

Abstract
In principle, serial femtosecond crystallography (SFX) could yield data sets that are completely free of the effects caused by slow, radiation‐induced chemical reactions, for example, oxygen additions, responsible for radiation damage. However, experimental evidence is presented here that SFX data sets obtained by techniques that expose different parts of the same specimen to single pulses of radiation do not have this property, even if the specimen in question is frozen. The diffraction image of each such crystal obtained with the first pulse of radiation is certain to represent the structure of a protein that has not been modified chemically, but all of the images obtained subsequently from the same crystal will represent structures that have been modified to a lesser or greater extent by oxygen additions because of the rapid diffusion of oxygenic free radicals through the specimen. The higher the level of oxygen additions a crystal suffers during data collection, the poorer the statistical quality of data set obtained from it will, and the higher the free R‐factors of the resulting structural model.
Keywords: X‐ray free‐electron laser, XFEL, serial femtosecond crystallography, SFX, radiation chemistry, free radical chain reaction, dirty diffraction data, dirty enzyme, cryroprotectants, free radical amplifiers
Introduction
The structural biology community has long sought to improve the quality of the data obtained from weakly diffracting protein crystals by developing ever more intense radiation sources, as well as other techniques that make it possible to increase the total X‐ray dose crystals are exposed to during data collection. Although significant improvements in both <I/σI> ratios and the resolutions of data sets have resulted in many cases, paradoxically, a corresponding improvement in the quality of the resulting models often has not.1 In fact, the lower the dose of X‐rays crystals are exposed to during data collection, the lower the free R‐factors of the resulting models are likely to be.1 In this regard, it is instructive to compare two models for cytochrome c peroxidase (CcP) reported for 3M23 and 3E2O.2, 3 The former is based on data collected using a severely attenuated synchrotron beam, and the latter derives from data collected using a beam about 100 times more intense. The resolution of the 3M23 data set is worse than that the 3E2O data set, 1.40‐Å versus 1.06‐Å, but the free R‐factor of the model obtained from it is much better, 13.4% versus 19.9%.2, 3 In addition, Burmeister has documented the effect of X‐ray dose on structure quality by comparing structures derived from a succession of data sets that were collected from a single crystal. The free R‐factor of model obtained using the first data set is much better than that of the model derived from the fifth such data set, 17.9% versus 28.9%.4 Both observations indicate that high X‐ray dose has a deleterious effect on structure quality, and there are now indications that this may be true also of structures obtained from pseudo‐oscillation data that have been collected from frozen protein crystals using serial femtosecond crystallographic (SFX) methods. A CcP model produced this way reported for 5EJX has a nominal resolution nearly as high as that of 3M23, 1.50‐Å, but its free R‐factor is much worse, 26.1% versus 13.4%.5
The relationship between X‐ray dose and free R‐factor is the topic of this study. It will be shown that high‐dosage data sets are often much less accurate than data sets collected at lower doses from the same crystals. A crystallographic explanation will be offered for why the true quality of high‐dosage data sets, as measured by model free R‐factors, is often not as good as expected based on the commonly used quality indexes for data sets, for example, resolution, merging R‐factor, and/or mean <I/σI> values.
In the past, most structural biologists have equated radiation damage with the general decrease in resolution that invariably accompanies it, and have paid no attention to the fact that the relative intensities of reflections often change as it occurs, with some reflections actually increasing in intensity. These relative alterations in intensities imply that the average structures of the unit cells in macromolecular crystals are altered by exposure to X‐rays in ways that cannot be represented as a simple, progressive increase in average B‐factor. In fact, I recently showed that exposure to X‐rays results in a dose‐dependent addition of oxygen atoms to the C‐H bonds of amino acids, the effects of which are clearly visible in electron density maps; alanines get transformed into serines, for example.6 Thus the structure of a protein crystal being used for data collection may be changing chemically during data collection in ways that make data reduction a problematic process.
The chemical reactions responsible for oxygen insertions and other kinds of damage of that sort are slow compared to the duration of the femtosecond pulses produced by X‐ray free electron lasers. Thus, there is reason to hope that the effects caused by radiation‐induced chemistry might be circumvented using SFX data collection methods, i.e. “diffraction‐before‐destruction” techniques.7, 8, 9 My purpose here is to show that one of the methods now being used to collect SFX data does not achieve this goal. The method in question involves exposing either different parts of a single large frozen crystal, typically 50 − 60 μm apart, or different small crystals within a single frozen sample to a single femtosecond pulse of X‐rays with long pauses between pulses so that the sample can be translated to bring fresh material into the beam. It will become clear that these pauses are so long that free radicals generated in one part of the crystal can diffuse to adjacent regions, and damage them before they are exposed to X‐rays. In fact, the experimental data available indicate that crystals from which data are collected in this way suffer from much more radiation‐induced damage than they would have if data had been collected from them using a lower‐dose, conventional synchrotron. The problem discussed here differs from another recent study on destruction‐while‐diffraction by X‐ray free‐electron laser.10
Results
Evidence for oxygen addition in SFX structures
The atomic‐resolution SFX model5 assigned the PDB accession number 5EJX was re‐refined using the data set deposited for 5EJX out to a resolution of 1.50‐Å resolution with H atoms included in riding positions. The refined model had R‐factor of 19.7% and free R‐factor of 25.0%. A σ A‐weighted (F obs‐F model, or ΔF) difference electron density map was then calculated to identify discrepancies between the structure of the crystal and the model proposed for it.11 That map includes many positive features having amplitudes above +3.0σ that are adjacent to, but slightly outside the H atoms of C‐H bonds (Fig. 1). The residual peaks that next to the Cα‐H bonds of Tyr71, Lys278, Asn200, Leu270, and Gln120, for example, can be explained by hypothesizing that O atoms have been inserted into the C‐H bonds in question (Fig. 1). The same is true of the positive peaks next to the C‐H bonds of the terminal methyl C atoms of Ala147, Thr234, and Leu232 (Fig. 1). Other such peaks are also evident next to methylene C‐H bonds, for example Lys243 (Fig. 1). Some other peaks that may represent oxygen insertions could be explained by proposing that the side chains with which they are associated have alternative conformations. However, when the model was modified this way, in subsequent cycles of model refinement, new large negative peaks often appeared in the ΔF maps obtained in the second C atoms next to the branched point of densities. It is important to point out that all these inserted O atoms were also clearly visible in σA‐weighted F‐map at above 0.8σ level (Fig. 1). In total, 23% of the protein residues in the structure corresponding to the 5EJX data set appear to have at least one O atom inserted, and many have multiple O atoms inserted.
Figure 1.
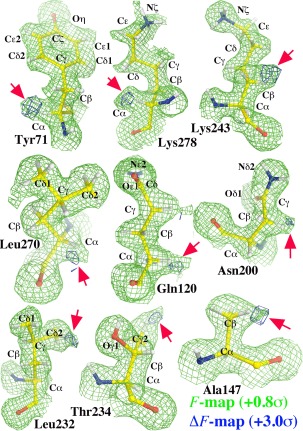
Conventional σ A‐weighted F‐map (contoured at +0.8σ, green) and ΔF‐map (at +3.0σ, blue) for inserted O atoms.
Using the same method, clear evidence for the additions of O atoms to the structures reported in the SFX data sets with PDB accession numbers of 3WG7 for cytochrome c oxidase (CcO) and of 4UB8/4UB6 for photosystem II (PSII) could be seen in both conventional ΔF and F maps following careful model re‐refinement.12, 13 Using a direct methods‐generated E‐map,6 it was estimated that about 30% of the residues in the CcO structure corresponding to the conventional synchrotron data set reported for 2DYR had multiple additions of O atoms.6, 14 However, peaks corresponding to added O atoms in the 2DYR structure were much smaller than those found in the structure corresponding to the 3ARC data set for PSII, even though the number of residues modified is about the same.6, 15 When CcO structures corresponding to both the SFX data set of 3WG7 and the conventional data set of 2DYR are modified by additions of O atoms, isomorphous difference Fourier maps between them show no peaks for added O atoms.10 The observations reported here argue strongly against the claims that have been made recently by several groups that data sets collected by SFX represent undamaged structures, which have collected SFX data sets using the same methodology of translating large frozen crystals.5, 12, 13 This conclusion is also supported by our recent quantum mechanism and molecular modeling calculations which bear on the issue of whether or not the PSII models derived from SFX data sets of 4UB8/4UB6 indeed represent the intermediate state its authors intended.13, 16
In addition to direct visualization of added O atoms to the protein residues described above and previously,6 other statistical properties such as model free R‐factors, data merging R‐factors, and the L‐test17 can provide clues about the degree to which protein structures change during data collection. As will shortly become obvious, the higher the level of oxidation suffered by a crystal of during data collection, the higher the free R‐factors for models being derived from those data, no matter whether they are SFX data or conventional synchrotron data, the higher the merging R‐factors (typically for SFX data sets only), and the poorer the L‐test scores.17
Correlation between free R‐factor and the level of O additions or X‐ray dose
Since the data sets and corresponding models for 3E2O, 3M23, and 5EJX, which describe the same CcP enzyme in the same space group, were obtained by the same laboratory,2, 3, 5 it is safe to assume that the efforts were made to refine these models were similar, and hence that their free R‐factors could correlate with the extent of the radiation‐induced modifications that occurred during data collection. My analysis shows that the level of O addition that accompanied the collection of the conventional synchrotron data set that gave rise to 3E2O is far lower than the level that occurred during the collection of the data set for 5EJX (Supporting Information Fig. S1), and that the level of radiation‐induced oxidation evident in 3M23, the data for which were collected at low dose, is too low to detect2, 3, 5 (the data set for 3M23 provides evidence for only a single O addition; it occurred at Cys128, and is almost certainly a consequence of the treatment of the corresponding crystals with 10 mM H2O2). Consistent with hypothesis, 5EJX, the structure with the highest level of oxygen addition, had the highest free R‐factor, 26.1% (at 1.50‐Å resolution), 3M23, the low dose structure, which shows the least evidence of oxygen addition, has the lowest free R value, 13.4% (at 1.40‐Å resolution), and 3E2O, which suffered an intermediate level of oxygen addition has free R‐factor in between, 19.9% (at 1.06‐Å resolution). This finding is consistent with what Meents and colleagues have reported for all the protein crystals they have studied.1
Rossmann recently expressed concern about the quality of the structure described by 4N4Z, which was obtained from a SFX data set that was reported to have an overall intensity merging R‐factor of 71%.18, 19 In fact, there is nothing special about that data set. All the SFX data sets collected using pseudo‐oscillation methods have unusually high merging R‐factors, which must have a correspondingly adverse impact on the free R‐factors of the structure derived from them.5, 12, 13 Merging R‐factors this high raise question about the validity of the assumption that the frames of data that were merged to generate such data sets all represent the same protein structure.
When the SFX data sets corresponding to 5EJX and 3WG7 were subjected to the L‐test,5, 12, 17 they were all found to exhibit the statistical properties expected for data sets collected from highly twinned crystals (Fig. 2). Since the space group of these particular crystals is P212121 in which twinning is not physically possible, this observation is likely to be a consequence of the fact that these data sets were constructed by merging frames of data obtained from crystals that differed significantly in structure, for example the original, unmodified crystal, and all the variants of that crystal progressively generated by the radiation damage it suffered during data collects (and in some instances the data may have been misprocessed in other ways). In addition, because X‐ray free‐electron laser pulse is generated by a highly stochastic self‐amplification self‐emission process having large intrinsic fluctuations, no two individual XFEL pulses would be alike in term of their intensity profiles. Thus, individual XFEL pulses may have recorded different dying processes of protein crystals. The effect of merging dissimilar intensity measurements is to create a more average intensity distribution, which was detected in the abnormal L‐test results discussed here.
Figure 2.
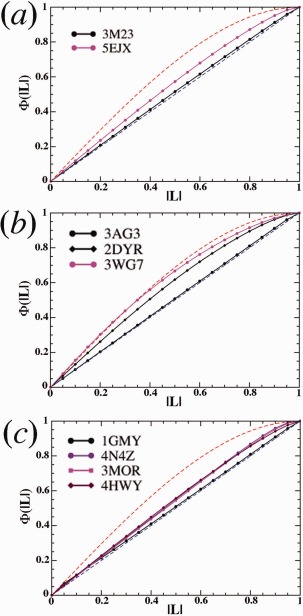
L‐tests for various data sets as indicated (see text). Theoretically nontwinned L‐test results are shown in blue dashed line, and theoretically perfectly twinned L‐test results are shown in red dashed curves. L‐test results for observed data are in solid lines. In case of 3AG3 in panel b and 1GMY in panel c, experimental results are overlaid on the top of the theoretically lines.
For comparison, the synchrotron data sets for 3M23, which represents the same CcP intermediate as 5EJX, and for 3AG3, which should correspond more or less exactly to 3WG7,2 show no signs of twinning at all in an L‐test (Fig. 2). Similarly, all the SFX data sets18, 20, 21 for 4N4Z, 3MOR, and 4HWY, which are not metalloproteins and have been collected in the same methodology, also exhibit highly twinning‐like properties, but the synchrotron data set22 of 1GMY shows no signs of aberrant intensity statistics (Fig. 2), from which the structure obtained was used to solve the related SFX structures by molecular replacement.
Discussion
The evidence presented in this study establishes that radiation‐induced addition of oxygen atoms to protein residues occurs when SFX data sets are collected using pseudo‐oscillation or spiral‐translation methods. The criterion often used by SFX investigators to determine the minimum separation between successive shots on large frozen crystals is preservation of resolution,5, 12, 13 an approach rendered questionable by the recent observation that oxygenic insertions do not necessarily result in a reduction in diffraction intensities.6 Because the total X‐ray dose in each femtosecond pulse is enormous, the concentration of oxygenic free radicals that is generated in the affected volume during a single pulse is many order magnitudes higher than that characteristic of crystals exposed to a conventional radiation source. Free radicals do not just disappear during data collection. They are either immobilized by interactions with protein residues such as at tertiary Cα‐H bonds, or continue to propagate through frozen samples. Moreover, the half‐life of free radicals increases with decreasing temperature. Worse, small diffusible organic molecules like ethylene glycol, glycerol, or other low‐molecular weight organic alcohols, which are sometimes used to prevent the formation of ice crystals in frozen samples, may amplify and propagate free radical chain reactions via reaction schemes like the following:1, 23
On average, each X‐ray photon absorbed by a 10% ethylene glycol solution can result in the generation of 400 molecules of H2 gas through free radical chain reactions of this type.1
Some investigators have used the appearance of model‐phased electron density maps to assess the quality of SFX data sets, but the validity of this approach is questionable. The fact that a model‐phased electron density map contains protein‐like features tells us nothing about the amount of useful structural information contained in the parent data set because the phases used will contribute much more to the appearance of such a map than the observed amplitudes.24, 25 The σA‐weighting function often used in computing model‐phased maps will reduce model bias when the data contain strong structural information, but it will not when data contain no structural information at all. A model‐phased electron density map computed using randomly assigned, positive‐only amplitudes will be reasonably well fit by the atomic model used to compute phases.24, 25
Finally, the phenomenon explored here can explain an observation that appeared to make no sense at all when it was initially made. As described in supporting information of a previous paper26 on quality of the models deposited in the PDB that derive data sets belonging to the space group of P212121, a very large fraction of those data sets exhibited some degree of twinning when subjected to the L‐test.17 It turned out that within this set of structures a correlation exists between their free R‐factors and the apparent degree of twinning L‐test scores in the corresponding data sets. The higher the apparent twinning‐like L‐test scores, the poorer the free R‐factor. In P212121, calculated intensities from any properly refined atomic models must display the intensity distribution predicted by Wilson,27 and thus cannot account fully for the measured data if it has twinning‐like statistical properties. This explains why high free‐R factors correlate with high apparent twinning‐like L‐test scores. This observation is consistent with what Meents et al. observed,1 and it now appears likely that O additions caused by exposure to radiation accounts for the apparent twinning‐like properties in these data sets.
Concluding Remarks
Arthur Kornberg once said that one should not waste clean thinking on dirty enzymes;28 and his adage is applicable to structural biology. Do not waste clean thinking on dirty diffraction data. Yet, structural biologists have unknowingly been working with diffraction data that was dirtier than they realized for the last few decades. It remains unclear how many conclusions about the mechanisms of enzymatic reactions and protein dynamics have been affected. For example, unrecognized compositional heterogeneity due to radiation‐induced additions of O atoms will often have been interpreted as evidence of conformational heterogeneity indicative of enzyme dynamics. Unrecognized radiation‐induced oxidation/reduction of redox enzymes is sure to mislead us about the actual oxidation states of enzyme intermediates. It is clear that the challenge of obtaining clean data sets for protein structure determination remains unmet, and that so far SFX has not yet solved this problem.
Methods
All diffraction data sets and corresponding models were retrieved from the PDB.29 Models were refined using Refmac5 and rebuilt with the graphics program Coot.30, 31 The statistical L‐test was carried out using the CCP4 package.17, 32 Figures were made using the program Pymol.33
Supporting information
Supporting Information
Acknowledgement
The author thanks Professor P. B. Moore for simulating discussion and for editing this manuscript.
References
- 1. Meents A, Gutmann S, Wagner A, Schulze‐Briese C (2010) Origin and temperature dependence of radiation damage in biological samples at cryogenic temperatures. Proc Natl Acad Sci USA 107:1094–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Meharenna YT, Doukov T, Li H, Soltis SM, Poulos TL (2010) Crystallographic and single‐crystal spectral analysis of the peroxidase ferryl intermediate. Biochemistry 49:2984–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meharenna YT, Oertel P, Bhaskar B, Poulos TL (2008) Engineering ascorbate peroxidase activity into cytochrome c peroxidase. Biochemistry 47:10324–10332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burmeister WP (2000) Structural changes in a cryo‐cooled protein crystal owing to radiation damage. Acta Cryst D56:328–341. [DOI] [PubMed] [Google Scholar]
- 5. Chreifi G, Baxter EL, Doukov T, Cohen AE, McPhillips SE, Song J, Meharenna YT, Soltis SM, Poulos TL (2016) Crystal structure of the pristine peroxidase ferryl center and its relevance to proton‐coupled electron transfer. Proc Natl Acad Sci USA 113:1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang J (2016) X‐ray radiation‐induced addition of oxygen atoms to protein residues. Protein Sci. 25:1407–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neutze R, Wouts R, van der Spoel D, Weckert E, Hajdu J (2000) Potential for biomolecular imaging with femtosecond X‐ray pulses. Nature 406:752–757. [DOI] [PubMed] [Google Scholar]
- 8. Chapman HN, Fromme P, Barty A, White TA, Kirian RA, Aquila A, Hunter MS, Schulz J, DePonte DP, Weierstall U, Doak RB, Maia FR, Martin AV, Schlichting I, Lomb L, Coppola N, Shoeman RL, Epp SW, Hartmann R, Rolles D, Rudenko A, Foucar L, Kimmel N, Weidenspointner G, Holl P, Liang M, Barthelmess M, Caleman C, Boutet S, Bogan MJ, Krzywinski J, Bostedt C, Bajt S, Gumprecht L, Rudek B, Erk B, Schmidt C, Homke A, Reich C, Pietschner D, Struder L, Hauser G, Gorke H, Ullrich J, Herrmann S, Schaller G, Schopper F, Soltau H, Kuhnel KU, Messerschmidt M, Bozek JD, Hau‐Riege SP, Frank M, Hampton CY, Sierra RG, Starodub D, Williams GJ, Hajdu J, Timneanu N, Seibert MM, Andreasson J, Rocker A, Jonsson O, Svenda M, Stern S, Nass K, Andritschke R, Schroter CD, Krasniqi F, Bott M, Schmidt KE, Wang X, Grotjohann I, Holton JM, Barends TR, Neutze R, Marchesini S, Fromme R, Schorb S, Rupp D, Adolph M, Gorkhover T, Andersson I, Hirsemann H, Potdevin G, Graafsma H, Nilsson B, Spence JC (2011) Femtosecond X‐ray protein nanocrystallography. Nature 470:73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Emma P, Akre R, Arthur J, Bionta R, Bostedt C, Bozek J, Brachmann A, Bucksbaum P, Coffee R, Decker FJ, Ding Y, Dowell D, Edstrom S, Fisher A, Frisch J, Gilevich S, Hastings J, Hays G, Hering P, Huang Z, Iverson R, Loos H, Messerschmidt M, Miahnahri A, Moeller S, Nuhn HD, Pile G, Ratner D, Rzepiela J, Schultz D, Smith T, Stefan P, Tompkins H, Turner J, Welch J, White W, Wu J, Yocky G, Galayda J (2010) First lasing and operation of an angstrom‐wavelength free‐electron laser. Nature Photonics 4:641–647. [Google Scholar]
- 10. Wang J (2016) Destruction‐and‐diffraction by X‐ray free electron laser. Protein Sci. In press, DOI: 10.1002/Pro.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Read RJ (1986) Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Cryst A42:140–149. [Google Scholar]
- 12. Hirata K, Shinzawa‐Itoh K, Yano N, Takemura S, Kato K, Hatanaka M, Muramoto K, Kawahara T, Tsukihara T, Yamashita E, Tono K, Ueno G, Hikima T, Murakami H, Inubushi Y, Yabashi M, Ishikawa T, Yamamoto M, Ogura T, Sugimoto H, Shen JR, Yoshikawa S, Ago H (2014) Determination of damage‐free crystal structure of an X‐ray‐sensitive protein using an XFEL. Nat Methods 11:734–736. [DOI] [PubMed] [Google Scholar]
- 13. Suga M, Akita F, Hirata K, Ueno G, Murakami H, Nakajima Y, Shimizu T, Yamashita K, Yamamoto M, Ago H, Shen JR (2015) Native structure of photosystem II at 1.95 A resolution viewed by femtosecond X‐ray pulses. Nature 517:99–103. [DOI] [PubMed] [Google Scholar]
- 14. Shinzawa‐Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S (2007) Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. Embo J 26:1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Umena Y, Kawakami K, Shen JR, Kamiya N (2011) Crystal structure of oxygen‐evolving photosystem II at a resolution of 1.9 A. Nature 473:55–60. [DOI] [PubMed] [Google Scholar]
- 16. Askerka M, Wang J, Vinyard DJ, Brudvig GW, Batista VS (2016) S3 State of the O2‐evolving complex of photosystem II: Insights from QM/MM, EXAFS, and femtosecond X‐ray diffraction. Biochemistry 55:981–984. [DOI] [PubMed] [Google Scholar]
- 17. Padilla JE, Yeates TO (2003) A statistic for local intensity differences: robustness to anisotropy and pseudo‐centering and utility for detecting twinning. Acta Cryst D59:1124–1130. [DOI] [PubMed] [Google Scholar]
- 18. Gati C, Bourenkov G, Klinge M, Rehders D, Stellato F, Oberthur D, Yefanov O, Sommer BP, Mogk S, Duszenko M, Betzel C, Schneider TR, Chapman HN, Redecke L (2014) Serial crystallography on in vivo grown microcrystals using synchrotron radiation. IUCrJ 1:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rossmann MG (2014) Serial crystallography using synchrotron radiation. IUCrJ 1:84–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koopmann R, Cupelli K, Redecke L, Nass K, Deponte DP, White TA, Stellato F, Rehders D, Liang M, Andreasson J, Aquila A, Bajt S, Barthelmess M, Barty A, Bogan MJ, Bostedt C, Boutet S, Bozek JD, Caleman C, Coppola N, Davidsson J, Doak RB, Ekeberg T, Epp SW, Erk B, Fleckenstein H, Foucar L, Graafsma H, Gumprecht L, Hajdu J, Hampton CY, Hartmann A, Hartmann R, Hauser G, Hirsemann H, Holl P, Hunter MS, Kassemeyer S, Kirian RA, Lomb L, Maia FR, Kimmel N, Martin AV, Messerschmidt M, Reich C, Rolles D, Rudek B, Rudenko A, Schlichting I, Schulz J, Seibert MM, Shoeman RL, Sierra RG, Soltau H, Stern S, Struder L, Timneanu N, Ullrich J, Wang X, Weidenspointner G, Weierstall U, Williams GJ, Wunderer CB, Fromme P, Spence JC, Stehle T, Chapman HN, Betzel C, Duszenko M (2012) In vivo protein crystallization opens new routes in structural biology. Nat Methods 9:259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Redecke L, Nass K, DePonte DP, White TA, Rehders D, Barty A, Stellato F, Liang M, Barends TR, Boutet S, Williams GJ, Messerschmidt M, Seibert MM, Aquila A, Arnlund D, Bajt S, Barth T, Bogan MJ, Caleman C, Chao TC, Doak RB, Fleckenstein H, Frank M, Fromme R, Galli L, Grotjohann I, Hunter MS, Johansson LC, Kassemeyer S, Katona G, Kirian RA, Koopmann R, Kupitz C, Lomb L, Martin AV, Mogk S, Neutze R, Shoeman RL, Steinbrener J, Timneanu N, Wang D, Weierstall U, Zatsepin NA, Spence JC, Fromme P, Schlichting I, Duszenko M, Betzel C, Chapman HN (2013) Natively inhibited Trypanosoma brucei cathepsin B structure determined by using an X‐ray laser. Science 339:227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Greenspan PD, Clark KL, Tommasi RA, Cowen SD, McQuire LW, Farley DL, van Duzer JH, Goldberg RL, Zhou H, Du Z, Fitt JJ, Coppa DE, Fang Z, Macchia W, Zhu L, Capparelli MP, Goldstein R, Wigg AM, Doughty JR, Bohacek RS, Knap AK (2001) Identification of dipeptidyl nitriles as potent and selective inhibitors of cathepsin B through structure‐based drug design. J Med Chem 44:4524–4534. [DOI] [PubMed] [Google Scholar]
- 23. Meents A, Dittrich B, Gutmann S (2009) A new aspect of specific radiation damage: hydrogen abstraction from organic molecules. J Synchrotron Radiat 16:183–190. [DOI] [PubMed] [Google Scholar]
- 24. Lattman E, DeRosier D (2008) Why phase errors affect the electron function more than amplitude errors. Acta Crystallographica Section a 64:341–344. [DOI] [PubMed] [Google Scholar]
- 25. Dodson E (2008) The befores and afters of molecular replacement. Acta Cryst D64:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J (2015) Estimation of the quality of refined protein crystal structures. Protein Sci 24:661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilson AJC (1949) The probability distribution of X‐ray intensities. Acta Cryst 2:318–321. [Google Scholar]
- 28. Kornberg A (2000) Ten commandments: lessons from the enzymology of DNA replication. J Bacteriol 182:3613–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berman HM, Kleywegt GJ, Nakamura H, Markley JL (2012) The Protein Data Bank at 40: reflecting on the past to prepare for the future. Structure 20:391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum‐likelihood method. Acta Cryst D53:240–255. [DOI] [PubMed] [Google Scholar]
- 31. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 32. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Cryst D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. The PyMOL Molecular Graphics System: 2002, version 1.8. Schrödinger, LLC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information