were predominant even at 0.5 nM Trl1, comprising 52% and 42% of the total labeled RNA in the AfuTrl1 and CimTrl1 reaction, respectively. (The values cited for the product distributions are the average of three independent Trl1 titration experiments.) From these data, we can estimate that 0.5 nM AfuTrl1 and CimTrl1 ligated 21 and 17 RNAs per enzyme, respectively, during the 30-min reaction.
FIGURE 3.

Trl1 titration. Reaction mixtures (20 µL) containing 50 mM Tris–HCl (pH 7.5), 2 mM DTT, 10 mM MgCl2, 100 µM ATP, 100 µM GTP, 20 nM 20-mer HORNA>p (depicted at the bottom), and 0.5. 1, 2, 5, 10, 20, 50, or 100 nM Trl1 (from left to right in each titration series) were incubated for 30 min at 22°C. Trl1 was omitted from control reactions in lanes (–). The products were analyzed by urea–PAGE. An autoradiograph of the gel is shown. The positions and identities of the HORNA>p substrate (*), 5′-phosphorylated kinase product (5′-P), and the ligation products (circle and multimers) are indicated.
Separation of the healing and sealing domains of AfuTrl1 and CimTrl1
Alignment of the primary structures of AfuTrl1 and CimTrl1 in Figure 4A highlights 615/834 positions of side chain identity/similarity. The N-terminal ligase domain (LIG) includes three landmark motifs that form the active site for adenylate transfer by all known clades of ATP-dependent polynucleotide ligases (Unciuleac et al. 2015). Motif I (KENG) contains the lysine nucleophile that forms the covalent LIG-AMP intermediate; motif IV (EGFV[V/I]R) includes a glutamate that coordinates a catalytic divalent cation; motif V (FFKYK) contains two lysines that coordinate the ATP phosphates and the 5′-PO4 of the polynucleotide substrate. The central kinase domain (KIN) contains a classic P-loop motif, GCGKT, that coordinates the β and γ phosphates of the NTP substrate for the polynucleotide kinase reaction (Das et al. 2014). The C-terminal cyclic phosphodiesterase domain (CPD) includes the motifs HVTL and HVTV characteristic of the 2H phosphoesterase superfamily. The histidines serve as general-acid-base catalysts of the conversion of RNA>p to RNA2′p (Remus et al. 2014). Based on the alignment, we split the fungal Trl1 proteins into two fragments at the sites indicated in Figure 4A. The putative N-terminal sealing domains AfuTrl1(1–380) (calculated mass 43.3 kDa) and CimTrl1(1–382) (43.5 kDa) will be referred to henceforth as Afu LIG and Cim LIG. The putative bifunctional healing domains AfuTrl1(381–834) (50.9 kDa) and CimTrl1(383–834) (50.9 kDa) are henceforth Afu KIN-CPD and Cim KIN-CPD.
FIGURE 4.

Separate healing and sealing domains of AfuTrl1 and CimTrl1. (A) Alignment of the primary structures of AfuTrl1 and CimTrl1. Positions of side chain identity/similarity are indicated by (•). Gaps in the alignment are denoted by dashes. Signature active site motifs of the ligase, kinase, and CPDase enzymes are shaded in gray boxes. Forward and reverse arrowheads indicate the boundaries of the N-terminal LIG and C-terminal KIN-CPD domain constructs, respectively. (B) Aliquots (5 µg) of the recombinant AfuTrl1 and CimTrl1 LIG and KIN-CPD preparations were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (kDa) of marker proteins are indicated on the left. (C) Reaction mixtures (20 µL) containing 50 mM Tris–HCl (pH 7.5), 2 mM DTT, 10 mM MgCl2, 100 µM ATP, 100 µM GTP, 20 nM 20-mer HORNA>p (depicted at the top), and 50 nM LIG, KIN-CPD, or full-length (FL) Trl1 proteins (as specified by + below the lanes) were incubated for 5 min at 22°C. The products were analyzed by urea–PAGE. An autoradiograph of the gel is shown. The positions and identities of the HORNA>p substrate (*), 5′-phosphorylated kinase product (5′-P), and ligation products (circle and multimers) are indicated.
Recombinant Afu and Cim LIG and KIN-CPD proteins were produced in E. coli as His10Smt3 fusions, recovered from soluble extracts by Ni-affinity chromatography, treated with Ulp1 to remove the His10Smt3 tag, and further purified via a second Ni-affinity step followed by gel filtration. The recombinant domains were monomeric by gel filtration. SDS–PAGE analysis of the Afu and Cim LIG and KIN-CPD preparations is shown in Figure 4B.
Reaction of the isolated Afu and Cim KIN-CPD domains with the 20-mer HORNA>p substrate resulted in its quantitative conversion to a 5′-PO4 product (Fig. 4C, denoted by the arrowhead). Reaction of HORNA>p with the isolated LIG domains elicited no change in its electrophoretic mobility. Efficient ligation was reconstituted in reaction mixtures containing the LIG and KIN-CPD domains (Fig. 4C).
Afu and Cim polynucleotide kinases prefer GTP as the phosphate donor
We reacted 0.2 µM Afu and Cim KIN-CPD domains with 20 nM 3′ 32P-labeled 10-mer HORNA>p substrate and a divalent cation for 2 min, in the absence of added NTP or in the presence of 0.1, 1, 10, or 100 µM ATP, GTP, CTP, or UTP. The reactions were quenched with EDTA, the products were analyzed by urea–PAGE, and the extents of 5′-phosphorylation were quantified (Fig. 5). No phosphorylation was detected absent an added NTP. Varying the GTP concentration by a factor of 1000 had virtually no effect on the yields of phosphorylated RNA generated by CimTrl1. The fact that the kinase reaction was saturated at 0.1 µM GTP (i.e., fivefold molar excess over the input RNA substrate) attests to the efficiency with which GTP is used as the phosphate donor by CimTrl1. In contrast, the extent of RNA phosphorylation decreased progressively as ATP, CTP, or UTP concentrations were back-titrated from 100 to 0.1 µM NTP (Fig. 5A). Similar results were obtained for AfuTrl1 (Fig. 5B). These experiments show that the GTP donor preference of budding yeast Trl1 kinase, described previously for KlaTrl1 (Remus and Shuman 2014) using the identical kinase substrate and assay format used here, is a feature shared with Trl1 enzymes from two clinically significant human fungal pathogens.
FIGURE 5.
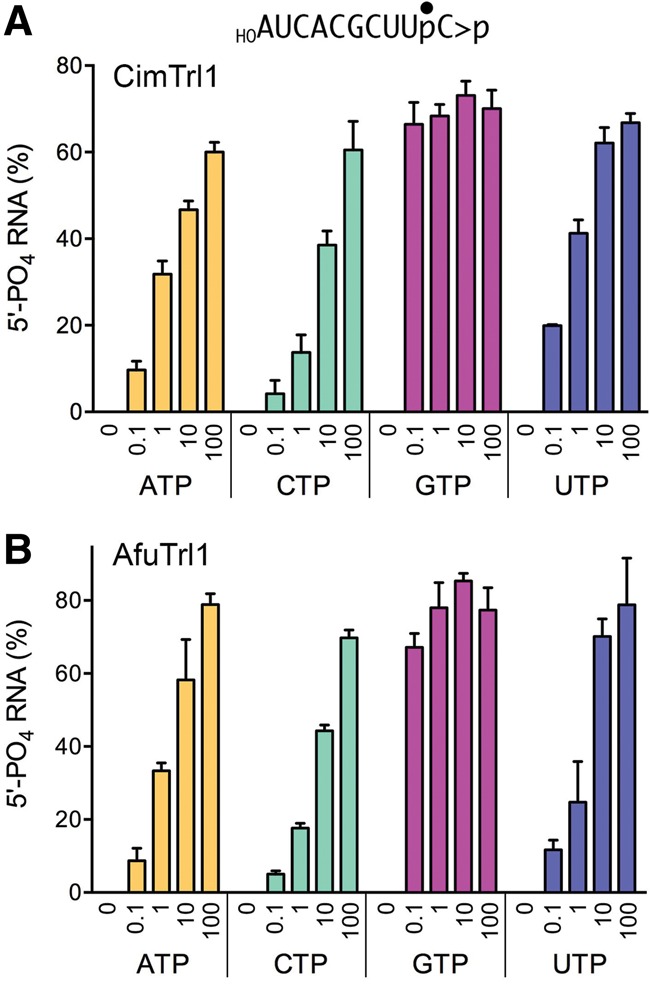
NTP donor specificity of the AfuTrl1 and CimTrl1 5′ kinase reaction. Reaction mixtures (20 µL) containing 50 mM Tris–HCl (pH 7.5), 2 mM DTT, 10 mM MgCl2, 20 nM 10-mer HORNA>p, 200 nM CimTrl1 (panel A) or AfuTrl1 (panel B) KIN-CPD enzyme, and 0, 0.1, 1, 10, or 100 µM of the indicated NTP (ATP, CTP, GTP, or UTP) were incubated at 22°C for 2 min. The products were analyzed by urea–PAGE. The extents of 5′ phosphorylation were quantified by scanning the gel and are plotted in bar graph format as a function of NTP concentration. Each datum is the average of three separate experiments ±S.E.M.
Probing CimTrl1 structure by chemical cross-linking
Chemical cross-linking of proteins in solution followed by mass spectrometry identification of the sites of cross-linking can afford insights into protein folding and a low-resolution map of protein–protein contacts (Yang et al. 2012). We applied this method here to probe cross-linkable sites within and between the catalytic domains of CimTrl1, using a series of homo-bifunctional N-hydroxysuccinimide (NHS) esters that react with lysine side chains. CimTrl1 (1 µM) was incubated with 500 µM disuccinimidyl tartarate (DST; spacer arm 6.4 Å) for 30 min at 22°C. Mass spectrometry after digestion with trypsin identified 893 spectra (hits) corresponding to 308 lysine-to-lysine cross-linked peptides. Of these, 125 cross-links were single-hits. The 834-aa CimTrl1 polypeptide includes 52 lysines, of which 47 were retrieved in at least one cross-linked peptide in the DST data set. Of the five lysines that were not cross-linked by DST (K115, K177, K318, K399, and K667), two are especially informative: K115 is the site of covalent adenylylation in the kinase domain and K399 is the active site lysine in the P-loop of the kinase domain. (An underlying assumption of the cross-linking analysis is that it primarily detects lysine–lysine pairs within a single CimTrl1 enzyme molecule. That this is likely to be the case was suggested by the paucity of self-to-self peptide cross-links in the DST data set. To wit, there were only two self-cross-linked peptides detected: K27–K27 [one hit] and K33–K33 [three hits].) For the purpose of discussion here, we tabulated only those DST cross-links (n = 47) that yielded five or more hits. The cross-links are grouped in Supplemental Figure S1 according to whether they are intradomain involving pairs of lysines within the ligase, kinase, and CPD modules (thereby providing information about the folds of the individual domains) and in Supplemental Figure S2 when they are interdomain.
Cross-linking was also performed using NHS esters with longer spacer arms: disuccinimidyl glutarate (DSG, spacer arm 7.7 Å) and disuccinimidyl suberimidate (DSS, spacer arm 11.4 Å). The DSG data set comprised 956 hits corresponding to 255 cross-linked peptides, of which 92 cross-linked peptides were single-hits. (The DSG data set included five self-to-self cross-linked peptides totaling 13 hits.) The DSS data set consisted of 1429 hits. There were no cross-links to the K115 or K399 active site lysines in the DST and DSS data sets. Supplemental Figures S1 and S2 list the DSG cross-links and DSS cross-links that scored five or more hits.
Cross-links of the CimTrl1 LIG, KIN, and CPD domains
The LIG component of CimTrl1 consists of two subdomains: (i) an adenylyltransferase module (aa 1–297) that extends from the N-terminus to just downstream from motif V, and which is credibly homologous to the equivalent N-terminal segment of T4 RNA ligase 1 (El Omari et al. 2005); and (ii) a flanking C-domain (aa 298–382) that is unique to the Trl1 clade of ATP-dependent ligases. The DST data highlight a network of chemically reactive and cross-linkable pairs of lysines within the adenylyltransferase module involving K27, K32, K33, K50, K57, K87, K155, and K289, which suggests that they are near each other in the Trl1 tertiary structure. The same adenylyltransferase cross-links are prominent in the DSS data set; several are well represented in the DSG data set. Use of the longer spacer NHS esters DSG and DSS revealed a network of cross-links within the adenylyltransferase module involving K57, K98, K155, and K166 and a pairwise cross-link of K235 to K242 that were not prominent in the DST data set (presumably because they were not accessible to the shorter spacer cross-linker). With respect to the internal organization of the LIG C-domain, the cross-linking underscored pairwise proximities of K340 and K360, K360 and K365, and (in the DSS data) K307 and K319, and K316 and K319. It is of particular interest to understand the arrangement of the adenylyltransferase and C-domain modules of the Trl1 LIG relatively to one another. Our results indicate that the two modules are disposed in such a way that permits DSG cross-linking of K57 in the adenylyltransferase to K340 and K360 of the C-module and of K242 in the adenylyltransferase to the same K340 and K360 residues of the C-module.
The bifunctional end-healing domain of CimTrl1 consists of kinase (KIN) (aa 383–638) and cyclic phosphodiesterase (CPD) (aa 629–834) modules that belong to the P-loop phosphotransferase and 2H phosphoesterase enzyme superfamilies, respectively. Our chemical cross-linking data feature a network of pairwise lysine interactions within the KIN domain involving K442, K453, K457, K482, K512, K598, and K606 (Supplemental Fig. S1). Indeed, the K453–K457 cross-link had the highest hit counts in the DSG (n = 79) and DSS (n = 112) data sets, consistent with the close spacing of K453 and K457 in the CimTrl1 primary structure 453KRERK457. (Note that there are two tryptic cleavage sites between K453 and K457, at R454 and R456.) Within the CPD domain, the DST data set included a network of cross-links among K635, K689, K703, and K693 and a pairwise cross-link of K743 to K790 (Supplemental Fig. S1).
Interdomain cross-links can provide clues to the spatial relations of the LIG, KIN, and CPD components of CimTrl1. Whereas many cross-links with ≥5 hits were detected between lysines in the LIG and KIN domains (Supplemental Fig. S2), the distribution of such LIG-to-KIN cross-links differed between the DST and the DSG data sets. DST treatment revealed cross-links to the kinase involving LIG adenylyltransferase module sites (K27, K32, K57, and K155) that were not prevalent in the DSG set; DSG treatment highlighted kinase cross-links to LIG C-domain sites (K319, K322, K340, and K360) that were not well represented in the DST set. Two LIG-KIN cross-links with ≥5 hits were detected with all three agents: K57–K442 and K307–K576.
Cross-linkable contacts of the LIG and CPD domains of CimTrl1 featured LIG sites K155, K242, K340, K360, and CPD sites K632, K673, K790 (Supplemental Fig. S2). Several cross-links between KIN and CPD domains with ≥5 hits were detected, albeit with distinct distributions in the DST and DSG data sets (Supplemental Fig. S2).
Probing AfuTrl1 by chemical cross-linking
Parallel cross-linking and mass spec studies were performed with AfuTrl1. The DSS data set comprised 493 hits corresponding to 182 cross-linked peptides, of which 96 were single-hits and 19 cross-linked peptides had ≥5 hits. The DSG set included 454 hits representing 157 cross-links, of which 73 were single-hits and 22 cross-linked peptides had ≥5 hits. The DST set yielded 424 hits from 157 cross-linked peptides, of which 77 were single-hits and 20 had ≥5 hits. AfuTrl1 has 52 lysines. No cross-links were detected to K117, the site of covalent adenylylation in the AfuTrl1 ligase domain; only one single-hit cross-link was detected to K397, the P-loop lysine of the kinase active site. The AfuTrl1 cross-links with ≥5 hits are compiled in Supplemental Figure S3. In brief, the cross-links were primarily between lysines within the LIG domain of AfuTrl1 and several of these were well represented with all three cross-linking agents. Several cross-links within the KIN domain were detected by DST treatment; only two cross-links with ≥5 hits were noted within the CPD domain. Cross-links between the domains of AfuTrl1 were sparse.
DISCUSSION
The present study extends our knowledge of tRNA splicing enzymology to two fungal pathogens—Aspergillus fumigatus and Coccidioides immitis—and provides a genetic and biochemical foundation for pursuing tRNA ligases as antifungal drug targets. The ability to produce catalytically active recombinant AfuTrl1 and CimTrl1, either as full-length proteins or as autonomous healing and sealing domains, sets the stage for in vitro screening for candidate inhibitors. Budding yeast strains that rely on fungal pathogen tRNA ligases for viability can enable cell-based screening for compounds that selectively block the growth of such strains, but do not impact the growth of a yeast strain that relies on RtcB for tRNA splicing (Tanaka et al. 2011b).
The biochemical properties of CimTrl1 and AfuTrl1 are similar to those of SceTrl1 and KlaTrl1, insofar as the pathogen Trl1 kinases also preferentially utilize GTP as the phosphate donor for the polynucleotide kinase reaction of the tRNA splicing pathway. Thus, the kinase itself might be amenable to selective inhibition. Still, it is the ligase component of fungal Trl1 that holds the most promise as an anti-infective target, in light of the absence of any homolog in mammalian proteomes and its distinctive chemical mechanism requiring a 2′-PO4 end for RNA strand joining.
The lack of a crystal structure of a fungal tRNA ligase is a major knowledge gap in tRNA metabolism and an impediment to inhibitor design and discovery. Attaining an atomic structure of Trl1 will provide key insights into the active site architectures and catalytic mechanisms of the CPD, kinase and ligase enzymes, and the structural basis for their distinctive substrate specificities. Whereas crystallization efforts are being pursued with the AfuTrl1 and CimTrl1 proteins described in this study, we opted in the short term for chemical cross-linking as a low-resolution probe of their structures in solution. The results presented here highlight the most prevalent cross-linked peptides (≥5 hits), which reflect both the accessibility of the involved lysines to the chemical modifier in solution and the accessibility of the cross-linked lysines to each other. To what degree can we draw inferences about shared structural features by comparing the cross-linking patterns of CimTrl1 and AfuTrl1? To this end, we are limited by the fact that many of the cross-links detected involve lysines that are not conserved in the two fungal enzymes. That said, there are 26 lysines that are shared in the CimTrl1 and AfuTrl1 primary structures and a subset of these are involved in prominent pairwise lysine cross-links common to both enzymes. These are located in the LIG domain and include AfuK59–K100/CimK57–K98, AfuK28–K59/CimK27–K57, AfuK59–K89/CimK57–K87, AfuK89–K290/CimK87–K289, and AfuK361–K366/CimK360–K365. The shared intra-LIG cross-links are illustrated in Figure 6.
FIGURE 6.

Shared cross-links within the LIG domains of CimTrl1 and AfuTrl1. Conserved lysines are indicated by ( | ) above the aligned primary structures of the Cim and Afu LIG domains. Nucleotidyltransferase motifs I, IV, and V are highlighted in gold. Shared lysine cross-links are indicated by red boxes and red lines connecting the lysine pairs.
To see whether any of the shared intra-LIG cross-links are structurally plausible, we submitted the amino acid sequences of the Cim and Afu LIG domains to the Phyre2 server (Kelley et al. 2015) for structural modeling. As expected, the top template “hit” was T4 Rnl1 (El Omari et al. 2005; pdb 2C5U) to which Phyre2 generated a tertiary structure model of Cim LIG from aa 10–297 and of Afu LIG from aa 8–299. In the Cim model, the K57 and K87 residues are near neighbors, with a Cα–Cα distance of 8.7 Å, in keeping with the high hit counts for the K57–K87 cross-link in the DST (n = 19), DSG (n = 20), and DSS (n = 44) data sets (Supplemental Fig. S1). In the Afu model, the corresponding K59 and K89 Cα atoms are also 8.7 Å apart, consistent with the prevalence of the K59–K89 cross-link in the DST (n = 18), DSG (n = 6), and DSS (n = 7) data sets (Supplemental Fig. S3). Cim residues K87 and K289 are modeled at a Cα–Cα distance of 11.1 Å, concordant with the presence of the K87–K289 cross-link in the DST (n = 6), DSG (n = 5), and DSS (n = 15) data sets. The equivalent Afu K100 and K292 amino acids are modeled at a Cα–Cα distance of 9.0 Å, consistent with a high hit count for the K89–K290 cross-link in the shortest-spacer DST data set (n = 17). In the Cim model, the K57 and K98 residues are more remotely situated, at a Cα–Cα distance of 15.6 Å, and this agrees with the observed capture of many K57–K98 cross-links by the longest-spacer DSS reagent (n = 44), but not by the shortest spacer DST reagent. In the Afu model, the equivalent K59–K100 pair is at a Cα–Cα distance of 15.1 Å, in accord with the abundance of K59–K100 cross-links in the DSS (n = 44) and DSG (n = 35) data sets, but relatively few hits in the DST data set (n = 6). The modeled Cα–Cα distances for the Cim K27–K57 pair and the Afu K28–K59 pair are 13.4 and 16.7 Å, respectively. Needless to say, validation of particular lysine cross-links as indicators/predictors of Trl1 domain folding and interdomain interactions must await atomic resolution structures of the domains, per se and in the context of full-length Trl1.
MATERIALS AND METHODS
Yeast expression plasmids for fungal tRNA ligases
The ORFs encoding AfuTrl1 (Genbank accession XP_752336), CimTrl1 (Genbank accession XP_001239237), and CalTrl1 (Genbank accession XP_720836) were PCR amplified from A. fumigatus genomic DNA (a gift of Drs. Bing Zhai and Tobias Hohl, MSKCC), C. immitis genomic DNA (a gift of Drs. Chiung-Yu Hung and Gary Cole, UT San Antonio), and C. albicans genomic DNA using primers that introduced a BamHI (AfuTRL1, CalTRL1) or NdeI (CimTRL1) site at the translation start codon and an NheI (AfuTRL1, CimTRL1) or XhoI (CalTRL1) site immediately downstream from the stop codon. The PCR products were digested with the aforementioned restriction enzymes and inserted into yeast expression vectors so as to place them under the transcriptional control of the yeast TPI1 promoter and yield pYN132-AfuTRL1 (CEN TRP1), pYB132-CimTRL1 (CEN TRP1), and pRS415-TPI-CalTRL1 (CEN LEU2). The plasmid inserts were sequenced to verify that no unwanted coding changes were introduced during PCR amplification and cloning.
Saccharomyces cerevisiae trl1Δ complementation by heterologous fungal tRNA ligases
We tested complementation by plasmid shuffle in an S. cerevisiae trl1Δ p(CEN URA3 TRL1) strain, which is unable to grow on medium containing 0.75 mg/mL FOA (5-fluoroorotic acid), a drug that selects against the URA3 plasmid (Sawaya et al. 2003). trl1Δ cells were transfected with CEN TRP1 plasmids bearing SceTRL1, AfuTRL1, or CimTRL1 genes, or a CEN LEU2 plasmid harboring CalTRL1. Individual Trp+ or Leu+ transformants were selected and streaked on agar medium containing FOA. All of the TRL1 transformants gave rise to FOA-resistant colonies after incubation for 3–4 d at 30°C. trl1Δ cells transformed with the empty CEN plasmid vectors failed to give rise to FOA-resistant colonies. Individual FOA-resistant colonies were grown to mid-log phase in YPD broth and adjusted to A600 of 0.1. Aliquots (3 µL) of serial 10-fold dilutions were spotted to YPD agar plates, which were incubated at 20°C, 25°C, 30°C, 34°C, and 37°C.
Trl1 purification
Recombinant full-length AfuTrl1, CimTrl1, and their respective LIG and KIN-CPD domains AfuTrl1(1–380), AfuTrl1(381–834), CimTrl1(1–382), and CimTrl1(383–834) were produced in E. coli as His10Smt3 fusions. pET28b-His10Smt3-Trl1 plasmids were transformed into E. coli BL21-CodonPlus(DE3). Cultures (4-L) derived from single transformants were grown at 37°C in terrific broth medium containing 50 µg/mL kanamycin until A600 reached 0.8. The cultures were chilled on ice for 30 min and then adjusted to 0.4 mM isopropyl β-d-thiogalactoside, 2% (v/v) ethanol. Incubation was continued at 17°C for 16 h with constant shaking. Cells were harvested by centrifugation and stored at −80°C. All subsequent procedures were performed at 4°C. The cell pellet was resuspended in 200 mL buffer A (50 mM Tris–HCl, pH 7.5, 1 M NaCl, 20 mM imidazole, 10% glycerol) and lysozyme was added to 0.2 mg/mL. After 30 min, the lysate was sonicated and insoluble material was removed by centrifugation at 20,000g for 45 min. The supernatant was mixed for 1 h with 10 mL of His60 Ni Superflow resin (QIAGEN) that had been equilibrated in buffer A. The resin was recovered by centrifugation and washed three times with 75 mL of buffer B (50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 10% glycerol) containing 40 mM imidazole, before a final wash step with 75 mL of 50 mM Tris–HCl, pH 7.5, 3 M KCl. The washed resin was then poured into a column. Bound proteins were eluted with 500 mM imidazole in buffer B. The elution profile was monitored by SDS–PAGE. Peak fractions containing His10Smt3-Trl1 proteins were treated with the Smt3-specifc protease Ulp1 (at a His10Smt3-Trl1:Ulp1 ratio of 500:1) during overnight dialysis against buffer B. The dialysate was mixed with 5 mL of His60 Ni Superflow resin that had been equilibrated in buffer B and the mixture was poured into a column. The column was washed with buffer B and bound material was eluted with 500 mM imidazole. The tag-free Trl1 proteins were recovered in the flow-through and wash fractions; the cleaved His10Smt3 tag was bound to the resin and recovered in the imidazole eluate. The Trl1 preparations were concentrated by centrifugal ultrafiltration to ∼10 mg/mL (in 2 mL) and then gel-filtered through a 120-mL 16/60 HiLoad Superdex 200 column equilibrated with buffer C (50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 1 mM DTT, 1 mM EDTA, 10% glycerol) at a flow rate of 1 mL/min, while collecting 5-mL fractions. The peak Trl1-containing fractions were pooled and concentrated by centrifugal ultrafiltration. Protein concentrations were determined by using the Biorad dye reagent with bovine serum albumin as the standard. The yields of the full-length Trl1 proteins were 5–10 mg; the yields of the LIG and KIN-CPD domains were 15 mg.
Preparation of 3′ 32P-labeled RNA>p substrates
HORNA3′p oligonucleotides labeled with 32P at the penultimate phosphate were prepared as described by T4 Rnl1-mediated addition of [5′-32P]pCp to a 19-mer, or 9-mer synthetic oligoribonucleotides. The resulting 20-mer and 10-mer HORNA3′p oligonucleotides were treated with E. coli RNA 3′-terminal phosphate cyclase (RtcA) and ATP to generate the 2′,3′-cyclic phosphate derivatives, HORNA>p, which were gel-purified, eluted from an excised gel slice, and recovered by ethanol precipitation.
Chemical cross-linking
Purified CimTrl1 or AfuTrl1 was dialyzed against buffer containing 50 mM HEPES–KOH (pH 7.5), 100 mM NaCl, and 10% glycerol. NHS esters disuccinimidyl glutarate (DSG), disuccinimidyl tartarate (DST), and disuccinimidyl suberate (DSS) (from Fisher Scientific) were dissolved in 100% DMSO to a final concentration of 100 mM. Cross-linking reactions (20 µL) containing 50 mM HEPES–KOH (pH 7.5), 100 mM NaCl, 10% glycerol, 2 µg (∼1 µM) CimTrl1, 0.5 mM DSG, DST, or DSS, and 10% DMSO were incubated at 22°C for 30 min, then quenched by the addition of 1 µL of 1 M Tris–HCl (pH 7.5). The samples were digested with trypsin and the tryptic peptides were purified using a 2-µL bed volume of Poros 50 R2 (Applied Biosystems) reversed-phase beads packed in Eppendorf gel-loading tips (Erdjument-Bromage et al. 1998; Sebastiaan Winkler et al. 2002).
Mass spectrometry analysis and pLink to identify cross-linked peptides
The digested peptides were diluted to 0.1% formic acid, and each sample was analyzed separately by microcapillary LC with tandem MS by using the NanoAcquity system (Waters) with a 100-µm inner diameter × 10-cm length C18 column (1.7 µm BEH130; Waters) configured with a 180-µm × 2-cm trap column coupled to a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific). A Proxeon nanoelectrospray source set at 1800 V and a 75-µm (with 10-µm orifice) fused silica nano-electrospray needle (New Objective) was used to complete the interface. One microliter of sample was loaded onto the trap column, washed with 3× loop volume of buffer A (0.1% formic acid), and the flow was reversed through the trap column and the peptides eluted with a 1%–50% acetonitrile with 0.1% formic acid gradient over 50 min at a flow rate of 300 nL/min over the C18 analytical column. The mass spectrometer was operated in automatic, data-dependent MS/MS acquisition mode with one MS full scan (370–1700 m/z) at 70,000 mass resolution and up to 10 concurrent MS/MS scans for the 10 most intense peaks selected from each survey scan. Survey scans were acquired in profile mode, and MS/MS scans were acquired in centroid mode at 17,500 resolution and an isolation window of 1.5 m/z. Automatic gain control was set to 1 × 106 for MS1 and 5 × 105 and 100 msec ionization time for MS2. Charge exclusion of 1, 2, 8, and >8 was enabled and dynamic exclusion was set at 15 sec. To analyze the cross-linked peptides, we used pLink (Yang et al. 2012). The raw MS data were analyzed using pLink search with the following parameters: precursor mass tolerance 50 ppm, fragment mass tolerance 10 ppm, cross-linker DSG, DSS, DST, cross-linking sites Lys and protein N terminus, xlink mass-shift 96.021 (DSG),138.068 (DSS), or 113.995 (DST), monolink mass-shift 114.032 (DSG), 156.079 (DSS), or 168.154 (DST), fixed modification Cys 57.02146, variable modification oxidized Met, deamidation Asn/Gln, protein N-acetyl, peptide length minimum four amino acids and maximum 100 amino acids per chain, peptide mass minimum 400 and maximum 10,000 Da per chain, enzyme trypsin, two missed cleavage sites per chain (four per cross-link). The reference protein sequences for CimTrl1 and AfuTrl1 were from Genbank accessions XP_001239237 and XP_752336, respectively.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (National Institute of General Medical Sciences) grant GM42498 (to S.S.). We are grateful to Dr. Ron Hendrickson and Elizabeth Chang of the MSKCC Microchemistry Core Laboratory for performing the mass spectrometry analysis in the Trl1 cross-linking experiments. The MSKCC Core Laboratory is supported by National Cancer Institute grant P30-CA008748.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.057455.116.
REFERENCES
- Apostol BL, Westaway SK, Abelson J, Greer CL. 1991. Deletion analysis of a multifunctional yeast tRNA ligase polypeptide: identification of essential and dispensable functional domains. J Biol Chem 266: 7445–7455. [PubMed] [Google Scholar]
- Chakravarty AK, Shuman S. 2012. The sequential 2′,3′ cyclic phosphodiesterase and 3′-phosphate/5′-OH ligation steps of the RtcB RNA splicing pathway are GTP-dependent. Nucleic Acids Res 40: 8558–8567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty AK, Subbotin R, Chait BT, Shuman S. 2012. RNA ligase RtcB splices 3′-phosphate and 5′-OH ends via covalent RtcB-(histidinyl)-GMP and polynucleotide-(3′)pp(5′)G intermediates. Proc Natl Acad Sci 109: 6072–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagenais TRT, Keller NP. 2009. Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin Microbiol Rev 22: 447–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das U, Wang LK, Smith P, Jacewicz A, Shuman S. 2014. Structures of bacterial polynucleotide kinase in a Michaelis complex with GTP•Mg2+ and 5′-OH oligonucleotide and a product complex with GDP•Mg2+ and 5′-PO4 oligonucleotide reveal a mechanism of general acid-base catalysis and the determinants of phosphoacceptor recognition. Nucleic Acids Res 42: 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai KK, Bingman CA, Phillips GN Jr, Raines RT. 2013. Structure of the noncanonical RNA ligase RtcB reveal the mechanism of histidine guanylylation. Biochemistry 52: 2518–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Omari K, Ren J, Bird LE, Bona MK, Klarmann G, LeGrice SFJ, Stammers DK. 2005. Molecular architecture and ligand recognition determinants for T4 RNA ligase. J Biol Chem 281: 1573–1579. [DOI] [PubMed] [Google Scholar]
- Englert M, Xia S, Okada C, Nakamura A, Tanavde V, Yao M, Eom SH, Koningsberg WH, Söll D, Wang J. 2012. Structural and mechanistic insights into guanylylation of RNA-splicing ligase RtcB joining RNA between 3′-terminal phosphate and 5′-OH. Proc Natl Acad Sci 109: 15235–15240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdjument-Bromage H, Lui M, Lacomis L, Grewal A, Annan RS, McNulty DE, Carr SA, Tempst P. 1998. Examination of micro-tip reversed-phase liquid chromatographic extraction of peptide pools for mass spectrometric analysis. J Chromatogr A 826: 167–181. [DOI] [PubMed] [Google Scholar]
- Goodyear D. 2014. Death dust: The valley-fever menace. The New Yorker (January 20 issue). [Google Scholar]
- Greer CL, Peebles CL, Gegenheimer P, Abelson J. 1983. Mechanism of action of a yeast RNA ligase in tRNA splicing. Cell 32: 537–546. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. 2015. The Phyre2 web portal for protein modeling prediction and analysis. Nat Protoc 10: 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maughan WP, Shuman S. 2015. Characterization of 3′-phosphate RNA ligase paralogs RtcB1, RtcB2, and RtcB3 from Myxococcus xanthus highlights a DNA and RNA 5′-phosphate capping activity of RtcB3. J Bacteriol 197: 3616–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar J, Schwer B, Schaffrath R, Shuman S. 2008. RNA repair: an antidote to cytotoxic eukaryal RNA damage. Mol Cell 31: 278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popow J, Englert M, Weitzer S, Schleiffer A, Mierzwa B, Mechtler K, Trowitzsch S, Will CL, Lürhmann R, Söll D, et al. 2011. HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science 331: 760–764. [DOI] [PubMed] [Google Scholar]
- Remus BS, Shuman S. 2013. A kinetic framework for tRNA ligase and enforcement of a 2′-phosphate requirement for ligation highlights the design logic of an RNA repair machine. RNA 19: 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus BS, Shuman S. 2014. Distinctive kinetics and substrate specificities of plant and fungal tRNA ligases. RNA 20: 462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus BS, Jacewicz A, Shuman S. 2014. Structure and mechanism of E. coli RNA 2′,3′-cyclic phosphodiesterase. RNA 20: 1697–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya R, Schwer B, Shuman S. 2003. Genetic and biochemical analysis of the functional domains of yeast tRNA ligase. J Biol Chem 278: 43928–43938. [DOI] [PubMed] [Google Scholar]
- Schwer B, Sawaya R, Ho CK, Shuman S. 2004. Portability and fidelity of RNA-repair systems. Proc Natl Acad Sci 101: 2788–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiaan Winkler G, Lacomis L, Philip J, Erdjument-Bromage H, Svejstrup JQ, Tempst P. 2002. Isolation and mass spectrometry of transcription factor complexes. Methods 26: 260–269. [DOI] [PubMed] [Google Scholar]
- Stockamp NW, Thompson GR III. 2016. Coccidioidomycosis. Infect Dis Clin North Am 30: 229–246. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Shuman S. 2011. RtcB is the RNA ligase component of an Escherichia coli RNA repair operon. J Biol Chem 286: 7727–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Chakravarty AK, Maughan B, Shuman S. 2011a. A novel mechanism of RNA repair by RtcB via sequential 2′,3′-cyclic phosphodiesterase and 3′-phosphate/5′-hydroxyl ligation reactions. J Biol Chem 286: 43134–43143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Meineke B, Shuman S. 2011b. RtcB, a novel RNA ligase, can catalyze tRNA splicing and HAC1 mRNA splicing in vivo. J Biol Chem 286: 30253–30257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unciuleac MC, Goldgur Y, Shuman S. 2015. Structure and two-metal mechanism of a eukaryal nick-sealing RNA ligase. Proc Natl Acad Sci 112: 13868–13873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Shuman S. 2005. Structure-function analysis of yeast tRNA ligase. RNA 11: 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Schwer B, Englert M, Beier H, Shuman S. 2006. Structure-function analysis of the kinase-CPD domain of yeast tRNA ligase (Trl1) and requirements for complementation of tRNA splicing by a plant Trl1 homolog. Nucleic Acids Res 34: 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westaway SK, Belford HG, Apostol BL, Abelson J, Greer CL. 1993. Novel activity of a yeast ligase deletion polypeptide: evidence for GTP-dependent tRNA splicing. J Biol Chem 268: 2435–2443. [PubMed] [Google Scholar]
- Yang B, Wu YJ, Zhu M, Fan SB, Lin J, Zhang K, Li S, Chi H, Li YX, Chen HF, et al. 2012. Identification of cross-linked peptides from complex samples. Nature Methods 9: 904–906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.