

Abstract
Spliced messages constitute one-fourth of expressed mRNAs in the yeast Saccharomyces cerevisiae, and most mRNAs in metazoans. Splicing requires 5′ splice site (5′SS), branch point (BP), and 3′ splice site (3′SS) elements, but the role of the BP in splicing control is poorly understood because BP identification remains difficult. We developed a high-throughput method, Branch-seq, to map BPs and 5′SSs of isolated RNA lariats. Applied to S. cerevisiae, Branch-seq detected 76% of expressed, annotated BPs and identified a comparable number of novel BPs. We performed RNA-seq to confirm associated 3′SS locations, identifying some 200 novel splice junctions, including an AT-AC intron. We show that several yeast introns use two or even three different BPs, with effects on 3′SS choice, protein coding potential, or RNA stability, and identify novel introns whose splicing changes during meiosis or in response to stress. Together, these findings show unanticipated complexity of splicing in yeast.
Keywords: branch point sequence, pre-mRNA splicing, stress response
INTRODUCTION
Pre-mRNA splicing is required for the expression of most eukaryotic genes and is often regulated. The first step of splicing involves selection of a specific base, usually an adenine, in the pre-mRNA as the branch point (BP) nucleophile and formation of an unusual 2′–5′ RNA linkage between the 2′ OH of the BP and the 5′SS (Wahl et al. 2009). This step is followed by ligation of the two exons and freeing of the intron in the form of a branched lariat (Padgett et al. 1984; Chapman and Boeke 1991). The lariat is rapidly debranched and degraded in most cases (Ruskin and Green 1985; Chapman and Boeke 1991; Friedman and Brewer 1995; Wahl et al. 2009; Awan et al. 2013; Folco and Reed 2014; Qin et al. 2016), making BP identification difficult.
Current BP annotations suggest that Saccharomyces introns almost always have a single BP. However, those annotations are based on computational predictions and lacked a comprehensive experimental basis (Padgett et al. 1984; Chapman and Boeke 1991; Spingola et al. 1999; Meyer et al. 2011) until very recently (Wahl et al. 2009; Awan et al. 2013; Qin et al. 2016). While computational predictions are useful, experimental identification of BP location is essential to understand the full repertoire of splicing decisions cells make. For instance, unusual BP placement far upstream of the second mutually exclusive exon of the mammalian α-tropomyosin gene is known to affect the outcome of splicing of α-tropomyosin, preventing splicing of the intervening intron (Padgett et al. 1984; Smith and Nadal-Ginard 1989; Chapman and Boeke 1991; Qin et al. 2016). BP position can also affect the choice of alternative 3′SS, including “NAGNAG” choice, a common type of alternative splicing (AS) in mammals (Ruskin and Green 1985; Chapman and Boeke 1991; Friedman and Brewer 1995; Bradley et al. 2012; Guo et al. 2012; Awan et al. 2013; Folco and Reed 2014).
Many types of AS involve regulated use of 3′SS (e.g., alternative 3′SS, exon skipping, mutually exclusive exons, alternative last exons, and intron retention). In general, the relative contribution of BP recognition versus 3′SS recognition to each of these types of AS is unknown. In budding yeast, the BP is arguably more critical than the 3′SS to the first step of splicing because the 3′SS does not have to be identified by the splicing machinery until after the first step of splicing (Séraphin and Kandels-Lewis 1993; Spingola et al. 1999; Meyer et al. 2011). In contrast, in metazoans (Aebi et al. 1986; Qin et al. 2016) and S. pombe (Reich et al. 1992; Mercer et al. 2015), recognition of the 3′SS often precedes the first step of splicing. A 3′SS without a BP is not sufficient for splicing, as mutating the annotated BP motif greatly reduces splicing of the transcript in yeast splicing reporters (Vijayraghavan et al. 1986; Rain 1997; Taggart et al. 2012; Awan et al. 2013; Bitton et al. 2014). Similarly, in humans, BP motif mutations can result in aberrant splicing or intron retention, which are associated with several diseases (Královicová et al. 2006).
Regulation of AS in yeast can occur in response to environmental cues. For example, amino acid starvation inhibits splicing of ribosomal protein genes and exposure to other stresses can decrease or increase the splicing of different subsets of genes (Pleiss et al. 2007). In the case of PTC7, a serine/threonine phosphatase, changes in the available carbon source alter AS, producing one protein isoform that localizes to the mitochondria and another that localizes to the nuclear envelope (Juneau et al. 2009). Nonsense-mediated mRNA decay (NMD), a process that degrades mRNAs that contain premature termination codons (PTCs), can alter protein production by degrading mRNAs. Several metazoan splicing factors autoregulate by AS of transcripts from their own loci to shift toward increased production of unstable, NMD-targeted isoforms when protein levels are high (Sureau 2001; Wollerton et al. 2004; Kawashima et al. 2014). NMD also occurs in yeast (Spingola et al. 1999; González et al. 2001) and can be coupled to splicing to regulate gene expression (Kawashima et al. 2014; Stepankiw et al. 2015).
Here we developed Branch-seq, a genome-wide technique to sequence lariat BPs and their associated 5′SS. We tested our method in S. cerevisiae, where every annotated intron has a confident BP prediction (Spingola et al. 1999; Meyer et al. 2011), allowing us to assess the accuracy and sensitivity of our method. Surprisingly, in addition to confirming the locations of most annotated BPs, we also identified many novel BPs and introns, whose splicing patterns we investigated.
RESULTS
Branch-seq accurately identifies locations of 75% of expressed, annotated BPs
Although the yeast genome sequence has been available since 1996 and studies have sought to comprehensively identify yeast introns and test those predictions (Spingola et al. 1999; Davis 2000; Volanakis et al. 2013), genome-wide assays are still discovering additional S. cerevisiae introns (Malone et al. 1991; Zhang et al. 2007; Kawashima et al. 2014; Qin et al. 2016). BP mapping has lagged behind intron detection largely because of the short life span and unique structure of lariat RNAs. BPs are typically verified using fairly laborious, low-throughput techniques such as primer extension, in vitro splicing, and RT-PCR across the lariat 5′SS–BP junction (Padgett et al. 1985; Vogel et al. 1997; Vierstra and Callis 1999), with alternative approaches developed only recently (Parker and Siliciano 1993; Taggart et al. 2012; Awan et al. 2013; Bitton et al. 2014; Mercer et al. 2015; Qin et al. 2016). Until very recently, budding yeast BPs had not been experimentally mapped genome wide (Qin et al. 2016).
To locate BPs, we developed Branch-seq, an untargeted, high-throughput method for identification of lariat BPs and their associated 5′SSs. Initially, lariats were stabilized in vivo by deleting DBR1, the debranching enzyme that linearizes lariats in the default intron decay pathway (Chapman and Boeke 1991). In the first step of Branch-seq, lariats were enriched from dbr1Δ total RNA using denaturing two-dimensional (2D) polyacrylamide gel electrophoresis. Because the mobility of lariat and other circular RNAs is retarded to different extents at different gel densities compared to linear RNA, lariat and circular RNAs run in an arc above the diagonal produced by linear RNAs (Chapman and Boeke 1991; Friedman and Brewer 1995; Beggs 2005; Awan et al. 2013; Stepankiw et al. 2015). A prominent above-diagonal arc was visible in 2D gel analysis of dbr1Δ RNA (Fig. 1A).
FIGURE 1.

Branch-seq accurately identifies BP locations on a genome-wide scale. (A) Schematic of the Branch-seq protocol. Steps labeled with “B” and “L” correspond to Branch-seq and Lariat-seq, respectively. (B) Branch-seq locates the annotated 5′SS (pink) and BP (blue) in the PCH2 intron (Sureau 2001; Wollerton et al. 2004; Robinson et al. 2011). Dashed lines show locations of 5′SS (GTATGT), BP (CACTAAC), and 3′SS (AG) sequences. Mismatches from consensus are underlined. BP nucleotide is red and bold. Mismatches in reads are indicated by small red, green, dark blue, and orange horizontal lines. Inset axes show read start locations for PCH2 intron 5′SS and BP reads where the 0 nt is the 5′SS or BP nucleotide, respectively. (C) Meta 5′SS and BP read start plots as in B but for all annotated 5′SS and BPs. Dotted vertical lines at ±2 nt. (D) Locations of BP peaks called by winBP and GEM-BP relative to annotated BP positions.
RNA was isolated from the top, middle, and bottom portions of the 2D gel arc to enrich for lariats of different sizes (Supplemental Fig. S1A). Lariats were linearized using recombinant DBR1 enzyme followed by preparation of libraries for paired-end Illumina sequencing. This strategy yields read pairs in which the 3′ mapping read corresponds to the BP, and the 5′ mapping read identifies the associated 5′SS, unlike other methods where the BP and 5′SS reads are uncoupled (Qin et al. 2016). The 3′ ends mostly correspond to BPs rather than 3′SSs because the lariat form stabilized in dbr1Δ yeast is one in which the intron sequence 3′ of the BP has been degraded (Supplemental Fig. S1B). To further characterize yeast introns, we performed a version of random hexamer-primed RNA-seq known as “Lariat-seq” (Awan et al. 2013), again using RNA isolated from a 2D gel arc (steps 2L and 3L) (discussed further below).
Branch-seq accurately identified annotated BPs and 5′SSs. Overall, ∼60% of mappable reads corresponded to annotated introns, and ∼75% of expressed yeast introns contained one or more read pairs. As an example, read pairs mapping to the intron of PCH2 are shown (Fig. 1B). The 5′ end reads mostly start exactly at the annotated 5′SS, which matches the /GTATGT yeast consensus, while 3′ end reads mostly start at the presumptive BP of this intron, a CACTAAC sequence near the intron 3′ end (differing only at the underlined C from the yeast BP consensus motif, TACTAAC, where the BP nucleotide is in bold). A meta-analysis of all annotated 5′SSs and BPs confirmed this pattern, with a sharp peak of 5′ end read starts at annotated 5′SSs, and a similarly sharp peak of 3′ end read starts at annotated BPs (Fig. 1C), with one case of support for a BP different than the annotated location (Supplemental Fig. S1F) as also recently reported by Qin and colleagues (Qin et al. 2016). A small secondary peak two bases upstream of the BP in the meta-analysis likely reflects shifted RT priming (Supplemental Fig. S1C,E; Supplemental Methods). These results support the utility of Branch-seq for systematic identification of yeast BPs and associated 5′SSs.
Branch-seq identifies novel BPs and associated 5′SSs
Application of two independent peak calling algorithms to Branch-seq data identified BP locations with high precision, yielding over 200 plausible novel BPs, an unexpectedly large number. First, we used a simple sliding window approach (winBP) to find peaks of high local read density without using any sequence information. Second, we adapted the existing GEM ChIP-seq peak caller (Guo et al. 2012) to identify BP peaks in software called GEM-BP (Supplemental Methods). GEM-BP uses the sharply peaked distribution of read starts at BPs and strong BP motifs in yeast to accurately call peaks. GEM-BP recovered 75% of expressed annotated BPs within 3 nt of their annotated locations, while winBP identified 59% of expressed annotated BPs, including two not found by GEM-BP, with somewhat lower precision (Table 1; Fig. 1D; Supplemental Table S1).
TABLE 1.
Summary of BP peak calling analysis
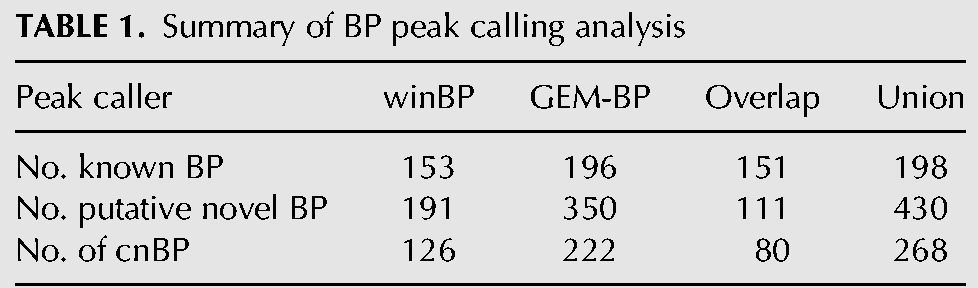
The BP motif is highly constrained in S. cerevisiae, with ∼90% of annotated BPs matching the TACTAAC motif perfectly (Spingola et al. 1999; Miura et al. 2006). Overall, GEM-BP peaks matched the consensus BP motif more frequently than winBP peaks, reflecting GEM-BP's use of a motif in its predictions. To maximize sensitivity, the union of peaks called by both approaches was used, producing a set of 430 putative novel BPs (Supplemental Table S2). We generated a high confidence set of novel BP peaks for further analyses using the paired-end sequencing information from Branch-seq, which provides a built-in quality control for BP identification. Requiring the presence of a typical 5′SS motif in the associated 5′ end reads yielded a set of 268 “confident novel BPs” (cnBPs), with an estimated false discovery rate (FDR) of 1.1% (Fig. 2A; Supplemental Table S3) (see Supplemental Methods for further discussion of possible artifacts), nearly doubling the number of BPs in budding yeast. Surprisingly, in a recent study Qin and colleagues found only 1/6th as many novel BPs, about half of which were also found in this study (Supplemental Methods; Qin et al. 2016). The remaining set of 162 putative novel BPs with atypical 5′SS in our study showed a modest bias toward the /GTATGT consensus (Supplemental Fig. S2B), suggesting the presence of additional novel BPs, but was not pursued further.
FIGURE 2.

Branch-seq locates hundreds of novel BPs. (A) Number of annotated BPs recovered by Branch-seq (light orange) compared to number of computationally predicted BPs (dark orange) (Meyer et al. 2011). The cnBPs (light green) are a subset of all novel BPs (dark green). (B) Genomic locations of the 268 cnBPs. Novel BPs located in CDS (C,D), introns (D), and of the TDA5 and RPL30 genes. Annotated TDA5 BP and 5′SS are blue. Potential AG 3′SS are depicted. 3′SS confirmed by entropy are indicated by asterisk (C,D). Potential BP-3′SS pairing indicated by matching colors (D). (E) Sequence motifs created by MEME of annotated BPs (left) and typical 5′SS cnBP (middle) recovered by Branch-seq and human BP motif (right) for comparison. Position 0 is the BP nucleotide.
Most of the 268 cnBPs were located in annotated exons, introns, or UTRs, but almost one-third were located outside of annotated transcripts, sometimes in regions antisense to annotated genes such as cryptic unstable transcripts (CUTs) and stable uncharacterized transcripts (SUTs) (Fig. 2B; Supplemental Fig. S2C). These observations suggest that Branch-seq can be used to extend annotation of genic as well as nongenic features in yeast. For example, in the second exon of the RPL30 gene, we observed a substantial peak of more than 100 Branch-seq reads at a variant BP motif, GGCTAAC, associated with a potential novel 5′SS, pointing to the presence of a second intron in this gene (Fig. 2C). The 5′ end reads associated with the cnBP in RPL30 began with the sequence GTAAGT, just one mismatch from the yeast 5′SS consensus (Fig. 2C). As another example, we observed three distinct peaks of Branch-seq reads in the intron and second exon of the TDA5 gene. These peaks corresponded to the annotated BP (TACTAAC) and to two other sites downstream in the transcript, which were associated with motifs related to the BP motif by one (AACTAAC) or two (GTCTAAC) mismatches (Fig. 2D). All three of these peaks were paired with reads mapping to the annotated 5′SS. These data suggest that alternative BPs are used in splicing of this intron, likely yielding at least two or three different 3′SS.
Comprehensive BP sequencing allowed us to identify BPs that deviated from the consensus. Of the 268 cnBP, 51 were a perfect match to the TACTAAC consensus motif and the remaining 217 had up to four mismatches, yielding a more degenerate motif when aligned (Fig. 2E). Interestingly, the −1 position (canonically “A” in yeast) appears to also tolerate “G,” as often seen in the mammalian BP motif (Mercer et al. 2015). The −5 to −3 positions also appear more degenerate, with “T” apparently tolerated at the −4 and −3 positions. Overall, the cnBP motif resembles known mammalian BP motifs CTRAC or minimally TNA (R = A or G, N = any base). The weaker cnBP consensus motif might reflect lower levels of splicing (Supplemental Fig. S3C; Supplemental Table S4) or more frequent regulation of novel BPs than of annotated BPs. As a group, the set of cnBPs lacked the peak of sequence conservation seen for the set of annotated BPs, suggesting that these BPs are often specific to S. cerevisiae (Supplemental Fig. S2A).
We compared our approach for BP detection to a recently described approach that used “lariat junction” (LJ) reads that originate from reverse transcription across the 5′SS to BP junction of the lariat (Taggart et al. 2012; Awan et al. 2013; Bitton et al. 2014). For this purpose, we identified Lariat-seq reads that were composed of a pair of segments that mapped near each other but in a discordant order and used the ends of these read segment pairs, incorporating RT errors at the 2′–5′ linkage (Supplemental Fig. S2D), to define 5′SS and BP locations (Fig. 3A; Supplemental Table S5). For example, we detected 23 reads that crossed the 5′SS (GTAAGT) and BP (CACTAAC) of a recently annotated intron in the BDF2 gene (Volanakis et al. 2013), which encodes a transcription factor (Fig. 3B). The yield of LJ reads was two orders of magnitude higher in Lariat-seq data (450 per 106 reads) than in conventional RNA-seq data (5.5 per 106), (Taggart et al. 2012; Awan et al. 2013). LJ reads confirmed 41 annotated BPs and 17 novel BPs (Supplemental Table S5), several of which overlapped with cnBPs identified by Branch-seq (Fig. 3C). Biases in the lariat sizes successfully recovered by each method likely limit the extent of overlap between cnBPs and LJ-supported BPs (Supplemental Fig. S3D).
FIGURE 3.
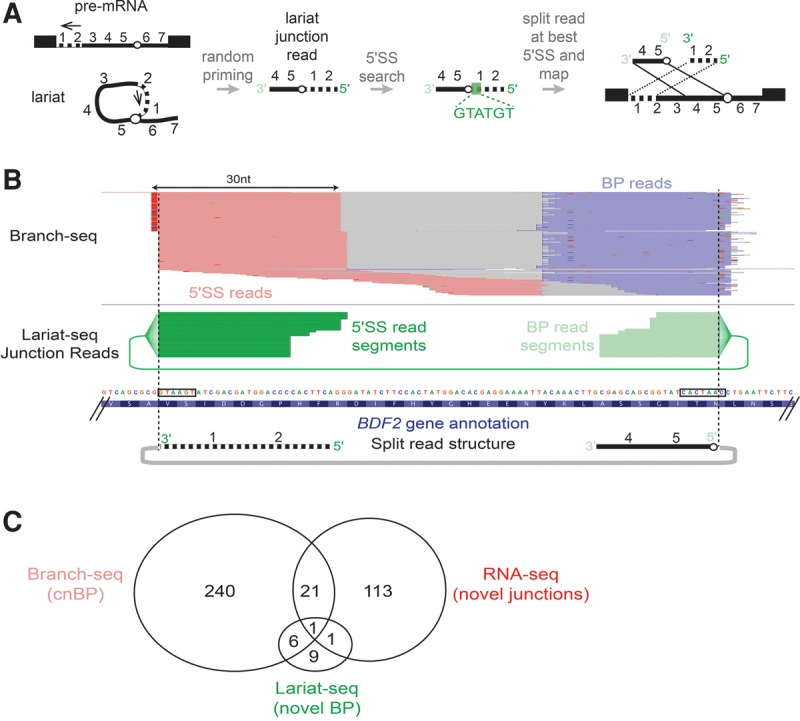
Lariat-set junction reads identify BP locations. (A) Schematic of lariat junction read mapping strategy. Green box indicates location of best 5′SS in lariat junction read. (B) Novel intron in BDF2 CDS is supported by Branch-seq reads (top, pink and blue as in Fig. 1) and Lariat-seq junction reads (middle, 5′SS read fragments in dark green, BP read fragments in light green). Black boxes denote novel 5′SS and BP sequences identified by Branch-seq and Lariat-seq reads. (C) Summary of overlaps among novel BPs identified by Lariat-seq JR reads, cnBP identified by Branch-seq, and novel splice junctions identified by RNA-seq.
Over 100 additional introns and splice sites in the yeast genome
The unexpectedly large number of new BPs identified by our approaches prompted us to further explore yeast splicing patterns using RNA-seq, which is complementary to Branch-seq in that it identifies the 3′SS as well as the 5′SS. We hypothesized that some cnBPs might fall inside unannotated introns that derive from spliced mRNAs that are quickly degraded by NMD. Therefore, we performed RNA-seq on a upf1Δ strain (which is defective for NMD), as well as wild-type (WT) and dbr1Δ strains, and used stringent criteria to define novel splice junctions from the data. Briefly, RNA-seq reads were mapped using TopHat, allowing mapping to novel junctions (Kim et al. 2013). For confident novel splice junction identification, we required relatively uniform read coverage across each exon–exon junction by imposing a minimum splice junction (SJ) entropy of at least two bits (Supplemental Fig. S4A; see Supplemental Methods; Graveley et al. 2011).
This approach yielded 136 unannotated splice junctions, 38 of which were also observed in a recent study (Supplemental Table S6; Kawashima et al. 2014). In all, 115 novel introns overlapped 88 annotated genes. Gene ontology analysis of this set yielded a bias for ribosomal protein genes, a class which is enriched for annotated introns as well (Spingola et al. 1999). Comparing the locations of cnBPs defined by Branch-seq (n = 268) and novel introns defined by RNA-seq (n = 136), we observed a degree of overlap (n = 22) that was significant (P < 0.001, binomial test) but relatively modest in magnitude (Fig. 3C), perhaps because the two protocols have different biases in the transcripts they capture. Novel splice junctions supported by RNA-seq are strongly biased toward shorter genes, while cnBPs are biased toward shorter introns but not toward shorter genes (Supplemental Fig. S4C). Modest overlap between RNA-seq and lariat-based sequencing was also recently observed in S. pombe (Stepankiw et al. 2015). The modest overlap between novel BPs and novel introns could also be explained by stalled splicing transcripts where the first, but not the second, step of splicing has been completed (and thus, no splice junction is created that could be detected by RNA-seq). Although we were unable to find evidence of stalled splicing in our sequencing data (Materials and Methods), it remains a possible explanation for the modest overlap observed.
The novel introns identified by RNA-seq showed several characteristics of known yeast introns. Their length distribution mirrored that of known introns (Fig. 4A). Their splice sites resembled those of annotated introns, but showed more deviation from the consensus, especially at +4 and +6 of the 5′SS, and they lacked a polypyrimidine tract at the 3′SS (Supplemental Fig. S4B), consistent with a recent report (Kawashima et al. 2014). The presence of weaker splice site motifs suggested that splicing of these introns might be less efficient and/or regulated, making detection more difficult. Recent evidence indicates that nuclear RNA decay of alternative isoforms can regulate expression of some genes in yeast, including BDF2 (Volanakis et al. 2013). We analyzed RNA-seq data from nuclear decay-deficient yeast from that study, but did not identify overlaps with the cnBPs we identified.
FIGURE 4.

RNA-seq discovers additional novel introns. (A) Length distribution of annotated (blue) and novel (red) splice junctions. Novel splice junctions include any junction with entropy ≥2 bits. (B) Conservation of splice sites for annotated splice sites (black) and novel splice sites located in annotated CDS (blue), introns (yellow), and outside of ORFs (green). Average predicted BP location for intronic 3′SS is denoted with dotted line, shading is ±1 SD (only plotting −30 to +30 nt around the splice site). For 5′SS, annotated n = 282, CDS n = 14, intron n = 19, intergenic n = 18. For 3′SS, annotated n = 282, CDS n = 34, intron n = 7, intergenic n = 18. (C) Effect on coding length of ORFs from splicing out of novel introns. Predicted change to the coding sequence of REC107 (D) and RUB1 (E) after splicing out novel introns. Red arrow indicates location of RUB1 protein cleavage prior to its addition to substrates. (F) RT-PCR sequence (black) aligns to annotated intron of RPL30 (light blue), SacCer3 genome assembly (Kent 2002). Colored triangles represent splice sites. Gray, annotated splice sites; red, AT-AC 5′SS; orange, AT-AC 3′SS 1; green, AT-AC 3′SS 2. Depending on which AC/ 3′SS is used, the second AUG is either 104 nt or 170 nt downstream from the truncated main ORF. (G) WebLogo of published U2-dependent AT-AC intron 5′SS and 3′SS. RPL30 AT-AC splice sites shown below (Sheth et al. 2006).
New splice sites have distinctive features and conservation patterns
The novel splice junctions identified by RNA-seq were associated with distinct patterns of evolutionary sequence conservation across yeast species compared to annotated introns, indicative of a level of evolutionary constraint that is above background but well below that of annotated introns (Fig. 4B). For novel splice sites that fell inside of annotated coding sequences, a decline in conservation was observed after the 5′SS that was more modest than for annotated introns. This pattern might be expected for an intron that is spliced in some species but retained (or alternatively spliced) in others, giving rise to a conservation pattern that is intermediate between those of typical exons and introns. Novel pairs of splice sites located in annotated introns had low conservation overall (expected in introns), but with elevated conservation ∼20 nt upstream of the 3′SS, in the vicinity of the predicted BP location (shaded yellow, Fig. 4B), supporting that at least some are functionally important.
Often, splicing of the novel introns shortened the predicted protein sequence by at least 50% (Fig. 4C), as in the case of REC107 (Fig. 4D), a gene involved early on in meiotic recombination (Malone et al. 1991). Some smaller changes may also alter function. For example, the spliced mRNA of the ubiquitin-related RUB1 gene encodes a protein differing in the C terminus (Fig. 4E), losing the C-terminal glycine that is used in ligation of RUB1 to its targets (Vierstra and Callis 1999), suggesting that splicing may produce an inactive or dominant negative protein isoform.
AT-AC splice sites are used in yeast
No endogenous /AT-AC/ introns have been reported in yeast, but an engineered /AT-AC/ intron can splice in vivo (Parker and Siliciano 1993). We identified RNA-seq SJ reads supporting the splicing of a novel, endogenous intron nested inside the annotated RPL30 intron, which had an /AT 5′SS that spliced to one of two different AC/ 3′SS, one of which had high SJ entropy (>3 bits). The unconventional AT-AC isoform with distal AC/ 3′SS was supported in the WT, dbr1Δ, and upf1Δ RNA-seq data sets (Supplemental Fig. S4A), and we confirmed both novel AT-AC splice junctions by RT-PCR and sequencing (Fig. 4F; Supplemental Fig. S5). By RNA-seq analysis, AT-AC splicing represents ∼1%–2% of mRNAs from this highly expressed gene locus (see Supplemental Text).
Yeast lack the distinct machinery of the minor spliceosome that splices most known /AT-AC/ introns in metazoans (Russell et al. 2006), and the 5′SS motif of the novel RPL30 introns bore no resemblance to the highly conserved /ATATCCTT consensus typical of animal and plant U12-type AT-AC introns. However, it also deviated quite substantially from the consensus of the few dozen known metazoan U2-type AT-AC introns (Sheth et al. 2006), which have a very strong /ATAAGT consensus (Fig. 4G), raising questions about the mechanism of its splicing (see Discussion).
Multi-BP introns occur in at least 12 genes
Branch-seq revealed 11 unconventional introns that make use of two BPs (Fig. 5A) and one intron that uses three BPs (Fig. 2D). In about half of these cases, the novel BP is located in an intron longer than 200 nt, but is much closer to the 5′SS than the annotated BP (Fig. 5A), consistent with preferential selection of small lariats by Branch-seq (Supplemental Fig. S3A,B,D). In one case a methylation guide snoRNA, snR18, is located between two BPs, and in two other cases an annotated putative ORF occurs between the BPs (Fig. 5B). In the case of snR18, use of the annotated 3′SS and upstream BP would shift the intron-encoded snoRNA from the lariat loop to the lariat tail. For introns with two BPs, the 3′ BP tends to match the consensus better than the 5′ BP (Fig. 5C), analogous to the situation in mammalian genes where upstream alternative cleavage and polyadenylation signals tend to be weaker than competing downstream signals (Tian and Manley 2013). There are additional introns that likely contain multiple BPs but were omitted from Figure 5A because they did not meet the cnBP criteria. Such is the case of MCR1 where a novel BP is paired with a novel 5′SS (GTACGC) that contains two mismatches from the consensus “GTATGT,” creating a novel intron that overlaps the annotated intron but uses alternative 5′ and 3′SS (Fig. 5D).
FIGURE 5.

Alternative BP usage reveals previously unknown nonsense-mediated mRNA decay splice isoform. (A) Distance from 5′SS to BP for first and second BPs in introns that use two BPs. (Red line) x = y. Two BPs in BBP (MSL5) were also reported by Qin et al. (2016) (B) Three genes from A where novel BP (red) is located close to the 5′SS and far from the annotated BP (blue). Intronic transcript position shown below each intron; direction indicated with white arrows. (C) Motif of upstream BP (top) and downstream BP (bottom) for 11 introns that use two BPs. (D) Branch-seq reads support a novel 5′SS-BP shifted pair in the MCR1 intron with an “atypical” 5′SS motif. (E) Branch-seq read coverage from the top, middle, and bottom sections of the 2D gel arc (Supplemental Fig. S1A) correspond to usage of the canonical LSM2 BP (blue dotted line and circle) and a “new” BP (red dotted line and circle). Potential alternative 3′SS usage would insert a premature termination codon (octagon stop sign). (F) RT-PCR and subsequent sequencing confirmed the novel LSM2 PTC isoform. (G) qPCR verification that LSM2 PTC isoform is up-regulated in upf1 null yeast.
Branch-seq identified two BPs in the LSM2 gene (Fig. 5E), which encodes an Sm-like protein that has both nuclear and cytoplasmic functions and plays a role in RNA processing and turnover (Beggs 2005). The novel BP in LSM2, AACTAAC, is upstream of the annotated BP and allows for splicing to a novel 3′SS, located between the annotated and novel BPs (Fig. 5E,F). The novel isoform, which was confirmed by RT-PCR and sequencing (Fig. 5F), contains a PTC in the newly included portion of the downstream exon, making it a potential NMD target (Fig. 5E). Isoform-specific primers used for qRT-PCR showed that the PTC isoform is up-regulated about threefold in upf1Δ yeast, with the annotated isoform remaining unchanged (Fig. 5F,G), implicating NMD in targeting of the novel isoform. Thus, it is possible that AS of the novel isoform is used to regulate the level of LSM2 message and protein.
Some novel introns show altered splicing during meiosis or in stress conditions
To investigate the regulation of novel AS, we mapped RNA-seq data from 18 different environmental growth conditions to annotated and novel exon junctions to assess intron retention (Fig. 6A; Waern and Snyder 2013). MISO (Katz et al. 2010) was used to quantify “percent spliced in” (PSI, representing the fraction of a gene's mRNAs of the longer isoform) across samples. Novel introns generally had low levels of splicing relative to previously annotated introns, with some exceptions (Fig. 6A). Even some novel splice sites that are poorly conserved in other yeast species, such as RPL43B and MTR2 (Supplemental Fig. S6), undergo splicing changes in response to environmental conditions. Overall, we observed increased intron retention during stationary phase as compared to all other environmental conditions analyzed (Fig. 6A). Additionally, growth in salt or juice conditions each have unique effects on the splicing profile. We also observed a group of genes that have substantial but not complete intron retention during most growth conditions (bottom of Fig. 6A).
FIGURE 6.

Rare splicing of novel retained introns mirrors splicing patterns of known introns. (A) Clustering of PSI values calculated by MISO for retained introns in RNA-seq data from 18 environmental conditions (Waern and Snyder 2013), including 136 novel introns. PSI ranges from 0 (complete splicing, purple) to 1 (complete retention, black). (*) Alternative splice site. (**) One splice site overlaps gene ORF listed. (***) Antisense to an annotated transcript. (****) Intron likely in unannotated UTR. (*****) Intron encompasses gene. (YLL056C) 5′UTR supported by RNA-seq. (IDP3) 5′SS inside ORF. (RFU1 and RSB1) 3′SS inside ORF. Conditions are listed in Supplemental Methods. (B) Clustering of PSI of retained introns and alternative splice sites from RNA-seq of a meiosis time course, rapamycin treatment, and deletion strains, including additional novel introns. (C) Clustering of retained intron PSI from ribosome footprint profiling data from a meiosis time course (Brar et al. 2012). (D) Sashimi plot (Katz et al. 2010) depiction of ribosome footprint profiling splice junction reads from B joining YNL194 and YNL195 transcripts at a few stages of meiosis.
To further explore alternative usage of novel introns, we performed additional RNA-seq analysis of a meiosis time course and following rapamycin treatment, conditions under which alternative splicing is known to occur in yeast (Munding et al. 2013), in biological duplicate. We used rRNA depletion (Ribo-Zero) rather than poly(A) selection to avoid the mRNA length biases that appear in poly(A)-selected yeast RNA-seq. In the resulting data, we detected 78 additional novel introns (Supplemental Fig. S4C), of which seven contained cnBPs. One of these novel introns occurs in the gene ESP1, whose protein product separase (aka separin) is required for sister chromatid separation and progression through meiosis (Buonomo et al. 2000). Splicing of this intron, though observed only rarely in the RNA-seq data, was validated by RT-PCR and sequencing and is predicted to alter function of the protein as it removes the catalytic domain. Analyzing intron retention/splicing and alternative splice site usage in these data, we observed 175 splicing events that changed significantly (Bayes factor >5 and absolute change in PSI > 0.1) during meiosis (16 in novel introns, including introns in the RPS29B, OSH2, and HPF1 genes) and 59 that changed following rapamycin treatment (six in novel introns) with an estimated FDR of 7% (Fig. 6B). These data indicate a wider scope of altered splicing in meiosis than previously appreciated (introns and PSI values for all samples/conditions are listed in Supplemental Tables S7–S9).
To assess translation of alternative isoforms, we analyzed ribosome footprint profiling data from a published meiosis time course (Brar et al. 2012), and observed several introns with differential representation in the translatome at different times during meiosis (Fig. 6C,D; Supplemental Fig. S7). These include the YNL194C-YNL195C splice fusion transcript (Miura et al. 2006), and YNL194C, encoding an integral membrane protein required for sporulation (Young et al. 2002). Changes in translation of these novel isoforms during meiosis suggest involvement in meiotic progression (Fig. 6C,D).
DISCUSSION
The Branch-seq approach introduced here allowed comprehensive identification of BPs and associated 5′SS from individual lariat RNAs with high precision (Fig. 1). These BPs revealed splicing events that may have regulatory capacity and could be of interest for further studies to characterize in greater detail. Examples included genes that make use of multiple BPs. For instance, alternative BP usage in the LSM2 pre-mRNA alters 3′SS usage, producing a message that is degraded by NMD, enabling alteration of expression level by modulation of splicing (Fig. 5), reminiscent of regulatory strategies used by a number of metazoan splicing factors (Ruskin and Green 1985; Chapman and Boeke 1991; Sureau 2001; Wollerton et al. 2004; Folco and Reed 2014). We also found that the EFB1 intron contains alternative BP whose use shifts the location of the snoRNA snR18 between the lariat loop and the lariat tail (Fig. 5B), potentially impacting the relative production of mature mRNA and mature snoRNA as seen for other snoRNAs (Spingola et al. 1999; Hirose et al. 2006; Meyer et al. 2011). We observed a significant but modest overlap between our novel BPs from Branch-seq and novel introns from RNA-seq. It is possible that some of the Branch-seq reads derive from stalled splicing products, where the first but not the second step of splicing has occurred.
Some novel introns were spliced at intermediate or even high levels (e.g., those in the RPL22 and RPL43B genes), but most appear to be spliced at lower levels than annotated introns in standard growth conditions (Fig. 6A; Supplemental Tables S7, S8). Considering all introns, we observed a large increase in intron retention during stationary growth that affected most known introns, as well as some novel introns such as the one in RPL43B (Fig. 6A; Supplemental Tables S7, S8). Increased retention of many introns was observed in salt stress, but most novel introns maintained relatively low levels of splicing across most of the environmental conditions examined. However, some novel introns, like PDC1, showed large changes in splicing during meiosis (Fig. 6C; Supplemental Table S9). Together, these patterns indicate that both global splicing changes and highly condition- and intron-specific changes in splicing occur in yeast.
Defining novel splice junctions from RNA-seq led to us find and validate an intron with AT-AC splice site dinucleotides, nested inside the annotated intron of RPL30. To our knowledge, this is the first AT-AC splice site intron reported in S. cerevisiae. Yeast lack the U12-type “minor” spliceosomal machinery, ruling out involvement of the minor spliceosome (Russell et al. 2006). The RPL30 /AT 5′SS does not resemble typical /AT 5′SS spliced by the major spliceosome (Fig. 4G), leaving open the question of whether this intron is indeed spliced by the major spliceosome or by some other mechanism (e.g., a protein enzyme or RNA-based self-splicing).
Methods such as Branch-seq provide an advantage over other methods to locate BPs that require either prior knowledge of 5′SS and 3′SS locations or sequencing across the length of an intron (Smith and Nadal-Ginard 1989; Taggart et al. 2012; Awan et al. 2013; Bitton et al. 2014; Mercer et al. 2015; Stepankiw et al. 2015). A recent study by Qin and colleagues developed another untargeted approach, LIT-seq, to sequence 5′SS and BP of yeast introns (Qin et al. 2016). While both Branch-seq and LIT-seq identified similar numbers of annotated branch points (198 and 238, respectively), there are key differences between the approaches and the results. In the LIT-seq protocol the genomically templated portion of the reads are limited to 21 nt, while longer reads can be used in Branch-seq. Possibly related to this, the fraction of reads mapping to introns was quite a bit lower for LIT-seq (25%) than for Branch-seq (60%). Furthermore, Branch-seq produces paired 5′SS-BP reads, keeping the information in a single lariat intact while 5′SS-BP pairings are lost in LIT-seq due to the uncoupling of the lariat termini during library construction. The higher mapping rate and 5′SS-BP pair information allowed identification of roughly six times as many confident novel BPs by Branch-seq than LIT-seq. However, because the current Branch-seq protocol uses a poly(A) tailing step, A-tailing of lariats could be observed by LIT-seq but not by Branch-seq, and LIT-seq is applicable to WT yeast. Thus, the two approaches have different strengths and weaknesses and may be complementary in some respects.
Recently, recurrent mutations in several core spliceosome components that recognize BP and intron 3′ ends, including U2 snRNP component SF3B1 and the U2AF1 and U2AF2 genes, have been observed in leukemias (Séraphin and Kandels-Lewis 1993; Quesada et al. 2012), raising interest in understanding details of BP and 3′SS recognition. Branch-seq is a powerful method for detection of BPs in small lariats and could be used to study the effects of perturbations of the core splicing machinery on BP selection by crossing mutants analogous to those seen in leukemia to dbr1Δ yeast. Applying Branch-seq to other organisms with small introns such as other fungi, plants or Drosophila could aid in detection of novel introns or regulatory mechanisms, such as recursive splicing (Aebi et al. 1986; Burnette et al. 2005) or stalled splicing (Reich et al. 1992; Dumesic et al. 2013).
MATERIALS AND METHODS
Yeast strains
Strains were grown in YPD (1% Yeast extract, 2% Peptone, 0.01% Adenine hemisulfate, and 2% Dextrose) at 30°C with vigorous shaking unless otherwise noted. The null strains were obtained from the deletion collection. WT yeast were treated with rapamycin as described (Vijayraghavan et al. 1986; Rain 1997; Munding et al. 2013) and for 1 h. WT (s288c): BY4742 Mat α his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0. dbr1Δ:BY4742 Mat α his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 YLK149C::KanMX4. upf1Δ: BY4742 Mat α his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 YMR080C::Kan. Meiotic time courses were performed as described (Berchowitz et al. 2013; see also Supplemental Methods).
2D PAGE gels
For all 2D polyacrylamide gels, RNA was mixed with an equal volume of denaturing loading dye and heated at 80°C–95°C prior to loading. For the Branch-seq gels, ultrapure sequagel reagents from National Diagnostics were used to pour 6% (first dimension: D1) and 20% (second dimension: D2) gels. These gels were poured with 1.5 mm spacers and ∼20 cm by ∼32 cm plates and a metal heat sink was used. D1 was poured the night before with 12 wells and stored at 4°C in Saran wrap to maintain moisture. D1 was run at 15W for 1 h and 45 min, stained with sybr gold, and imaged on a Safe Light. D2 was poured while D1 was running using a comb with one large well. After removing the D2 comb, running buffer (TBE) was added to the well to aid in D1 gel insertion. A single lane of the D1 gels was cut out with a clean razor and slid into the D2 gel using tweezers and a razor blade, taking care to minimize the number of air bubbles between the D1 and D2 gel interface. Additional loading dye was added on top of the D1 gel slice in the D2 gel for easy visualization of running of the D2 gel. D2 was run at 30W for 6 h and 30 min. Gels were stained with Sybr gold. Slices were excised, and in the case of Supplemental Fig. S1A, were frozen at −20°C. The gels used for Lariat-seq were precast mini gels from Invitrogen where D1: 6% and D2: 10% where the wells were manually cut out to make one large well. RNA was eluted from 2D gels using 12 mL PAGE elution buffer (30 mM Tris–HCl [pH 7.5], 300 mM NaCl, and 3 mM EDTA) (Ooi et al. 2001; Pleiss et al. 2007) and rotated overnight at 4°C. RNA was precipitated with isopropanol and glycogen.
Debranching and poly(A) tailing
Debranching was performed similar to http://www.ncbi.nlm.nih.gov/pubmed/11586896 protocol. Briefly, RNA and debranching enzymes were incubated for 1 h at 30°C in debranching buffer (5× debranching buffer: 100 nM Hepes, 625 mM KCl, 2.5 mM MgCl2, 5 mM DTT, 50% glycerol). Prior to debranching of the top, middle, and bottom fractions of lariats, radiolabeled FTZ lariat RNA was spiked into each sample to confirm debranching via gel electrophoresis (see Supplemental Methods). Samples were phenol chloroform extracted after debranching and ethanol precipitated. Debranched lariat RNA was poly(A) tailed using Escherichia coli poly(A) polymerase from NEB for 10 min at 37°C and subsequently phenol chloroform extracted and isopropanol precipitated.
RNA isolation, library preparation, and sequencing
RNA isolation with TRIzol and library preparation for Illumina sequencing were performed as described in Supplemental Methods.
Stalled splicing analysis
We looked for evidence of stalled splicing in our data by comparing the read density at the 5′SS-intron boundary to the read density in the lariat tail in our WT RNA-seq data. Using the 3′SS-exon boundary instead of the lariat tail would have been preferable, but given we did not know the location of the 3′SS for many of the novel BPs, that approach was impractical. We reasoned that stalled splicing should yield a higher relative density of lariat tail reads to 5′SS-intron boundary reads, and this comparison should be able to distinguish pre-mRNAs, stalled splicing intermediates, and spliced mRNAs from each other. However, we did not observe evidence that this pattern reached statistical significance, even for the BDF2 transcript, which has been previously reported as an example of stalled splicing by Volanakis et al. (2013). As a result, we cannot rule out the possibility that stalled splicing is occurring on the transcripts that contain novel BPs.
DATA DEPOSITION
The data from this study have been submitted to the NCBI Gene Expression Omnibus under accession GSE68022. GEM-BP (GEM 2.6) software can be downloaded from http://cgs.csail.mit.edu/gem/. GEM-BP source code can be found in the Supplemental Material and downloaded from https://github.com/gifford-lab/GEM. Code to find BPs from lariat junction reads can be found in the Supplemental Material and downloaded from https://github.com/jpaggi/findbps.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Andy Berglund for initial ideas related to Branch-seq, the R. Reed laboratory for coupled in vitro splicing and translation reagents and protocols used in development of Branch-seq, and Stephen Bell for assistance with the meiosis time course experiments. We thank Yarden Katz for primer design assistance, Shijie Zhao for sequence analyses, Daniel Dominguez for validating the ESP1 intron, David Weinberg for personal communication, Josh Arribere for sporulating dbr1Δ yeast, the R. Sauer and J. King laboratories for assistance with protein purification, Thomas Hansen, Angela Brooks, and members of the Burge laboratory for helpful discussions, and the MIT BioMicro Center for sequencing assistance. This work was supported by a National Institutes of Health training grant, a National Science Foundation equipment grant (no. 0821391), and a research grant from the National Institutes of Health (C.B.B.).
Author contributions: G.M.G., E.T.W., and C.B.B. designed the study. D.V.P. performed the meiosis time course experiments; G.M.G. performed all other experiments. G.M.G. and J.M.P. analyzed the data. Y.G. and D.G. contributed to analysis tools. B.Z. processed the ribosome footprint data. G.M.G., J.M.P., Y.G., B.Z., W.V.G., and C.B.B. wrote the manuscript.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.057216.116.
REFERENCES
- Aebi M, Hornig H, Padgett RA, Reiser J, Weissmann C. 1986. Sequence requirements for splicing of higher eukaryotic nuclear pre-mRNA. Cell 47: 555–565. [DOI] [PubMed] [Google Scholar]
- Awan AR, Manfredo A, Pleiss JA. 2013. Lariat sequencing in a unicellular yeast identifies regulated alternative splicing of exons that are evolutionarily conserved with humans. Proc Natl Acad Sci 110: 12762–12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs JD. 2005. Lsm proteins and RNA processing. Biochem Soc Trans 33: 433–438. [DOI] [PubMed] [Google Scholar]
- Berchowitz LE, Gajadhar AS, van Werven FJ, De Rosa AA, Samoylova ML, Brar GA, Xu Y, Xiao C, Futcher B, Weissman JS, et al. 2013. A developmentally regulated translational control pathway establishes the meiotic chromosome segregation pattern. Genes Dev 27: 2147–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitton DA, Rallis C, Jeffares DC, Smith GC, Chen YYC, Codlin S, Marguerat S, Bahler J. 2014. LaSSO, a strategy for genome-wide mapping of intronic lariats and branch points using RNA-seq. Genome Res 24: 1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley RK, Merkin J, Lambert NJ, Burge CB. 2012. Alternative splicing of RNA triplets is often regulated and accelerates proteome evolution. PLoS Biol 10: e1001229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS. 2012. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335: 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonomo SB, Clyne RK, Fuchs J, Loidl J, Uhlmann F, Nasmyth K. 2000. Disjunction of homologous chromosomes in meiosis I depends on proteolytic cleavage of the meiotic cohesin Rec8 by separin. Cell 103: 387–398. [DOI] [PubMed] [Google Scholar]
- Burnette JM, Miyamoto-Sato E, Schaub MA, Conklin J, Lopez AJ. 2005. Subdivision of large introns in Drosophila by recursive splicing at nonexonic elements. Genetics 170: 661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman KB, Boeke JD. 1991. Isolation and characterization of the gene encoding yeast debranching enzyme. Cell 65: 483–492. [DOI] [PubMed] [Google Scholar]
- Davis CA. 2000. Test of intron predictions reveals novel splice sites, alternatively spliced mRNAs and new introns in meiotically regulated genes of yeast. Nucleic Acids Res 28: 1700–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic PA, Natarajan P, Chen C, Drinnenberg IA, Schiller BJ, Thompson J, Moresco JJ, Yates JR, Bartel DP, Madhani HD. 2013. Stalled spliceosomes are a signal for RNAi-mediated genome defense. Cell 152: 957–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folco EG, Reed R. 2014. In vitro systems for coupling RNAP II transcription to splicing and polyadenylation. Methods Mol Biol 1126: 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman KL, Brewer BJ. 1995. Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol 262: 613–627. [DOI] [PubMed] [Google Scholar]
- González CI, Wang W, Peltz SW. 2001. Nonsense-mediated mRNA decay in Saccharomyces cerevisiae: a quality control mechanism that degrades transcripts harboring premature termination codons. Cold Spring Harb Symp Quant Biol 66: 321–328. [DOI] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Mahony S, Gifford DK. 2012. High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput Biol 8: e1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose T, Ideue T, Nagai M, Hagiwara M, Shu M-D, Steitz JA. 2006. A spliceosomal intron binding protein, IBP160, links position-dependent assembly of intron-encoded box C/D snoRNP to pre-mRNA splicing. Mol Cell 23: 673–684. [DOI] [PubMed] [Google Scholar]
- Juneau K, Nislow C, Davis RW. 2009. Alternative splicing of PTC7 in Saccharomyces cerevisiae determines protein localization. Genetics 183: 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB. 2010. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7: 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Douglass S, Gabunilas J, Pellegrini M, Chanfreau GF. 2014. Widespread use of non-productive alternative splice sites in Saccharomyces cerevisiae. PLoS Genet 10: e1004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Královicová J, Lei H, Vorechovský I. 2006. Phenotypic consequences of branch point substitutions. Hum Mutat 27: 803–813. [DOI] [PubMed] [Google Scholar]
- Malone RE, Bullard S, Hermiston M, Rieger R, Cool M, Galbraith A. 1991. Isolation of mutants defective in early steps of meiotic recombination in the yeast Saccharomyces cerevisiae. Genetics 128: 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Clark MB, Andersen SB, Brunck ME, Haerty W, Crawford J, Taft RJ, Nielsen LK, Dinger ME, Mattick JS. 2015. Genome-wide discovery of human splicing branchpoints. Genome Res 25: 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Plass M, Pérez-Valle J, Eyras E, Vilardell J. 2011. Deciphering 3′ss selection in the yeast genome reveals an RNA thermosensor that mediates alternative splicing. Mol Cell 43: 1033–1039. [DOI] [PubMed] [Google Scholar]
- Miura F, Kawaguchi N, Sese J, Toyoda A, Hattori M, Morishita S, Ito T. 2006. A large-scale full-length cDNA analysis to explore the budding yeast transcriptome. Proc Natl Acad Sci 103: 17846–17851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munding EM, Shiue L, Katzman S, Donohue JP, Ares M. 2013. Competition between pre-mRNAs for the splicing machinery drives global regulation of splicing. Mol Cell 51: 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi SL, Dann C, Nam K, Leahy DJ, Damha MJ, Boeke JD. 2001. RNA lariat debranching enzyme. Methods Enzymol 342: 233–248. [DOI] [PubMed] [Google Scholar]
- Padgett RA, Konarska MM, Grabowski PJ, Hardy SF, Sharp PA. 1984. Lariat RNA's as intermediates and products in the splicing of messenger RNA precursors. Science 225: 898–903. [DOI] [PubMed] [Google Scholar]
- Padgett RA, Konarska MM, Aebi M, Hornig H, Weissmann C, Sharp PA. 1985. Nonconsensus branch-site sequences in the in vitro splicing of transcripts of mutant rabbit β-globin genes. Proc Acad Natl Sci 82: 8349–8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Siliciano PG. 1993. Evidence for an essential non-Watson-Crick interaction between the first and last nucleotides of a nuclear pre-mRNA intron. Nature 361: 660–662. [DOI] [PubMed] [Google Scholar]
- Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. 2007. Rapid, transcript-specific changes in splicing in response to environmental stress. Mol Cell 27: 928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin D, Huang L, Wlodaver A, Andrade J, Staley JP. 2016. Sequencing of lariat termini in S. cerevisiae reveals 5′ splice sites, branch points, and novel splicing events. RNA 22: 237–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, Ramsay AJ, Beà S, Pinyol M, Martínez-Trillos A, et al. 2012. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 44: 47–52. [DOI] [PubMed] [Google Scholar]
- Rain JC. 1997. In vivo commitment to splicing in yeast involves the nucleotide upstream from the branch site conserved sequence and the Mud2 protein. EMBO J 16: 1759–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich CI, VanHoy RW, Porter GL, Wise JA. 1992. Mutations at the 3′ splice site can be suppressed by compensatory base changes in U1 snRNA in fission yeast. Cell 69: 1159–1169. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin B, Green MR. 1985. An RNA processing activity that debranches RNA lariats. Science 229: 135–140. [DOI] [PubMed] [Google Scholar]
- Russell AG, Charette JM, Spencer DF, Gray MW. 2006. An early evolutionary origin for the minor spliceosome. Nature 443: 863–866. [DOI] [PubMed] [Google Scholar]
- Séraphin B, Kandels-Lewis S. 1993. 3′ splice site recognition in S. cerevisiae does not require base pairing with U1 snRNA. Cell 73: 803–812. [DOI] [PubMed] [Google Scholar]
- Sheth N, Roca X, Hastings ML, Roeder T, Krainer AR, Sachidanandam R. 2006. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res 34: 3955–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CW, Nadal-Ginard B. 1989. Mutually exclusive splicing of alpha-tropomyosin exons enforced by an unusual lariat branch point location: implications for constitutive splicing. Cell 56: 749–758. [DOI] [PubMed] [Google Scholar]
- Spingola M, Grate L, Haussler D, Ares M. 1999. Genome-wide bioinformatic and molecular analysis of introns in Saccharomyces cerevisiae. RNA 5: 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepankiw N, Raghavan M, Fogarty EA, Grimson A, Pleiss JA. 2015. Widespread alternative and aberrant splicing revealed by lariat sequencing. Nucleic Acids Res 43: 8488–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureau A. 2001. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J 20: 1785–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart AJ, DeSimone AM, Shih JS, Filloux ME, Fairbrother WG. 2012. Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat Struct Mol Biol 19: 719–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Manley JL. 2013. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci 38: 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra RD, Callis J. 1999. Polypeptide tags, ubiquitous modifiers for plant protein regulation. Plant Mol Biol 41: 435–442. [DOI] [PubMed] [Google Scholar]
- Vijayraghavan U, Parker R, Tamm J, Iimura Y, Rossi J, Abelson J, Guthrie C. 1986. Mutations in conserved intron sequences affect multiple steps in the yeast splicing pathway, particularly assembly of the spliceosome. EMBO J 5: 1683–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J, Hess WR, Börner T. 1997. Precise branch point mapping and quantification of splicing intermediates. Nucleic Acids Res 25: 2030–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volanakis A, Passoni M, Hector RD, Shah S, Kilchert C, Granneman S, Vasiljeva L. 2013. Spliceosome-mediated decay (SMD) regulates expression of nonintronic genes in budding yeast. Genes Dev 27: 2025–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waern K, Snyder M. 2013. Extensive transcript diversity and novel upstream open reading frame regulation in yeast. G3 (Bethesda) 3: 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, Smith CWJ. 2004. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol Cell 13: 91–100. [DOI] [PubMed] [Google Scholar]
- Young ME, Karpova TS, Brugger B, Moschenross DM, Wang GK, Schneiter R, Wieland FT, Cooper JA. 2002. The Sur7p family defines novel cortical domains in Saccharomyces cerevisiae, affects sphingolipid metabolism, and is involved in sporulation. Mol Cell Biol 22: 927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Hesselberth JR, Fields S. 2007. Genome-wide identification of spliced introns using a tiling microarray. Genome Res 17: 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.