

Abstract
The rpsO-pnp operon encodes ribosomal protein S15 and polynucleotide phosphorylase, a major 3′–5′ exoribonuclease involved in mRNA decay in Escherichia coli. The gene for the SraG small RNA is located between the coding regions of the rpsO and pnp genes, and it is transcribed in the opposite direction relative to the two genes. No function has been assigned to SraG. Multiple levels of post-transcriptional regulation have been demonstrated for the rpsO-pnp operon. Here we show that SraG is a new factor affecting pnp expression. SraG overexpression results in a reduction of pnp expression and a destabilization of pnp mRNA; in contrast, inhibition of SraG transcription results in a higher level of the pnp transcript. Furthermore, in vitro experiments indicate that SraG inhibits translation initiation of pnp. Together, these observations demonstrate that SraG participates in the post-transcriptional control of pnp by a direct antisense interaction between SraG and PNPase RNAs. Our data reveal a new level of regulation in the expression of this major exoribonuclease.
Keywords: antisense RNA, PNPase, post-transcriptional regulation, SraG
INTRODUCTION
Polynucleotide phosphorylase (PNPase) is a 3′–5′ exoribonuclease that is conserved from bacteria to plants and metazoans (Zuo and Deutscher 2001). PNPase plays many roles in RNA metabolism. Together with RNase II, it is responsible for eliminating the small nonfunctional RNA fragments generated by endonucleases (Andrade et al. 2009). In addition, it is required in many RNA processing reactions to generate mature transcripts and in mRNA surveillance mechanisms to eliminate aberrant transcripts (Braun et al. 1996; Reuven et al. 1997). PNPase is a phosphorylase that uses a phosphate residue to attack the single-stranded 3′-end of RNAs and to liberate a nucleoside-diphosphate. Alone it is capable of degrading secondary structures of moderate stability that are resistant to RNase II. PNPase exists as a homotrimeric complex, and it is part of the degradosome in Escherichia coli (Carpousis et al. 1994). Its association with the RhlB helicase, another component of the degradosome, allows PNPase to disrupt very stable secondary structures that cannot be degraded by the free enzyme in E. coli (Régnier and Hajnsdorf 2009). PNPase degrades certain small RNAs (sRNA) (Andrade and Arraiano 2008; Andrade et al. 2012), but it can also interact with sRNAs in a nondestructive mode together with Hfq in E. coli or in complex with Rsr protein in Deinococcus radiodurans, where Y RNA act as adaptors between the two proteins (Chen et al. 2013; Bandyra et al. 2016).
PNPase also has a template-independent 3′-oligonucleotide polymerase activity in vitro (Grunberg-Manago et al. 1955). PNPase activity is dependent on the metabolic state of the cell since it is inhibited by magnesium-chelated citrate and ATP, while it is activated by c-di-GMP (Del Favero et al. 2008; Nurmohamed et al. 2011; Tuckerman et al. 2011).
PNPase is not necessary for growth at optimal temperatures, but it is essential for growth of E. coli, Bacillus subtilis, and Yersinia enterocolitica at low temperatures (Luttinger et al. 1996; Goverde et al. 1998; Zangrossi et al. 2000). PNPase is a cold-shock protein (CSP) that is required for selective degradation of CSP mRNAs, allowing the transition from the acclimatization phase to cell growth resumption after cold shock (Yamanaka and Inouye 2001). PNPase-deficient cells are less viable than wild-type to H2O2-mediated killing (Wu et al. 2009). It can also be noted that PNPase is necessary for bacterial virulence in some well-known pathogens, including the Salmonellae and the Yersiniae (Clements et al. 2002; Rosenzweig et al. 2007; Numata et al. 2014). In addition, it has been proposed that the pool of nucleoside diphosphates generated by PNPase-mediated degradation of RNA plays an important role in the appearance of spontaneous mutations in E. coli (Becket et al. 2012).
The pnp gene is part of the rpsO-pnp operon (Fig. 1A). Transcription from a promoter (P1), upstream of rpsO, encoding S15, generates two transcripts: a monocistronic rpsO transcript, which terminates at a Rho-independent terminator (T1) between the two open reading frames (ORFs), and a polycistronic rpsO-pnp transcript, which terminates at T2 downstream from the pnp gene (Fig. 1A). In addition, a monocistronic pnp mRNA is initiated at a promoter (P2) located downstream from the rpsO terminator (Fig. 1A). The rpsO-pnp intergenic region is crucial for the expression and stability of the rpsO and pnp transcripts.
FIGURE 1.

Two forms of SraG RNA. (A) Genomic context of sraG. The gene encoding SraG (shaded in gray) is located between the coding regions of the rpsO and pnp genes (encoding ribosomal protein S15 and PNPase, respectively). The sraG and pnp genes are expressed divergently but the 5′-ends of these transcripts overlap. Promoters P1, P2, and P, as well as terminators T1, T2, and T for rpsO, pnp, and sraG, respectively, are indicated. Note that T1 and T are the same bidirectional terminator. (B) Two forms of SraG. Wild-type N3433 and bacteria lacking SraG (mutation in the −10 sequence of SraG promoter [PSraGmut] identified in this study) were harvested in exponential and stationary phase and RNA extracted and separated on an acrylamide gel. Northern blots were probed to detect SraG RNA using an antisense RNA probe equivalent to positions 43–179 of the full size SraG identified here and to 5S RNA. Size of the SraG transcripts was estimated by comparison with radiolabeled markers. (C) Schematic representation of the processing of the rpsO and pnp primary transcripts. The monocistronic rpsO, pnp (1), and sraG (3) transcripts and the dicistronic rpsO-pnp mRNA (2) are shown with the black circle indicating their 5′-triphosphate extremities corresponding to P1 (rpsO promoter), P2 (pnp promoter), and P (sraG promoter). Processing sites by RNase E (RE) and RNase III (RIII) on the primary transcripts are indicated by gray and black scissors, respectively. Data on SraG are from experiments shown in Supplemental Figure S5A–C. Potential hairpins within the transcripts are shown: T1 and T2 are the rpsO and pnp terminators, and R is the stem–loop in the 5′-region of pnp subject to RNase III cleavage. (D) SraG primary transcript is 216 nt long. The sequence corresponds to the (+) strand (as shown in A) from the beginning of the pnp ORF to the rpsO termination codon. The sequence highlighted in gray corresponds to the SraG primary transcript identified by cRT-PCR. A total of 76 RT-PCR sraG clones were sequenced. Vertical arrows denote extremities located by cRT-PCR. Arrows in boxes 1 and 2 correspond to the positions of 5′-ends and in boxes 3 and 4 to 3′-extremities. In most cases, numerous clones were detected at each single position. The transcription initiation site (underlined T in box 1) was only detected in samples after polyphosphatase treatment (see Supplemental Fig. S2). Both the previously annotated 5′-extremity and the newly described +1 transcription start site are shown in bold and underlined. The −10 and the −35 sequences are underlined. The 3′-extremity of the terminator, which is the major 3′-extremity, is shown in bold and underlined, but a few clones harbor 3′-ends downstream, indicating that some read-through occurs. The 190-nt long RNA should correspond to a population of RNA molecules, with different 5′- and 3′-extremities. While a fraction of these RNA fragments, which starts at PSraG or very close (panel D, box 1), have their 3′-ends in box 3 (176- to 185-nt long fragments), the rest of the shorter SraG transcripts could correspond to a 5′-end in box 2 (panel D) and a stop at the terminator (174- to 189-nt long fragments) (panel D, box 4). These RNA fragments of ∼190 nt are hereafter collectively designated as SraGp. Boxes TTG and TAA in gray indicate the pnp START codon and rpsO STOP codon located on the (−) strand, respectively. The pnp transcription initiation site and the pnp ribosome-binding site (RBS) located on the (−) strand are also indicated in gray.
The general model for mRNA degradation in E. coli postulates that transcripts are irreversibly inactivated by an endonucleolytic cleavage near their 5′-end, predominantly by RNase E, followed by a wave of further endonucleolytic cuts and 3′–5′ exonucleolytic degradation of the generated mRNA fragments (Carpousis et al. 2009). However, the degradation of the rpsO and pnp transcripts does not fit to this model. RNase E initiates the decay of the rpsO transcript by cleaving near its 3′ extremity, upstream of the transcription terminator (T1) (Fig. 1C [1]). It also cleaves the polycistronic rpsO-pnp transcript at the T1 site and downstream from the rpsO terminator hairpin, generating an upstream rpsO transcript identical to the one derived from the monocistronic transcript (Fig. 1C [2]; Régnier and Hajnsdorf 1991). Degradation of pnp mRNA is initiated by RNase III, which very efficiently cleaves both the pnp monocistronic transcript and the dicistronic rpsO-pnp mRNA within a long stem–loop included in the pnp mRNA leader that is downstream from the T1 terminator (Fig. 1C [1,2]). These cleavages trigger the PNPase-mediated degradation of the processed 5′-end of the pnp mRNA and the rapid attack by RNase E at the beginning of the pnp coding sequence, which leads to the irreversible inactivation of the pnp message and a decreased synthesis of polynucleotide phosphorylase (Fig. 1C; Portier et al. 1987; Hajnsdorf et al. 1994a; Jarrige et al. 2001; Carzaniga et al. 2009). Translation of pnp is also tightly regulated. In the absence of RNase III cleavage, PNPase controls its own expression at the translational level, and this repression does not require the catalytic activity of the enzyme (Carzaniga et al. 2015). This RNase III-independent negative control may result from a competition between PNPase and S1 for pnp mRNA binding (Carzaniga et al. 2015). Higher expression of PNPase after a cold shock results from both a slight transcriptional induction from the P2 promoter and the stabilization of the pnp transcript (Zangrossi et al. 2000). The destabilizing effect of PNPase on its own mRNA is also less efficient at low temperatures, leading to a decrease in repression and an increase in the expression level (Beran and Simons 2001; Mathy et al. 2001). In addition, CsrA binds two sites in the pnp 5′UTR, after its processing by RNase III and PNPase, where one CsrA site overlaps the pnp Shine-Dalgarno sequence (SD) such that CsrA binding inhibits pnp translation (Park et al. 2015).
Although an antisense RNA (asRNA) has been described, called SraG, which is encoded between rpsO and pnp genes and convergently transcribed toward rpsO, no evidence of an effect on rpsO or pnp regulation by this noncoding RNA was reported (Fig. 1A; Argaman et al. 2001). Most of the sRNAs discovered in bacteria modulate gene expression at the post-transcriptional level by base pairing to mRNAs. This class of sRNAs includes asRNA transcribed from the complementary DNA strand of their targets and sRNAs transcribed at loci distant from their targets. These latter sRNAs usually require the Hfq protein to interact with their targets (Vogel and Luisi 2011). In an hfq null mutant, the pnp mRNA level was not affected, suggesting that control by a trans-acting Hfq-dependent small RNA is unlikely (Le Derout et al. 2010). However, the SraG transcript, which originates within the upstream untranslated sequence of pnp, could act as an asRNA controlling expression of either or both rpsO and pnp because it is complementary with the intergenic RNA sequences, which control the expression of S15 and PNPase (Fig. 1A).
Many of the first cis-acting asRNAs identified were regulators of plasmid copy number and replication (Brantl 2007). Some of the best known examples of antisense RNA regulation in E. coli come from the type I toxin–antitoxin (TA) systems, where the antitoxin is an asRNA and the toxin promotes cell killing, cell stasis or long-term cell persistence (Gerdes et al. 2005; Gerdes and Wagner 2007). In each of these cases, the asRNA affects the translation of its target (Wagner et al. 1992; Kawano et al. 2007). In other cases, the primary role of asRNAs is to either positively or negatively alter the RNA stability. For example, overexpression of the GadY sRNA results in the processing of the gadXW mRNA, giving rise to separate gadX and gadW transcripts that are more stable and that accumulate to higher levels than the full-length mRNA (Opdyke et al. 2004). The RyeA/SdsR sRNA pair is cleaved by RNase III and then degraded, with the result that SdsR expression is inversely correlated with RyeA expression (Vogel et al. 2003). Another possible mechanism of gene expression inhibition by asRNAs involves transcriptional inhibition of the genes encoded on the opposite strand by converging transcriptional interference (Thomason and Storz 2010).
To investigate a possible role of SraG on the expression and/or stability of rpsO and pnp, we analyzed the expression and function of SraG. We found that SraG RNA is transcribed from a different promoter than that previously described, producing a 216-nucleotide (nt) transcript and an ∼190-nt transcript. SraG RNA overproduction affected both the stability and the translation of pnp. In vitro experiments showed that interaction of SraG with pnp mRNA modifies RNase III cleavage of both SraG and pnp, and it does not allow the 30S ribosome to interact physically with the Shine-Dalgarno sequence. In addition, we looked for other possible targets of SraG, but we were unable to find other genes potentially regulated by SraG. Altogether, we show that SraG acts as an asRNA to pnp mRNA to modulate its expression.
RESULTS
Characterization of the SraG sRNA
SraG has been described as a 174-nt transcript that is cleaved to produce a 147-nt processed RNA that is potentially polyadenylated at its 3′ extremity (Argaman et al. 2001). We first examined the abundance of both transcripts by Northern blots and unexpectedly detected bands of ∼215 and ∼190 nt, i.e., distinctly longer than the previously described transcripts (Fig. 1B). Transcripts of the same size were reproducibly found in exponential and stationary phase and in N3433 and MG1655 strains (Fig. 1B; Supplemental Fig. S1). We performed circular RT-PCR (cRT-PCR) and found no clones starting at the previously annotated transcription initiation site (Fig. 1D). Comparisons of RNA treated or not by polyphosphatase allowed us to identify the SraG transcription initiation site at 42 nt upstream of the previously annotated 5′-extremity of SraG (Fig. 1D, box 1 marked with asterisk; Supplemental Fig. S2). Interestingly, this transcription-initiation start (PSraG, P in Fig. 1A) was originally predicted by bioinformatic analysis (Argaman et al. 2001). Most transcripts were found to harbor 3′-extremities at the previously described terminators (box 4, Fig. 1D). Altogether, this indicated that the longest transcript detected on Northern blots was a primary transcript of 216 nt starting at PSraG and ending at the terminator. The 190-nt long RNA corresponded to a population of RNA molecules with different 5′ and 3′ extremities. These RNA fragments of ∼190 nt are hereafter collectively designated as SraGp. They might result from maturation events at either the 5′- or 3′-end of the 216-nt primary transcript.
Mutation of the −10 box of PSraG (giving PSraGmut) eliminated the 216-nt long SraG and decreased by ninefold the level of SraGp (Fig. 1B), confirming that PSraG promoted transcription of SraG and that the 190-long SraGp RNA was produced by processing of this primary transcript. The residual SraG signal detected when PSraG was inactivated could correspond to the maturation of a longer transcript predicted to initiate 339 nucleotides upstream of PSraG.
RNase III, RNase E, and PNPase are involved in maturation and degradation of SraG
We investigated whether the endoribonucleases involved in maturation and degradation of E. coli mRNAs and sRNAs, namely RNase III (rnc), RNase E (rne), and RNase G (rng), were involved in SraG sRNA processing to generate the SraGp forms (Fig. 1B). Isogenic strains deficient for each of these RNases were used as well as strains with combinations of double and triple mutations. The effect of RNase E inactivation was examined by using the rne3071ts allele. For comparison, RNAs from all strains were prepared from bacteria grown at 30°C and shifted to 42°C to inactivate the thermosensitive RNase E. SraG and SraGp both accumulated in rne and rnc mutants, suggesting that both RNase III and RNase E played a role in SraG degradation (Fig. 2A). However, the relative intensity of the two bands did not change, which therefore excluded the direct involvement of RNase III or RNase E in the cleavage of the primary transcript to generate SraGp. Similarly, none of the exoribonucleases RNase II, PNPase, or RNase R individually modified the ratio between SraG and SraGp (Fig. 2B, data not shown). We observed in the rnc and rne mutants additional shorter bands of ∼160, 147, and 120 nt (Fig. 2A,B). While in the pnp mutant a significant accumulation of fragments of ∼147, 120, and 110 nt were observed, and their level was even higher in the double rnc-pnp mutant, suggesting that RNase III was not responsible for their generation and that the rnc effect was superimposed on the pnp effect (Fig. 2B). Only the 160-nt fragment was absent in the double rnc-pnp mutant, suggesting a processing by PNPase of SraG in the rnc mutant.
FIGURE 2.
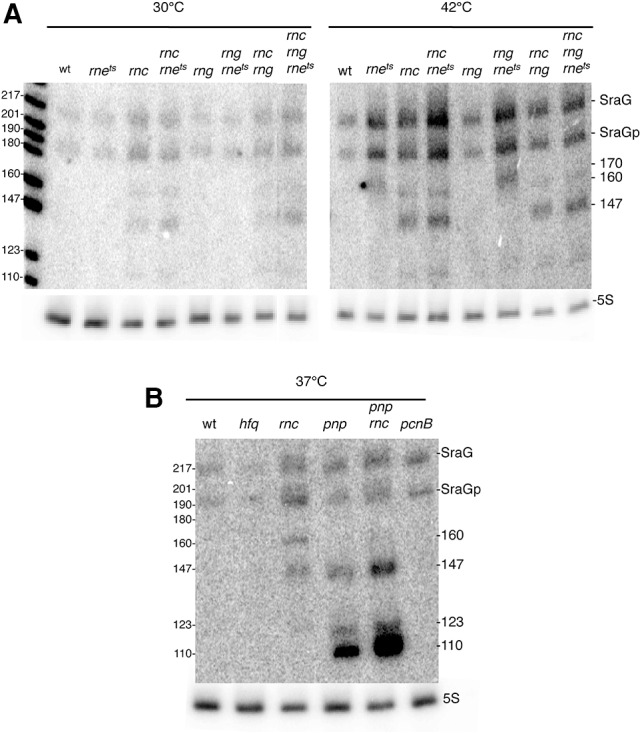
RNase III, RNase E, and PNPase are involved in SraG degradation. Northern blot analysis of SraG sRNA. N3433 strains carrying mutations in the genes indicated at the top of the autoradiograph were grown at 30°C in LB medium and shifted to 42°C for 15 min (A) or grown at 37°C (B). Total RNA (10 µg) was separated on acrylamide gels and analyzed by Northern blotting. Blots were probed for SraG and 5S rRNA.
We also tested the role of other RNases such as RNase BN (elaC or rbn), RNase P (rnpA), and YbeY (ybeY), which is a metal-dependent single-strand-specific endoribonuclease (Davies et al. 2010; Pandey et al. 2011). Their inactivation had no effect on the level or the balance between SraG and SraGp (Supplemental Fig. S3), indicating that none of these RNases were involved in SraG maturation and degradation.
Finally, we investigated whether other factors, such as Hfq (hfq), poly(A) polymerase (PAP I) (pcnB), and YhbJ (rapZ), which is involved in the regulation of GlmS (Gopel et al. 2013), affected the level of SraG and/or SraGp production (Massé et al. 2003; Reichenbach et al. 2008). YhbJ inactivation had no effect. Hfq and PAP I inactivation had only a small effect on the level of SraG and on its processing (0.94- and 1.27-fold compared to wt, respectively) (Fig. 2B; Supplemental Fig. S3A). The cRT-PCR experiments showed that 24 of the 76 clones with 3′-extremities close to the terminator (Fig. 1D, box 4) contained one to four noncoded adenosine residues, which were presumably added by PAP I. We concluded that polyadenylation of SraG did not facilitate the exoribonucleolytic degradation of the sRNA.
In conclusion, none of the mutations tested affected the relative abundance of the long and processed SraGp forms, implying that none of the tested RNases was responsible for the maturation events producing the 190-nt transcripts. The only conditions where the ratio between SraG and SraGp (usually 50% of each) was modified were the replacement of the TTG pnp start codon for a TGA stop codon or when the stringent response was provoked. This suggested that pnp translation could modify SraG maturation. This point will be discussed later.
Regulation of sraG expression
To attempt to characterize the biological role of SraG, we first investigated the regulation of its transcription. Both SraG forms are poorly expressed compared to rpsO and pnp mRNAs in LB medium in both exponential and stationary phases and in minimal media (data not shown). It has been reported that the levels of both 174- and 146-nt SraG were higher at 42°C and 15°C (Argaman et al. 2001). We have confirmed this observation for the 216-nt and 190-nt long SraGs detected in our experiments (data not shown). It has also been reported that SraG is up-regulated at the exterior of a biofilm when compared to the interior, suggesting that SraG expression may be regulated by differential stress conditions like nutrient availability (Ito et al. 2009). In order to investigate a possible role of nutrient starvation, we induced the stringent response by adding serine hydroxamate (SHX). SHX acts as a competitive inhibitor of serine aminoacid-tRNA synthetase, and thereby it induces the stringent response by starving ribosomes for tRNASer. We found that addition of SHX in the exponential phase for 30 min increased the relative level of the long SraG by about twofold. This suggested that SraG transcription and/or stability, as well as SraGp production/stability, may be modulated as a consequence of the induction of the stringent response (Supplemental Fig. S1).
To examine the potential role of SraG as an asRNA affecting rpsO and/or pnp expression, the SraG sRNA was ectopically expressed from the inducible PLacO-1 modified promoter (Guillier and Gottesman 2006). SraG DNA, starting at the transcription start site and ending at the transcription terminator identified by cRT-PCR, was cloned into the pBRplac plasmid (to give pBRSraG). As a control, we cloned the SraG short form that had 42 nt deleted from the 5′-end (pBRSraG-S); this corresponded to the previously annotated 5′-extremity described by Argaman et al. (2001). Overexpression of SraG or SraG-S had no effect on growth in rich medium in either liquid culture or on plates incubated at 37°C or 18°C, nor in response to heat shock or cold shock, nor on motility or biofilm formation (data not shown). Overexpression for 20 min resulted in about a 30-fold and 40-fold enhancement of SraG and SraG-S, respectively (Supplemental Fig. S4). Surprisingly, very low levels of SraGp relative to SraG (respectively indicated by [**] and [*] in Supplemental Fig. S4) were detected when the sRNA was ectopically transcribed from a foreign promoter. Also, the levels of plasmid-expressed SraG and SraG-S were five and three times more abundant, respectively, in the rnc mutant (Supplemental Fig. S4). In the case of full-size SraG, but not for SraG-S, this increase in the level of RNA was associated with a longer half-life in the rnc mutant (Table 1).
TABLE 1.
Overexpression of the 216-nt SraG transcript (SraG) and SraG-S has opposite effects on the stability of the pnp messenger, without impact on rpsO
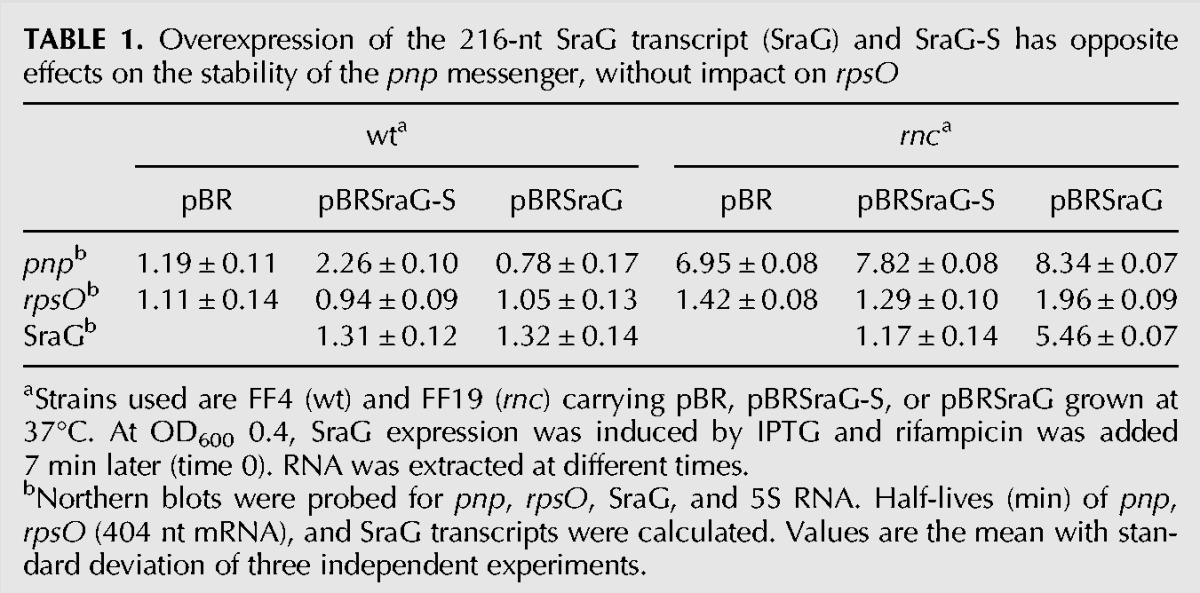
SraG overexpression affects maturation of the rpsO-pnp mRNA and the levels of pnp mRNA
The levels of rpsO and pnp mRNAs were determined 7 min after addition of IPTG to cells containing the empty plasmid or the SraG-expressing constructs to limit the level of SraG overexpression (Fig. 3A). SraG had no significant effect on the abundance of the rpsO transcripts (404 nt and 500 nt) (Fig. 3A, fragments 0 and 1; Table 1). The levels of the rpsO transcript were lower in the rnc mutant. This is presumably because the level of PNPase was higher in the rnc strain and because PNPase was responsible for the poly(A)-dependent degradation of the rpsO transcript (Hajnsdorf et al. 1994b; Robert-Le Meur and Portier 1994). This decrease was partly rescued by overproduction of either SraG or SraG-S, suggesting that they could negatively control the level of PNPase. However, there was no significant modification of rpsO stability upon SraG overproduction in the wt or in the rnc mutant (Table 1). This indicated that the possible pairing of SraG-S or SraG to rpsO and rpsO-pnp transcripts did not affect the RNase E cleavage of the mRNA upstream of the terminator T1 (Fig. 1C), which is the limiting step in the degradation of the rpsO transcript (Régnier and Hajnsdorf 1991; Hajnsdorf et al. 1994b). In addition, asRNA overproduction did not significantly affect RNase III cleavage efficiency at the RIII1 site (Figs. 1C, 3A, fragment 1).
FIGURE 3.
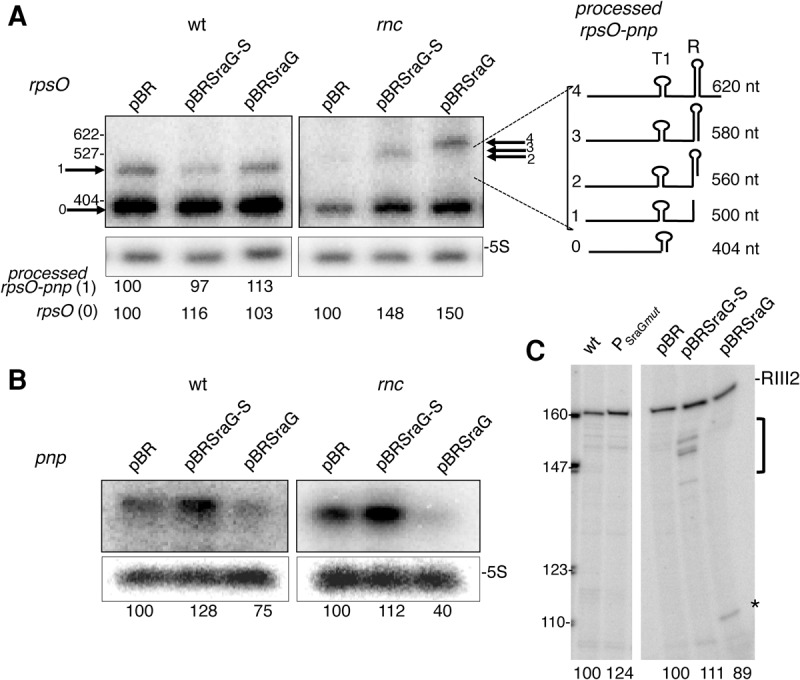
Overexpression of SraG affects the abundance of pnp, but not rpsO mRNAs. Northern blot analysis of rpsO (A) and pnp (B) RNA in FF4 (wt) and FF19 (rnc) containing the pBRplac (pBR), pBR-SraG (pSraG), and pBR-SraG short (pSraG-S) plasmids. Expression of SraG was induced by adding IPTG (100 µM) for 7 min. Radioactivity corresponding to each transcript was quantified and normalized relative to 5S and compared to the strain carrying the empty pBRplac plasmid. The mean value (in arbitrary units) of three independent experiments relative to the corresponding pBR containing cells is indicated below the gel. The Northern blot showing pnp detection in the rnc mutant was underexposed compared to wt to allow comparison. In A, the arrows indicate the various processed forms containing rpsO derived from the dicistronic transcript, which are numbered according to their size and schematically represented on the right side of the Northern blot. Cleavage by RNase III on the left side of the R secondary structure (RIII1 site) generated rpsO-pnp processed form 1 is detected in the wt strain. In the rnc strain, longer transcripts (2,3,4) are detected corresponding to cleavages within or downstream from the R secondary structure. (C) Total RNA from N3433 (wt), N3433 lacking SraG (mutation in SraG promoter PSraGmut) (left panel), and FF4 transformed with pSraG, pSraG-S, and empty pBRplac vector (same RNA preparations as in A and B) were used as templates for reverse transcription primed with the radiolabeled RNX1 primer. Reverse transcripts corresponding to the RNase III2 cleavage site (Fig. 1) are indicated, the asterisk (*) and bracket indicate other reverse transcriptase stops. Radioactivity corresponding to RNase III-processed pnp transcript was quantified and compared to the wt strain without plasmid or carrying the empty pBRplac plasmid. These values (in arbitrary units) are indicated below the gel.
In the absence of RNase III, the rpsO-pnp transcript was cleaved downstream from the hairpin recognized by RNase III (Fig. 3A giving the 560-nt processed fragment 2) (Régnier and Grunberg-Manago 1990). Overproduction of either SraG or SraG-S produced cleavages at new positions that generated the 580-nt and 620-nt processed rpsO-pnp transcripts (Fig. 3A, processed forms 3 and 4).
SraG-S and SraG have opposite effects on the accumulation of pnp mRNA. SraG-S overproduction slightly increased the level of pnp mRNA in the wt and the rnc mutant, presumably as a consequence of an increased stability (Fig. 3B,C; Table 1). On the other hand, SraG overproduction decreased the level of pnp and its half-life in the wt (Fig. 3B,C), although in the rnc mutant, the decreased level of pnp mRNA did not correlate with the half-life of the pnp transcript (Fig. 3B; Table 1). When SraG was overexpressed, RNase III still cleaved pnp RNA at the RNase III2 downstream site (Fig. 1C), but new stops of reverse transcriptase were observed, which could correspond to additional cleavages by RNase III or by another RNase (Fig. 3C).
SraG overexpression decreases pnp expression
We analyzed the effect of SraG-S and SraG overproduction on pnp expression by adding increasing amounts of IPTG at the beginning of the culture. Expression of pnp was determined by using a translational pnp-lacZ fusion and by Western blot analysis. Figure 4 shows that under these conditions SraG overexpression decreased slightly the expression of pnp-lacZ and PNPase, while SraG-S had no effect on β-galactosidase activity but increased by twofold the level of PNPase. The effect of SraG overproduction on pnp-lacZ expression was slightly stronger in the rnc mutant (about twofold decrease) when pnp mRNA was 10 times more abundant than in the wt, while SraG-S had nearly no effect. We concluded that the 5′-part of the sRNA was essential to modulate PNPase expression and pnp mRNA stability. Hfq inactivation had no effect on SraG and SraG-S control of pnp-lacZ expression (data not shown).
FIGURE 4.
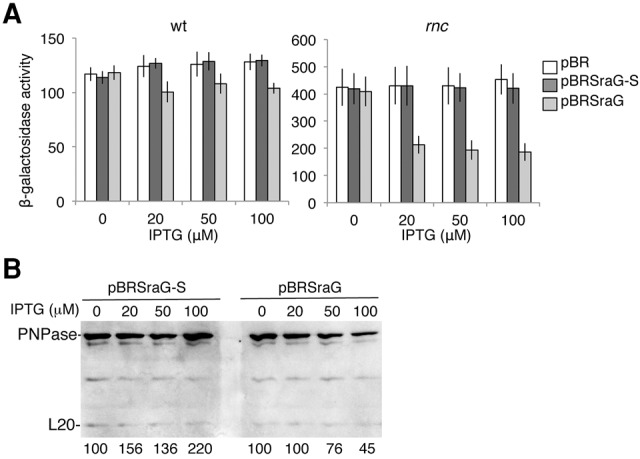
Overexpression of SraG represses pnp expression. Isogenic FF4 (wt) and FF19 (rnc) strains carrying the pnp-lacZ fusion were transformed with the pBRplac (pBR), pBR-SraG (pBRSraG), and pBR-SraG short (pBRSraG-S) plasmids. SraG and SraG-S synthesis were induced by addition of IPTG at the beginning of the cultures. Samples were taken in the middle of the exponential phase to determine β-galactosidase activity and PNPase levels. (A) β-Galactosidase activity is the mean value of three independent cultures with standard deviation. (B) Western blot analysis of PNPase and L20 has been made on FF4 strain (wt). Quantification of PNPase normalized to L20 is indicated below the gel.
Binding of SraG to pnp induces a new RNase III cleavage of the mRNA in vitro
These results suggested that upon SraG overexpression, new RNase III cleavages might occur when pnp is hybridized with SraGs and/or that translation initiation could be modified. To test these hypotheses, we performed in vitro experiments. RNase III cleavage is the crucial first step in the control of PNPase expression. It cleaves the stem loop (R) present in the pnp leader sequence at two sites, RIII1 and RIII2, located 39 and 77 nt downstream from the P2 transcription start site (Fig. 1C). These cleavages trigger pnp mRNA decay resulting in a decreased synthesis of PNPase. Since SraG was antisense to the 5′ region of pnp carrying the RNase III processing sites, base pairing of pnp UTR with SraG could interfere with RNase III processing of pnp. SraG could also be a substrate of RNase III because endogenous and overexpressed SraG were more abundant in the rnc strain and the overexpressed SraG was more stable in rnc than in wt strains with half-lives of 5.46 min and 1.32 min, respectively (Table 1; Supplemental Fig. S4).
We examined whether SraG affected the cleavage of pnp mRNA by RNase III in vitro and whether SraG was itself a substrate for RNase III. We used a pnp RNA fragment (pnp*, Fig. 5), which starts at the transcription start site P2 and extends into the open reading frame. RNase III was shown to cleave the pnp mRNA at two sites, RIII1 and RIII2, both in vivo and in vitro (Fig. 5A,B; Supplemental Fig. S5E; Hajnsdorf et al. 1994a). SraG and SraG-S alone were cleaved by RNase III at sites called RIII1′ and RIII2′, which are the mirror of sites RIII1 and RIII2 on the pnp transcript (Fig. 5B; Supplemental Fig. S5B,C,E). This indicated that SraG formed the same stem–loop structure as pnp mRNA, as predicted by several RNA folding programs/algorithms. SraG was also cleaved by RNase III at a site RIII3′ in the SraG terminator, which was a sequence outside the duplex with pnp mRNA (Figs. 1C, 5B; Supplemental Fig. S5B,C,E). RNase III was also tested on the pnp and SraG RNAs together, under conditions where all the molecules were fully engaged in a duplex (Supplemental Fig. S5A). After duplex formation with SraG or SraG-S, RNase III cleaved the pnp RNA at a new site close to its 5′-end (RIIIA) (Fig. 5A,B; Supplemental Fig. S5E). RNase III enzyme can cut both strands of double-stranded substrates (Court et al. 2013); accordingly a cleavage was also detected on SraG and SraG-S (site RIIIA′) (Fig. 5B; Supplemental Fig. S5B,D,E). In addition, SraG and SraG-S in the duplex were also cleaved by RNase III at sites near their 5′ extremities (sites RIIIB′ and C′ for SraG and RIIID′ for SraG-S), while no corresponding cleavages in the pnp RNA were observed (Fig. 5B; Supplemental Fig. S5B,D,E). These results showed that RNase III cleaved in vitro both the SraG-pnp and SraG-S-pnp duplexes at new positions. Among them, one site was close to the 5′-end of the pnp mRNA, in a region predicted to be single stranded in the transcript. We propose that this cleavage by RNase III in the pnp-SraG duplex may facilitate further cleavage by another ribonuclease, such as RNase E, as previously shown (Hajnsdorf et al. 1994a). The SraG RIII3′ cleavage site, outside the duplex, was as efficiently cleaved in the absence or presence of pnp mRNA (Fig. 5B; Supplemental Fig. S5B,E). However, these results do not explain why SraG, but not SraG-S, induced a decrease in the level of pnp mRNA (Fig. 3) and PNPase expression (Fig. 4) in the wt strain.
FIGURE 5.

Both SraG and SraG-S promote new RNase III cleavages in pnp mRNA in vitro. (A) RNase III hydrolysis of 5′-end-labeled pnp mRNA (pnp*) free or in the presence of a molar ratio of SraG or SraG-S. Increasing amounts of RNase III (10−6–5.10−3 units) were added to the pnp* RNA alone or in complex with SraG or SraG-S. Lanes T1 and NaOH indicate RNase T1 and alkaline ladders, respectively, of pnp*. See Supplemental Figure S5 for cleavages on the SraG RNAs. (B) Schematic representation of the RNase III processing sites on pnp (light gray), SraG (black), and SraG-S (dark gray), and pnp-SraG and pnp-SraG-S duplexes. Light gray crosses represent cleavages observed on both the single RNA molecules and when they are in duplex, black crosses indicate cleavages only observed after duplex formation. A dot represents the 5′-triphosphate extremity of the various RNA molecules.
Binding of SraG to pnp mRNA prevents the formation of the translational initiation complex
SraG binding to the pnp leader region was close to but did not overlap the Shine-Dalgarno sequence (Fig. 1C,D), so it was not obvious that SraG binding should affect pnp mRNA translation. In order to look for an SraG-dependent inhibition of pnp translation, we used a toe-printing assay. In the presence of initiator tRNAfMet, 30S ribosomal subunits blocked reverse transcription of the pnp RNA at 15 nt from the pnp initiation codon (UUG) (Fig. 6, lanes 11,16). This signal provided a measure for the formation of the initiation complex, since it was dependent on both 30S subunits and initiator tRNA. Performing the toe-print reaction with SraG RNAs in the absence of 30S/tRNAfMet allowed us to detect the 5′-end of SraGs hybridized to the pnp RNA (Fig. 6, lanes 2–5, 7–10). The addition of increasing amounts of SraG in the reaction with 30S and tRNAfMet resulted in the disappearance of the pnp toe-print. However, the presence of SraG-S did not affect the pnp toe-print. This result showed that SraG (but not SraG-S) annealing to the messenger interfered with the formation of the ternary initiation complex by preventing the 30S subunit from loading onto the pnp transcript, and thus this impeded pnp translational initiation. As a consequence, SraG could specifically affect the level of pnp RNA as shown in Figure 3B when SraG was overproduced because untranslated mRNAs are usually subject to degradation.
FIGURE 6.
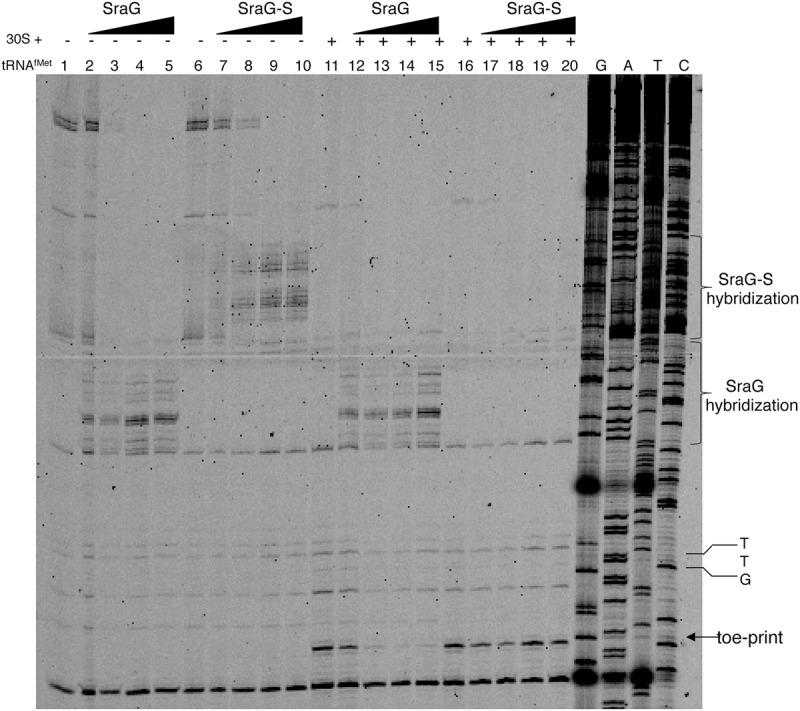
SraG blocks 30S binding to the pnp 5′-leader in vitro. In vitro synthesized pnp RNA fragment (positions 1–234 relative to the transcription start) was used in a toe-print assay in the presence and absence of SraG and SraG-S RNAs. pnp RNA was incubated with increasing concentrations of SraG or SraG-S (as a control RNA) giving a molar ratio sRNA:pnp RNA of 0 (lanes 1, 6, 11, 16), 0.5 (lanes 2, 7, 12, 17), 1 (lanes 3, 8, 13, 18), 2 (lanes 4, 9, 14, 19), and 5 (lanes 5, 10, 15, 20). Addition of tRNAfMet and 30S ribosomal subunits is indicated by (+) (lanes 11–20). GATC lanes are sequencing ladders obtained by asymmetric PCR using the same labeled primer. The TTG start codon of pnp is shown. The arrow indicates the 30S toe-print on the pnp RNA.
A possible reciprocal regulation between PNPase and SraG
So far we have shown that SraG modifies (i) rpsO-pnp maturation in the absence of RNase III; (ii) the decay-rate of pnp mRNA and its cleavage by RNase III; and (iii) the initiation of pnp translation in vitro (Figs. 3–5; Table 1) and hence the PNPase level. We next investigated whether pnp transcription or translation affected sraG expression. To gain insight into a possible reciprocal regulation of SraG by pnp, we performed a Northern blot analysis with RNA harvested from a wt strain and strains where the pnp P2 promoter was inactivated (P2mut) or the TTG pnp start codon was mutated to TGA (TTGmut). We also checked the effect of inhibiting SraG expression (PSraGmut with a mutation in the −10 box of PSraG) on pnp expression. None of the substitutions affected the overall secondary structure of SraG and/or the 5′-part of the pnp mRNA (mFold predictions) so that pnp autoregulation would not be affected. The mutation in PSraGmut was close to, but did not overlap, the pnp Shine-Dalgarno (Fig. 1D; Portier and Régnier 1984). Figure 7A shows that initiation of pnp transcription at P2 had no impact on the levels of SraG since SraG and SraGp were equally abundant in the wild-type strain and in the P2mut. Both strains exhibited similar levels of pnp mRNA since pnp was still transcribed from the P1 promoter as a dicistronic rpsO-pnp transcript. The PSraG mutation eliminated SraG expression and led to a 30% increase in the amount of pnp RNA and the level of PNPase. In the absence of pnp translation (strain TTGmut), the pnp mRNA was degraded. This mutation almost eliminated production of SraGp, but abundant shorter transcripts were generated. Such a difference was not observed with a pnp null mutant that was disrupted by a Tn5 transposable element in the coding sequence (Fig. 2B). This indicated that pnp translation was correlated with, or was required for, the processing of SraG into SraGp. Another line of evidence supported this observation; addition of SHX, which inhibits translation and induces the stringent response, also reduced processing of SraG (Supplemental Fig. S1). How this control was achieved remains to be determined.
FIGURE 7.
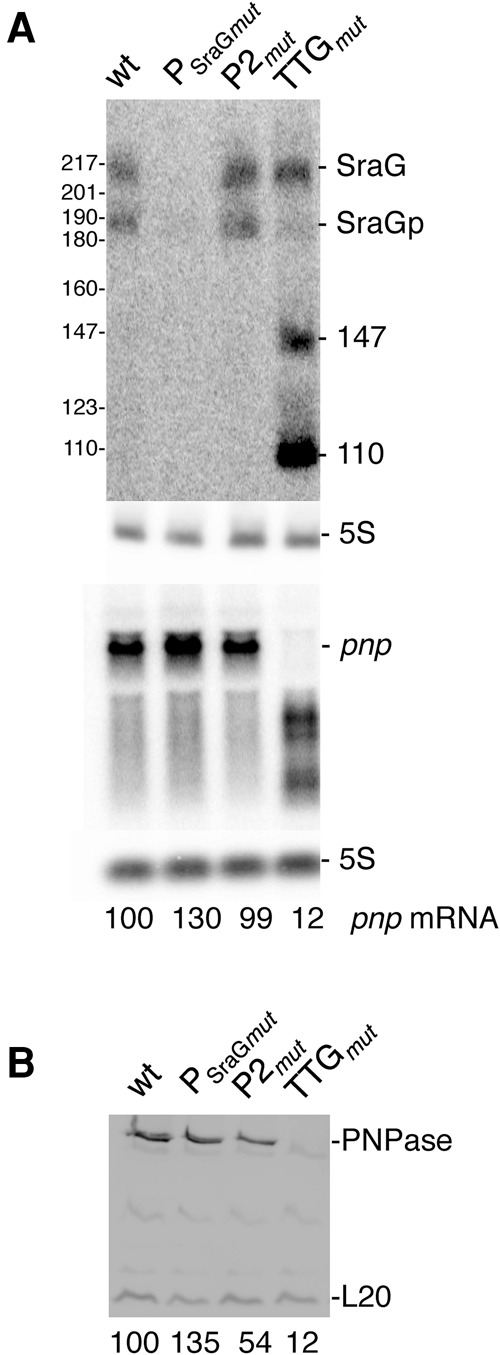
Reciprocal regulation of pnp and SraG expressions. (A) Northern blot analysis of SraG and pnp mRNA and (B) Western blot analysis of PNPase in N3433 wild-type, sraG (PSraGmut) and pnp mutants (P2mut and TTGmut). Quantifications of pnp radioactivity and PNPase levels (arbitrary units) normalized relative to 5S and L20, respectively, are indicated below the gels.
Interestingly, in the SraG promoter mutant (PSraGmut), there was a small (1.3-fold) increase in pnp mRNA and PNPase levels (Fig. 7), which was probably caused by the absence of the negative regulation by SraG on pnp maturation and/or translation. Altogether, these results showed that expression of pnp and SraG mutually affected each other with, on the one hand, SraG controlling pnp maturation/translation and, on the other hand, pnp translation affecting SraG processing to SraGp.
Does SraG have any other roles?
We wanted to determine whether SraG could also control expression of other targets in E. coli. One characteristic of sRNAs acting in trans in E. coli is a requirement for Hfq. Hfq has been shown to stabilize many sRNAs and to facilitate their interaction with their mRNA target (Vogel and Luisi 2011), although in some cases Hfq may be dispensable (Darfeuille et al. 2007). We investigated the ability of SraG and SraG-S to bind purified Hfq. The dissociation constants for Hfq on SraG and SraG-S were 0.49 nM and 1.08 nM, respectively, as assessed by gel retardation assay with purified Hfq (Supplemental Fig. S6; Hajnsdorf and Régnier 2000). These binding constants are consistent with previous results showing that Hfq immunoprecipitates with SraG sRNA (Zhang et al. 2003). However, as shown above, Hfq deletion had no impact on either the level of SraG or on its processing (Fig. 2B). Moreover, Hfq deletion did not affect the level of pnp mRNA (Le Derout et al. 2010) nor SraG-mediated inhibition of pnp expression, as assessed by pnp-lacZ expression (data not shown).
SraG of Yersinia pseudotuberculosis has been shown to regulate the YPK_1206-1205 operon (Lu et al. 2012). We investigated a possible role of SraG on RNA targets transcribed from elsewhere on the E. coli chromosome by using various strategies. We first showed that there was no difference in the functional expression of transcripts by looking at the pattern of total protein labeling in bacterial cultures after SraG or SraG-S overexpression for 1–24 min (Supplemental Fig. S7). To look for specific targets, we performed a computational search for potential SraG and SraG-S targets and conducted a microarray analysis of RNAs prepared after overexpression of SraG or SraG-S for 7 min in exponential phase (see Materials and Methods). Under these conditions, the level of pnp was reduced after overexpression of SraG but not that of SraG-S, which validated the experimental approach. Ten potential targets, highly ranked by both methods, were further tested; alx, araH, dadA, dsbC, fadH, efeO, ydiY, fhuC, deaD, dksA. However, none of them exhibited a significant variation in levels of mRNA by Northern blot analysis after 7 min of SraG overexpression (data not shown). Taken together, our findings show that SraG function seems to be restricted to controlling pnp in E. coli, at least under our experimental conditions. However, it was still possible that low abundance mRNAs and/or genes only expressed under specific (stress) conditions could be targeted.
Finally, we addressed a possible indirect effect of reducing levels of PNPase by SraG overproduction since a previous report showed that loss-of-function mutations in pnp resulted in an increased level of tRNAIle precursors (Maes et al. 2012). Various amounts of IPTG were added at the beginning of cultures of the strain carrying the pBRSraG plasmid to produce a graduated variation in pnp expression. Supplemental Figure S8 shows that the steady-state level of tRNAIle precursor was unaffected in cells expressing SraG-S from the plasmid, while a slight increase was detected in cells expressing SraG. This finding was in agreement with a reduction in PNPase activity resulting from an increased expression of SraG.
DISCUSSION
We demonstrate here that the SraG sRNA, which is transcribed from the antisense strand in the intergenic region between rpsO and pnp, is longer than was previously annotated at the 5′-end. We have located the promoter driving its expression in vivo and confirmed by mutagenesis that the shorter form of SraG (SraGp) is a processed product of the primary transcript. Processing of the primary sraG transcript occurs at both the 5′- and 3′-ends. We show that overexpression of SraG decreases the level of the pnp transcript. Consistently, mutating the PSraG promoter on the chromosome results in an increase in the levels of pnp mRNA and in PNPase, and we show that addition of SraG prevents the formation of the pnp translation initiation complex in vitro and also induces cleavages at new positions within the pnp mRNA in the region base paired with SraG. By systematically comparing the effects of the expression of full-size SraG and the RNA transcribed from the previously annotated 5′-end (42 nt downstream), we show that the 5′-part of the sRNA is essential to modulate PNPase expression and pnp mRNA stability. In conclusion, SraG RNA controls PNPase expression by a direct antisense interaction, which affects the processing and the translation of the pnp messenger. However, SraG overexpression does not completely shut off pnp expression. For example, it is unable to mimic the cold-sensitive phenotype of a pnp mutant. But our results demonstrate that SraG is a new player that fine-tunes PNPase regulation.
Antisense regulation of an endoribonuclease (SymE) that forms the toxin component of the SymE/SymR TA type I pair has been characterized (Kawano et al. 2007), but the antisense regulation of the expression of PNPase is, as far as we know, the first example of such a mode of regulation of a major exoribonuclease conserved from bacteria to plants.
Half of the SraG RNA exists in a form lacking ∼30 nt from either the 5′- or the 3′-ends. The long and short forms are always observed and in approximately equal molar amounts. The identity of the nuclease(s) responsible for the generation of these truncated forms is still unknown. We tested various ribonucleases (RNase E, RNase III, RNase G, RNase II, PNPase, RNase R, RNase BN, and RNase P), and other factors [poly(A) polymerase, Hfq, YbeY, YhbJ] that are known to modify processing and accumulation of sRNA, and we were unable to define a nucleolytic activity generating SraGp. This recalls the situation for the GadY-dependent processing of gadX-gadW where, in addition to RNase III, another unidentified endoribonuclease is involved in generating the monocistronic gadX and gadW transcripts (Opdyke et al. 2011). However, events occurring in cis, such as premature termination of SraG or transcriptional interference between SraG and pnp, could be involved in SraGp production since only a low level of SraGp is detected from ectopically overexpressed SraG (Supplemental Fig. S4). Similarly, the level of SraGp is decreased when pnp translation initiation is inhibited by mutating the pnp start codon (Supplemental Figs. S4, S7). Likewise, inhibiting translation by SHX also reduces processing of SraG to SraGp (Supplemental Fig. S1). One attractive hypothesis would be a competition between pnp translation initiation and the interaction between pnp mRNA-SraG. We propose that initiation of pnp translation requires unfolding of the 5′UTR of the messenger, which enables an initial interaction to occur between the two RNA molecules. Once the ribosome leaves the SD sequence, this interaction can extend back to the 5′-end of SraG. Consequently, the 5′ part of the SraG sRNA is paired very close to the pnp SD sequence (Fig. 1D), and it can further inhibit initiation of pnp translation, as shown by in vitro toe-printing (Fig. 6). Another consequence of the pairing between pnp mRNA and SraG could be that SraG adopts an alternative structure allowing its cleavage by an RNase, which remains to be identified, and could require a ribosome-associated factor. This cleavage would remove the 5′-end of SraG and generate an RNA unable to regulate the expression of pnp. In the absence of translation, the pnp mRNA is degraded (Fig. 7B), SraG cannot interact with this messenger and remains in its initial structure, which does not allow the processing, giving rise to SraGp. In addition to an effect on pnp translation, the generation of SraGp could also depend upon PNPase catalytic activity (Fig. 2).
RNase III is the primary actor in pnp regulation since it initiates a cascade of events involving RNase E and PNPase (requiring its catalytic activity) that leads to the degradation of the pnp messenger. Cleavage by RNase III is also required for CsrA to repress pnp translation (Park et al. 2015). On the other hand, when RNase III is absent, PNPase can inhibit its own translation by competing with the binding of S1 on its own mRNA (Carzaniga et al. 2015). In the case of SraG, we observed regulation of pnp in the presence or absence of RNase III; this indicates that CsrA and SraG regulation might be independent. The role of SraG sRNA as another player in pnp regulation was not suspected before, probably because of the annotation error of its transcription start site. But a previously described mutation reducing PNPase autoregulation was located in the −10 sequence of the SraG promoter we described here, implying that SraG expression does impact pnp expression in vivo (Robert-Le Meur and Portier 1992).
Both SraG and pnp transcripts are more abundant in the absence of RNase III: pnp because of the absence of RNase III-dependent PNPase autoregulation and CsrA repression, and SraG because of its higher stability; SraG inhibition of pnp expression was strongest in the rnc strain (Fig. 3B). These results suggest that the SraG-mediated regulation of pnp may act as an additional factor in the control of PNPase expression to supplement its autoregulation and CsrA-dependent control of translation. The physiological role of SraG might be more important in conditions where RNase III activity is down-regulated, e.g., in response to stresses (Kim et al. 2008). This interplay in the control of SraG processing and PNPase expression thus contributes to PNPase homeostasis.
Potential SraG homologs located between the pnp and rpsO genes exist in various enterobacteria, such as Salmonella typhimurium, Shigella flexneri, and Klebsiella pneumoniae (Hershberg et al. 2003; Sridhar et al. 2009). All the Yersinia species exhibit the rpsO-pnp synteny (i.e., gene co-localization), but in Yersinia pestis an insertion element is annotated in the intergenic region of the rpsO-pnp operon. In Listeria monocytogenes, despite the same localization between the pnpA and rpsO genes, the RliD sRNA does not share high-sequence homology with SraG (Mandin et al. 2007). In Enterococcus faecalis, ref41 is transcribed from the opposite strand to the highly conserved pnpA gene, and it includes a potential translatable 43 amino acids ORF (Fouquier d'Herouel et al. 2011). The synteny of rpsO and pnp genes exists in the majority of bacteria (Supplemental Fig. S9). However, there is no colocalization in the order of Campylobacterales (Epsilon proteobacteria) and in many species of the Firmicutes. The regulation of pnp expression and a possible function of the sRNA has not been investigated in any of these bacteria.
The function of SraG was studied in Yersinia pseudotuberculosis. While no effect on pnp was reported, SraG was shown to negatively regulate the YPK_1206-1205 operon, which encodes genes that have no homologs outside the Yersinia genus (Lu et al. 2012). Other potential candidates were identified by proteomic analysis of the wt and ΔsraG mutant in this species but without further confirmation. Although we tried various strategies, we were unable to identify other putative SraG targets in E. coli. SraG may have additional targets in E. coli, which were not detected in our experimental conditions.
In spite of the redundancy in 3′–5′ exoribonucleases in E. coli, a pnp mutant exhibits specific phenotypes as compared to mutants deficient in RNase II or RNase R. In addition, PNPase is a component of the E. coli degradosome. A pnp mutant accumulates numerous RNA fragments produced by endoribonucleic cleavages due to a major defect in poly(A)-dependent degradation (Braun et al. 1996; Coburn and Mackie 1998). PNPase is involved in the turnover of sRNAs, the modulation of RNase II expression and in the processing of RliD-CRISPR in Listeria (Zilhao et al. 1996; Andrade and Arraiano 2008; De Lay and Gottesman 2011; Andrade et al. 2012; Sesto et al. 2014). PNPase enzymatic activity is dependent on magnesium-chelated citrate, ATP and c-di-GMP, thereby creating a network between metabolism and RNA turnover (Del Favero et al. 2008; Nurmohamed et al. 2011; Tuckerman et al. 2011). PNPase availability is a determinant for growth when bacteria are submitted to cold-shock, and for survival under oxidative stress, for UV radiation and for mitomycin C exposure (Zangrossi et al. 2000; Yamanaka and Inouye 2001; Haddad et al. 2009; Wu et al. 2009; Rath et al. 2012). PNPase is a global regulator of virulence and persistency in Salmonella enterica, and it has an impact on the physiology of Campylobacter jejuni (Clements et al. 2002; Haddad et al. 2012). It is clear that PNPase has specific roles in bacteria that cannot be fulfilled by the other 3′–5′ exoribonucleases. A tight control of the level of PNPase seems to be required for its pluripotent role in bacteria, and the results reported here indicate that SraG has a role in this fine-tuning in E. coli.
MATERIALS AND METHODS
Bacterial strains and plasmids
Bacterial strains, primers, and plasmids used in this work are listed in Supplemental Table S1. All constructions and mutations, described in the Supplemental Material, were confirmed by sequencing. Strains are derived from E. coli K12 MG1655 and N3433. Strains were grown in Luria-Bertani (LB) medium at 37°C or 30°C with appropriate antibiotics when needed.
β-Galactosidase activity
Twenty to 100 µM IPTG was added either at the beginning of the culture after diluting an overnight preculture to OD600 = 0.025 or during the exponential phase in cells containing pBRSraG or pBRSraG-S. Expression of the lacZ fusions was measured during the exponential phase. The cells were lysed in 800 µL Z buffer with 15 µL 0.1% SDS and 30 µL chloroform. β-Galactosidase activity was assayed as described by Miller (1972).
RNA extraction
Aliquots of 10 mL of cultures were mixed with 10 mL cold ethanol and stored at −20°C. After centrifugation, pellets were resuspended in 750 µL lysis buffer (0.5% SDS, 20 mM sodium acetate, 10 mM EDTA), 500 µL hot acid phenol (pH 4.5) was added, and samples were incubated at 65°C for 10 min. This step was repeated twice followed by a final extraction with chloroform. RNA were precipitated with ethanol, pellets were washed with 70% ethanol and resuspended in water. To determine RNA stability, rifampicin (500 µg/mL) was added to bacteria (OD650 = 0.4) and samples were taken at different times.
Northern blot analysis
RNA samples were separated on 6% polyacrylamide-urea gel and blotted to Hybond N+ membranes (Hajnsdorf et al. 1994b) to detect SraG and rpsO transcripts or on 1% agarose-formaldehyde gel (Hajnsdorf et al. 1994a) to detect pnp mRNA. Riboprobes were generated by in vitro transcription of PCR fragments with T7 RNA polymerase in the presence of [α-32P]UTP (Hajnsdorf and Régnier 2000). Primers used to generate PCR fragments with a T7 promoter are listed in Supplemental Table S1. 5S rRNA was used as a loading control.
In vitro processing by RNase III
DNA templates carrying a T7 promoter sequence for in vitro transcription were generated by PCR using the primers mT7-pnp1 and RNX1 for pnp, T7mSraG2 and sraGter2 for SraG-S, and T7mSraG1 and sraGter2 for SraG (Supplemental Table S1). RNA substrates were obtained by transcription of the PCR fragments using T7 RNA polymerase and purified on denaturing 6% polyacrylamide-urea gels (Folichon et al. 2005). pnp, SraG, and SraG-S RNA sizes were 237, 216, and 177 nt, respectively, with additional GGG at the 5′-end. 5′-end labeling was performed with [γ-32P]-ATP and T4 polynucleotide kinase (Folichon et al. 2005), 3′-end-labeling was performed with [32P]-pCp and T4 RNA ligase in 50 mM HEPES (pH 7.5), 20 mM MgCl2, 3 mM DTT, 0.1 mM ATP, 10% DMSO, 10 mg/mL BSA at 16°C for 16 h. The purified labeled RNAs in 1× TE were denaturated and incubated 30 min at 37°C with the unlabeled complementary strand to allow them to anneal. Hybridization efficiency between pnp mRNA and SraG RNAs was controlled on nondenaturing polyacrylamide gels (Folichon et al. 2005). RNase III digestion was conducted in 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 10 mM MgCl2, 0.1 mM DTT with increasing concentrations of RNase III (Epicentre) at 37°C for 30 min. After precipitation, addition of loading buffer and heat denaturation, samples were analyzed on 6% polyacrylamide-urea gels. Cleavage positions were identified by running radiolabeled DNA fragments, and RNase T1 and alkaline ladders of the same end-labeled RNA (Folichon et al. 2003).
Mapping of 5′- and 3′-extremities of RNA
To map RNase III cleavages on pnp mRNA, reverse transcription was performed with the radiolabeled RNX1 oligonucleotide and 5 µg total RNA by using Superscript II RT (Invitrogen). cDNAs were separated on a 6% polyacrylamide-urea gel.
To map 5′- and 3′-extremities of SraG, circular RT-PCR was performed with total RNA extracted from N3433 transformed by the pB15.6 plasmid to increase the gene dosage of the whole nusA-infB and rpsO-pnp operons (Plumbridge and Springer 1983; Braun et al. 1996). Plasmid DNA contamination was removed by DNase RQ1 digestion according to Promega's instructions. RNAs were re-extracted with phenol–chloroform and precipitated with ethanol. Five micrograms of RNA was treated with 20 units of 5′-polyphosphatase (Epicentre) to convert 5′-triphosphorylated RNA into 5′-monophosphorylated RNA. Five hundred nanograms of RNA treated or untreated by 5′-polyphosphatase were circularized with T4 RNA ligase (Biolabs) in denaturating buffer (50 mM HEPES, pH 7.5, 20 mM MgCl2, 3 mM DTT, 0.1 mM ATP, 10% DMSO, 10 µg/µL BSA) and subjected to RT-PCR across the 5′/3′ junction using the m423–442 primer to prime RT, then the oligonucleotide pair m1 and rpsO441 was used with the Triple Master Polymerase (Eppendorf) to generate PCR products, which were purified on agarose or acrylamide gels (Supplemental Fig. S2) and cloned into TOPO vector (Zero Blunt TOPOII Cloning, Invitrogen). Altogether, a total of 93 clones were sequenced and analyzed.
Toe-print analysis
The RNA fragments are the same as those used in the RNase III in vitro maturation assay. 1.2 pmol unlabeled pnp mRNA fragment (237 nt) and 4.8 pmol 5′-Cy5-end-labeled primer RNX1, an oligonucleotide complementary to nts 62–79 of pnp ORF were mixed with various amounts of SraG-S or SraG RNAs (0, 0.6, 1.2, 2.4, or 6 pmol). Two hundred and fifty micromolars dNTPs and 2.5 µM of initiator tRNAfMet were added with 0.5 µM 30S subunits, and the mixtures were incubated at 37°C for 10 min. One unit AMV RT (Finnzymes) was then added and cDNA synthesis was performed at 37°C for 20 min (Coornaert et al. 2013). A sequencing ladder was produced by asymmetric PCR with the pnp PCR fragment (15 fmol/µL) amplified in the presence of 5′-Cy5-end-labeled primer RNX1 and one of the 4 ddNTP mixes (at a ratio ddNTP:dNTP of 25:1 except for ddGTP which was at a ratio of 2.5:1). cDNAs and sequence ladders were separated on a 6% polyacrylamide-urea gel and visualized using a Typhoon fluorescent scanner set up for Cy5 detection.
Computational SraG target search
We performed a target search for SraG using the RNAup program from the Vienna package (http://www.tbi.univie.ac.at/RNA/RNAup.html). The SraG and SraG-S sequences were compared to each annotated E. coli CDS plus 50 nt 5′ to include the putative 5′UTR. Predicted sRNA/target pairs were ranked by free energy values, as estimated by RNAup.
Transcriptome analysis
Three independent cultures of FF4 cells transformed with the pBRplac (pBR), pBR-SraG (pSraG), and pBr-SraG short (pSraG-S) plasmids were grown to exponential phase. SraG and SraG-S synthesis was induced with 100 µM IPTG addition at OD600 = 0.4 for 7 min. RNA was extracted and incubated with DNase RQ1 (1 U/3 µg RNA). cDNAs were generated and labeled with either Cy3 or Cy5-Mono-Reactive Dye (Amersham) using the FairPlay III Microarrays Labeling Kit (Stratagene). Transcriptome experiments were performed with E. coli Gene Expression Microarrays 8 × 15 K (Agilent). Results were analyzed using two different statistical tests: Bonferroni and BH.
Western blotting
Frozen cells were resuspended in 0.1 M Tris-HCl (pH 8), 0.4 M NaCl, 0.1 mM EDTA, 1 mM 2-β-mercaptoethanol and lysed by sonication on ice. Protein concentrations were determined by the BCA protein assay (Pearce). Cellular proteins (10 µg) were separated on a 10% SDS–PAGE gel and transferred to Hybond C-super (Amersham) by electroblotting. Membranes were probed at the same time with 1/10,000 dilution of polyclonal PNPase antibodies and of polyclonal L20 antibodies followed by incubation with hRP anti-rabbit IgG in 5% milk. The membrane was developed using the kit Super Signal West Femto (Thermo Scientific).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are indebted to D. Gautheret for comments and suggestions and to J. Plumbridge and K. Tanner for discussions and critical reading of the manuscript. We thank C. Chiaruttini for advice, L20 antibodies, and providing materials to perform the toe-print experiment, C. Guerrier-Takada and M.P. Deutscher for strains, and A.J. Carpousis for PNPase antibodies. The DNA array analysis was performed by the ArraySud Plateform. This work was supported by the Centre National de la Recherche Scientifique (UPR9073 then FRE3630 and now UMR8261), University Paris-Diderot and Agence Nationale de la Recherche (asSUPYCO, ANR-12-BSV6-0007-03), and by the “Initiative d'Excellence” program from the French State (Grant “DYNAMO,” ANR-11-LABX-0011).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.055236.115.
REFERENCES
- Andrade JM, Arraiano CM. 2008. PNPase is a key player in the regulation of small RNAs that control the expression of outer membrane proteins. RNA 14: 543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade JM, Pobre V, Silva IJ, Domingues S, Arraiano CM. 2009. The role of 3′-5′ exoribonucleases in RNA degradation. Prog Mol Biol Transl Sci 85: 187–229. [DOI] [PubMed] [Google Scholar]
- Andrade JM, Pobre V, Matos AM, Arraiano CM. 2012. The crucial role of PNPase in the degradation of small RNAs that are not associated with Hfq. RNA 18: 844–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EGH, Margalit H, Altuvia S. 2001. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol 11: 941–950. [DOI] [PubMed] [Google Scholar]
- Bandyra KJ, Sinha D, Syrjanen J, Luisi BF, De Lay NR. 2016. The ribonuclease polynucleotide phosphorylase can interact with small regulatory RNAs in both protective and degradative modes. RNA 22: 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becket E, Tse L, Yung M, Cosico A, Miller JH. 2012. Polynucleotide phosphorylase plays an important role in the generation of spontaneous mutations in Escherichia coli. J Bacteriol 194: 5613–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beran RK, Simons RW. 2001. Cold-temperature induction of Escherichia coli polynucleotide phosphorylase occurs by reversal of its autoregulation. Mol Microbiol 39: 112–125. [DOI] [PubMed] [Google Scholar]
- Brantl S. 2007. Regulatory mechanisms employed by cis-encoded antisense RNAs. Curr Opin Microbiol 10: 102–109. [DOI] [PubMed] [Google Scholar]
- Braun F, Hajnsdorf E, Régnier P. 1996. Polynucleotide phosphorylase is required for the rapid degradation of the RNase E-processed rpsO mRNA of Escherichia coli devoid of its 3′ hairpin. Mol Microbiol 19: 997–1005. [DOI] [PubMed] [Google Scholar]
- Carpousis AJ, van Houwe G, Ehretsmann C, Krisch HM. 1994. Co-purification of E. coli RNase E and PNPase: evidence for a specific association between two enzymes important in RNA processing and degradation. Cell 76: 889–900. [DOI] [PubMed] [Google Scholar]
- Carpousis AJ, Luisi BF, McDowall KJ. 2009. Endonucleolytic initiation of mRNA decay in Escherichia coli. Prog Mol Biol Transl Sci 85: 91–135. [DOI] [PubMed] [Google Scholar]
- Carzaniga T, Briani F, Zangrossi S, Merlino G, Marchi P, Dehò G. 2009. Autogenous regulation of Escherichia coli polynucleotide phosphorylase expression revisited. J Bacteriol 191: 1738–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carzaniga T, Dehò G, Briani F. 2015. RNase III-independent autogenous regulation of Escherichia coli polynucleotide phosphorylase via translational repression. J Bacteriol 197: 1931–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Taylor DW, Fowler CC, Galan JE, Wang HW, Wolin SL. 2013. An RNA degradation machine sculpted by Ro autoantigen and noncoding RNA. Cell 153: 166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements MO, Eriksson S, Thompson A, Lucchini S, Hinton JC, Normark S, Rhen M. 2002. Polynucleotide phosphorylase is a global regulator of virulence and persistency in Salmonella enterica. Proc Natl Acad Sci 99: 8784–8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn GA, Mackie GA. 1998. Reconstitution of the degradation of the mRNA for ribosomal protein S20 with purified enzymes. J Mol Biol 279: 1061–1074. [DOI] [PubMed] [Google Scholar]
- Coornaert A, Chiaruttini C, Springer M, Guillier M. 2013. Post-transcriptional control of the Escherichia coli PhoQ-PhoP two-component system by multiple sRNAs involves a novel pairing region of GcvB. PLoS Genet 9: e1003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court DL, Gan J, Liang YH, Shaw GX, Tropea JE, Costantino N, Waugh DS, Ji X. 2013. RNase III: genetics and function; structure and mechanism. Annu Rev Genet 47: 405–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darfeuille F, Unoson C, Vogel J, Wagner EG. 2007. An antisense RNA inhibits translation by competing with standby ribosomes. Mol Cell 26: 381–392. [DOI] [PubMed] [Google Scholar]
- Davies BW, Kohrer C, Jacob AI, Simmons LA, Zhu J, Aleman LM, Rajbhandary UL, Walker GC. 2010. Role of Escherichia coli YbeY, a highly conserved protein, in rRNA processing. Mol Microbiol 78: 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lay N, Gottesman S. 2011. Role of polynucleotide phosphorylase in sRNA function in Escherichia coli. RNA 17: 1172–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Favero M, Mazzantini E, Briani F, Zangrossi S, Tortora P, Dehò G. 2008. Regulation of Escherichia coli polynucleotide phosphorylase by ATP. J Biol Chem 283: 27355–27359. [DOI] [PubMed] [Google Scholar]
- Folichon M, Arluison V, Pellegrini O, Huntzinger E, Régnier P, Hajnsdorf E. 2003. The poly(A) binding protein Hfq protects RNA from RNase E and exoribonucleolytic degradation. Nucleic Acids Res 31: 7302–7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folichon M, Allemand F, Régnier P, Hajnsdorf E. 2005. Stimulation of poly(A) synthesis by E. coli poly(A)polymerase I is correlated with Hfq binding to poly(A) tails. FEBS J 272: 454–463. [DOI] [PubMed] [Google Scholar]
- Fouquier d'Herouel A, Wessner F, Halpern D, Ly-Vu J, Kennedy SP, Serror P, Aurell E, Repoila F. 2011. A simple and efficient method to search for selected primary transcripts: non-coding and antisense RNAs in the human pathogen Enterococcus faecalis. Nucleic Acids Res 39: e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes K, Wagner EG. 2007. RNA antitoxins. Curr Opin Microbiol 10: 117–124. [DOI] [PubMed] [Google Scholar]
- Gerdes K, Christensen SK, Løbner-Olesen A. 2005. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol 3: 371–382. [DOI] [PubMed] [Google Scholar]
- Gopel Y, Papenfort K, Reichenbach B, Vogel J, Görke B. 2013. Targeted decay of a regulatory small RNA by an adaptor protein for RNase E and counteraction by an anti-adaptor RNA. Genes Dev 27: 552–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goverde RL, Huis in't Veld JH, Kusters JG, Mooi FR. 1998. The psychrotrophic bacterium Yersinia enterocolitica requires expression of pnp, the gene for polynucleotide phosphorylase, for growth at low temperature (5°C). Mol Microbiol 28: 555–569. [DOI] [PubMed] [Google Scholar]
- Grunberg-Manago M, Oritz PJ, Ochoa S. 1955. Enzymatic synthesis of nucleic acid like polynucleotides. Science 122: 907–910. [DOI] [PubMed] [Google Scholar]
- Guillier M, Gottesman S. 2006. Remodelling of the Escherichia coli outer membrane by two small regulatory RNAs. Mol Microbiol 59: 231–247. [DOI] [PubMed] [Google Scholar]
- Haddad N, Burns CM, Bolla JM, Prévost H, Federighi M, Drider D, Cappelier JM. 2009. Long-term survival of Campylobacter jejuni at low temperatures is dependent on polynucleotide phosphorylase activity. Appl Environ Microbiol 75: 7310–7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad N, Tresse O, Rivoal K, Chevret D, Nonglaton Q, Burns CM, Prévost H, Cappelier JM. 2012. Polynucleotide phosphorylase has an impact on cell biology of Campylobacter jejuni. Front Cell Infect Microbiol 2: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnsdorf E, Régnier P. 2000. Host factor Hfq of Escherichia coli stimulates elongation of poly(A) tails by poly(A)polymerase I. Proc Natl Acad Sci 97: 1501–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnsdorf E, Carpousis AJ, Régnier P. 1994a. Nucleolytic inactivation and degradation of the RNase III processed pnp message encoding polynucleotide phosphorylase of Escherichia coli. J Mol Biol 239: 439–454. [DOI] [PubMed] [Google Scholar]
- Hajnsdorf E, Steier O, Coscoy L, Teysset L, Régnier P. 1994b. Roles of RNase E, RNase II and PNPase in the degradation of the rpsO transcripts of Escherichia coli: stabilizing function of RNase II and evidence for efficient degradation in an ams-rnb-pnp mutant. EMBO J 13: 3368–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Altuvia S, Margalit H. 2003. A survey of small RNA-encoding genes in Escherichia coli. Nucleic Acids Res 31: 1813–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A, May T, Taniuchi A, Kawata K, Okabe S. 2009. Localized expression profiles of rpoS in Escherichia coli biofilms. Biotechnol Bioeng 103: 975–983. [DOI] [PubMed] [Google Scholar]
- Jarrige AC, Mathy N, Portier C. 2001. PNPase autocontrols its expression by degrading a double-stranded structure in the pnp mRNA leader. EMBO J 20: 6845–6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano M, Aravind L, Storz G. 2007. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol Microbiol 64: 738–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Manasherob R, Cohen SN. 2008. YmdB: a stress-responsive ribonuclease-binding regulator of E. coli RNase III activity. Genes Dev 22: 3497–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Derout J, Boni IV, Regnier P, Hajnsdorf E. 2010. Hfq affects mRNA levels independently of degradation. BMC Mol Biol 11: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Zhang Y, Li L, Hu Y, Huang L, Li Y, Rayner S, Chen S. 2012. Small non-coding RNA SraG regulates the operon YPK_1206-1205 in Yersinia pseudotuberculosis. FEMS Microbiol Lett 331: 37–43. [DOI] [PubMed] [Google Scholar]
- Luttinger A, Hahn J, Dubnau D. 1996. Polynucleotide phosphorylase is necessary for competence development in Bacillus subtilis. Mol Microbiol 19: 343–356. [DOI] [PubMed] [Google Scholar]
- Maes A, Gracia C, Hajnsdorf E, Régnier P. 2012. Search for poly(A) polymerase targets in E. coli reveals its implication in surveillance of Glu tRNA processing and degradation of stable RNAs. Mol Microbiol 83: 436–451. [DOI] [PubMed] [Google Scholar]
- Mandin P, Repoila F, Vergassola M, Geissmann T, Cossart P. 2007. Identification of new noncoding RNAs in Listeria monocytogenes and prediction of mRNA targets. Nucleic Acids Res 35: 962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massé E, Escorcia FE, Gottesman S. 2003. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev 17: 2374–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathy N, Jarrige AC, Robert-Le Meur M, Portier C. 2001. Increased expression of Escherichia coli polynucleotide phosphorylase at low temperatures is linked to a decrease in the efficiency of autocontrol. J Bacteriol 183: 3848–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- Numata S, Nagata M, Mao H, Sekimizu K, Kaito C. 2014. CvfA protein and polynucleotide phosphorylase act in an opposing manner to regulate Staphylococcus aureus virulence. J Biol Chem 289: 8420–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurmohamed S, Vincent HA, Titman CM, Chandran V, Pears MR, Du D, Griffin JL, Callaghan AJ, Luisi BF. 2011. Polynucleotide phosphorylase activity may be modulated by metabolites in Escherichia coli. J Biol Chem 286: 14315–14323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opdyke JA, Kang JG, Storz G. 2004. GadY, a small-RNA regulator of acid response genes in Escherichia coli. J Bacteriol 186: 6698–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opdyke JA, Fozo EM, Hemm MR, Storz G. 2011. RNase III participates in GadY-dependent cleavage of the gadX-gadW mRNA. J Mol Biol 406: 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SP, Minesinger BK, Kumar J, Walker GC. 2011. A highly conserved protein of unknown function in Sinorhizobium meliloti affects sRNA regulation similar to Hfq. Nucleic Acids Res 39: 4691–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Yakhnin H, Connolly M, Romeo T, Babitzke P. 2015. CsrA participates in a PNPase autoregulatory mechanism by selectively repressing translation of pnp transcripts that have been previously processed by RNase III and PNPase. J Bacteriol 197: 3751–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumbridge JA, Springer M. 1983. Organization of the Escherichia coli chromosome around the genes for translation initiation factor IF2 (inf2) and a transcription terminator factor (nusA). J Mol Biol 167: 227–243. [DOI] [PubMed] [Google Scholar]
- Portier C, Régnier P. 1984. Expression of the rpsO and pnp genes: structural analysis of a DNA fragment carrying their control regions. Nucleic Acids Res 112: 6091–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portier C, Dondon L, Grunberg-Manago M, Régnier P. 1987. The first step in the functional inactivation of the Escherichia coli polynucleotide phosphorylase messenger is a ribonuclease III processing at the 5′ end. EMBO J 6: 2165–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath D, Mangoli SH, Pagedar AR, Jawali N. 2012. Involvement of pnp in survival of UV radiation in Escherichia coli K-12. Microbiology 158 (Pt 5): 1196–1205. [DOI] [PubMed] [Google Scholar]
- Régnier P, Grunberg-Manago M. 1990. RNase III cleavages in non-coding leaders of Escherichia coli transcripts control mRNA stability and genetic expression. Biochimie 72: 825–834. [DOI] [PubMed] [Google Scholar]
- Régnier P, Hajnsdorf E. 1991. Decay of mRNA encoding ribosomal protein S15 of Escherichia coli is initiated by an RNaseE-dependent endonucleolytic cleavage that removes the 3′ stabilizing stem and loop structure. J Mol Biol 217: 283–292. [DOI] [PubMed] [Google Scholar]
- Régnier P, Hajnsdorf E. 2009. Poly(A)-assisted RNA decay and modulators of RNA stability. Prog Mol Biol Transl Sci 85: 137–185. [DOI] [PubMed] [Google Scholar]
- Reichenbach B, Maes A, Kalamorz F, Hajnsdorf E, Görke B. 2008. The small RNA GlmY acts upstream of the sRNA GlmZ in the activation of glmS expression and is subject to regulation by polyadenylation in Escherichia coli. Nucleic Acids Res 36: 2570–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuven NB, Zhou Z, Deutscher MP. 1997. Functional overlap of tRNA nucleotidyltransferase, poly(A)polymearse I and polynucleotide phosphorylase. J Biol Chem 272: 33255–33259. [DOI] [PubMed] [Google Scholar]
- Robert-Le Meur M, Portier C. 1992. E. coli polynucleotide phosphorylase expression is autoregulated through an RNase III-dependent mechanism. EMBO J 11: 2633–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert-Le Meur M, Portier C. 1994. Polynucleotide phosphorylase of Escherichia coli induces the degradation of its RNase III processed messenger by preventing its translation. Nucleic Acids Res 22: 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig JA, Chromy B, Echeverry A, Yang J, Adkins B, Plano GV, McCutchen-Maloney S, Schesser K. 2007. Polynucleotide phosphorylase independently controls virulence factor expression levels and export in Yersinia spp. FEMS Microbiol Lett 270: 255–264. [DOI] [PubMed] [Google Scholar]
- Sesto N, Touchon M, Andrade JM, Kondo J, Rocha EP, Arraiano CM, Archambaud C, Westhof E, Romby P, Cossart P. 2014. A PNPase dependent CRISPR System in Listeria. PLoS Genet 10: e1004065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar J, Kumar SS, Rafi ZA. 2009. Small RNA identification in Enterobacteriaceae using synteny and genomic backbone retention II. OMICS 13: 261–284. [DOI] [PubMed] [Google Scholar]
- Thomason MK, Storz G. 2010. Bacterial antisense RNAs: how many are there, and what are they doing? Annu Rev Genet 44: 167–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuckerman JR, Gonzalez G, Gilles-Gonzalez MA. 2011. Cyclic di-GMP activation of polynucleotide phosphorylase signal-dependent RNA processing. J Mol Biol 407: 633–639. [DOI] [PubMed] [Google Scholar]
- Vogel J, Luisi BF. 2011. Hfq and its constellation of RNA. Nat Rev Microbiol 9: 578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J, Bartels V, Tang TH, Churakov G, Slagter-Jager JG, Huttenhofer A, Wagner EG. 2003. RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res 31: 6435–6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EGH, Blomberg P, Nordström K. 1992. Replication control in plasmid R1: duplex formation between the antisense RNA, CopA and its target CopT is not required for inhibition of RepA synthesis. EMBO J 11: 1195–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Jiang Z, Liu M, Gong X, Wu S, Burns CM, Li Z. 2009. Polynucleotide phosphorylase protects Escherichia coli against oxidative stress. Biochemistry 48: 2012–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Inouye M. 2001. Selective mRNA degradation by polynucleotide phosphorylase in cold shock adaptation in Escherichia coli. J Bacteriol 183: 2808–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zangrossi S, Briani F, Ghisotti D, Regonesi ME, Tortora P, Dehò G. 2000. Transcriptional and post-transcriptional control of polynucleotide phosphorylase during cold acclimation in Escherichia coli. Mol Microbiol 36: 1470–1480. [DOI] [PubMed] [Google Scholar]
- Zhang A, Wassarman KM, Rosenow C, Tjaden BC, Storz G, Gottesman S. 2003. Global analysis of small RNA and mRNA targets of Hfq. Mol Microbiol 50: 1111–1124. [DOI] [PubMed] [Google Scholar]
- Zilhao R, Cairrao F, Régnier P, Arraiano CM. 1996. PNPase modulates RNase II expression in Escherichia coli: implications for mRNA decay and cell metabolism. Mol Microbiol 20: 1033–1042. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Deutscher MP. 2001. Exoribonuclease superfamilies: structural analysis and phylogenetic distribution. Nucleic Acids Res 29: 1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.