

Abstract
Urea amidolyase (UAL) is a multifunctional biotin‐dependent enzyme that contributes to both bacterial and fungal pathogenicity by catalyzing the ATP‐dependent cleavage of urea into ammonia and CO2. UAL is comprised of two enzymatic components: urea carboxylase (UC) and allophanate hydrolase (AH). These enzyme activities are encoded on separate but proximally related genes in prokaryotes while, in most fungi, they are encoded by a single gene that produces a fusion enzyme on a single polypeptide chain. It is unclear whether the UC and AH activities are connected through substrate channeling or other forms of direct communication. Here, we use multiple biochemical approaches to demonstrate that there is no substrate channeling or interdomain/intersubunit communication between UC and AH. Neither stable nor transient interactions can be detected between prokaryotic UC and AH and the catalytic efficiencies of UC and AH are independent of one another. Furthermore, an artificial fusion of UC and AH does not significantly alter the AH enzyme activity or catalytic efficiency. These results support the surprising functional independence of AH from UC in both the prokaryotic and fungal UAL enzymes and serve as an important reminder that the evolution of multifunctional enzymes through gene fusion events does not always correlate with enhanced catalytic function.
Keywords: multifunctional, multidomain, substrate channeling, biotin, evolution, enzyme
Introduction
Many multifunctional enzymes and multienzyme complexes have evolved to efficiently perform sequential catalytic reactions via the modular assembly of multiple catalytic domains within a single enzyme complex. The advantages of such systems are numerous.1 Most notably, the efficient channeling of substrates and/or intermediates between active sites and the coordination of catalytic turnover between disparate domains is a well‐accepted rationale for the evolution of multicomponent enzyme systems.2
The enzyme urea amidolyase (UAL) offers a relatively straightforward system in which to further explore multifunctional enzyme catalysis. In Candida albicans, which can cause lethal systemic infections in immunocompromised patients, UAL is a virulence factor regulating the yeast to hyphae switch.3, 4 In the pathogenic bacterium, Granulibacter bethesdensis, degradation of urea by UAL facilitates survival in macrophages and neutrophils, allowing Granulibacter to persist in patients with chronic granulomatous disease.5
UAL hydrolyzes urea into NH3 and CO2 in a two‐step, biotin‐dependent process catalyzed by two separate enzyme activities: urea carboxylase (UC) and allophanate hydrolase (AH)6 (Scheme 1). UC transfers a carboxyl group from bicarbonate to urea, forming allophanate (carbamoylcarbamate). This takes place in two separate catalytic domains of UC: the biotin carboxylase (BC) domain, where a tethered biotin cofactor is carboxylated by bicarbonate with concomitant ATP cleavage, and the carboxyltransferase (CT) domain, where a carboxyl group is transferred from carboxybiotin to urea, forming allophanate. Allophanate is subsequently hydrolyzed to ammonia and CO2 by AH. In many fungi, including Saccharomyces cerevisiae and C. albicans, the UC and AH activities are both contained within a single polypeptide chain whereas, in bacteria, UC and AH exist as two physically distinct enzymes.7, 8, 9 Recently, the X‐ray crystal structures of bacterial and fungal AH10, 11, 12 and fungal UC13 were determined in isolation, but the spatial relationship and the mechanism of intermediate transfer between UC and AH has not been described in detail for either fungal or bacterial UAL.
Scheme 1.
UAL hydrolyzes urea into NH3 and CO2 in a two‐step, biotin‐dependent process.
UC and AH display a close evolutionary and functional association. They catalyze consecutive reactions in the urea degradation pathway. Phylogenetic analysis indicates that, in 14 out of 17 bacteria studied, genes encoding UC and AH are localized adjacent to one another.7 It has been suggested that UC and AH co‐evolved in bacteria and, following horizontal gene transfer, subsequently fused into a single UAL gene in fungi.7 Furthermore, while UC and AH exist as separate enzymes in prokaryotes, the ATP cleavage reaction catalyzed by UC is reported to be tightly coupled with ammonia release from AH when the enzymes are assayed in tandem.9 This suggests that allophanate is efficiently transferred from UC to AH and, consequently, it can be hypothesized that UC and AH functionally interact to directly channel the allophanate intermediate between active sites. However, the hypothesis of substrate channeling between UC and AH has never been carefully tested.
Here, we investigate the potential for communication between the AH and UC enzyme components of both prokaryotic and fungal UAL. In addition, we use the method developed by Geck and Kirsch to investigate substrate channeling through competition with inactivated enzyme.14, 15 The results do not support substrate channeling: no stable complex was detected between the two enzymes and kinetic assays offered no evidence for transient interactions. The addition of two potential scaffolding proteins does not assist complex formation nor facilitate substrate channeling in vitro. UC and AH from G. bethesdensis were genetically recombined to generate a single, fused polypeptide chain and neither the fused G. bethesdensis UAL nor the full‐length UAL from S. cerevisiae and C. albicans enhanced the overall catalytic efficiency relative to the individual bacterial enzymes assayed in tandem. Taken together, our results do not support substrate channeling between UC and AH in either prokaryotic or fungal UAL, suggesting that the coevolution and fusion of these two enzyme activities into a multifunctional enzyme was not driven by the pressure to maximize the efficiency of catalytic turnover.
Results
To determine whether the allophanate intermediate is channeled from UC to AH, we investigated complex formation, enzyme activities and direct channeling in the bacterial UC‐AH dual enzyme system and in the intact fungal UAL enzyme.
PsUC and PsAH do not form a stable complex in vitro
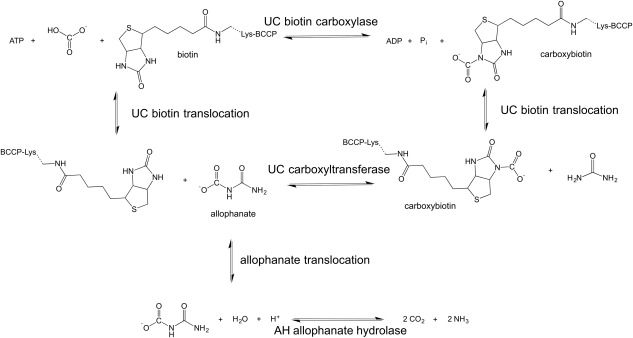
To directly test whether UC and AH associate in vitro, His‐tagged Pseudomonas syringae UC (PsUC) and AH (PsAH) were separately purified and were coapplied to a size‐exclusion column. No mobility shift was observed for either of the individual enzymes [Fig. 1(A)]. Furthermore, untagged PsAH did not copurify with His‐tagged PsUC, nor did untagged PsUC co‐purify with His‐tagged PsAH [Fig. 1(B)], consistent with the absence of a stable complex between UC and AH in vitro. Given that the formation of some protein complexes have been shown to be substrate‐induced,17 copurification experiments were repeated with purified His‐tagged and His‐tag‐cleaved PsAH and PsUC in the presence of all reaction substrates. The salt concentration was also reduced from 300 mM NaCl to 50 mM NaCl to minimize the possibility that the buffer ionic strength might interfere with protein–protein interactions.18, 19 Even in the presence of substrates and low salt concentrations, no co–purification was observed between the two enzymes [Fig. 1(C)].
Figure 1.
Urea carboxylase and allophanate hydrolase do not form a stable complex in vitro. (A) Representative chromatograms from size exclusion chromatography. In each case, a 200 μL injection of 1 mg mL−1 PsAH (dotted lines), 1 mg mL−1 PsUC (dashed lines) or preincubated 1 mg mL−1 PsAH/PsUC (solid lines) was applied to a superpose 6 HR 10/30 column. The elution peak at 15 mL corresponds to the predicted molecular weight for both the wild‐type PsAH dimer (MW = 130 kDa), and the PsUC monomer (MW = 130 kDa). Coapplication of both PsAH and PsUC on the size exclusion column does not alter the elution profile, indicating that a stable complex is not formed between PsAH and PsUC. The column was calibrated with molecular weight standards as previously described.16 (B) SDS‐PAGE demonstrates that PsUC and PsAH do not copurify as a complex. The soluble cell lysates loaded onto the Ni2+‐NTA affinity column, prior to purification, are denoted as “Ni2+‐NTA load” and the corresponding samples eluted from the column are denoted as “Ni2+‐NTA elution”. The lanes correspond to the overexpression of (His)8‐tagged UC (His‐UC), (His)8‐tagged AH (His‐AH), untagged UC (UC) or untagged AH (AH). The corresponding mixtures of (His)8‐tagged and untagged lysates are also indicated. In all cases, the untagged protein does not copurify with the tagged protein. (C) The copurification of AH and UC is not dependent on changes in ionic strength or the presence of substrates. Soluble cell lysates were loaded onto and eluted from the Ni2+‐NTA affinity column in a buffer that included 50 mM NaCl, 8 mM MgSO4, 1 mM ATP, 8 mM NaHCO3, and 20 mM urea. The proteins loaded onto the Ni2+‐column are shown as “Ni2+‐NTA load” and the proteins eluted from the column are shown as “Ni2+‐NTA elution”. The samples are labeled as indicated in panel B.
PsAH and PsUC do not influence each other's enzyme activities
The k cat and K M of an enzyme may be altered by the formation of a protein complex.20, 21 For example, if PsUC and PsAH coordinate their activities during catalysis through either intermediate channeling or direct physical interactions, the presence of one enzyme could alter substrate access or catalytic turnover of the other, and thus alter its kinetic parameters. To assess this possibility, the overall kinetic parameters were determined for UC, AH, and UAL cloned from several species (Table 1). The breakdown of ATP by PsUC was measured at varying concentrations of urea, both in the presence and absence of a 10‐fold molar excess of PsAH. Similarly, the allophanate hydrolase activity of PsAH was measured at varying allophanate concentrations, both in the presence and absence of a 10‐fold molar excess of PsUC. In neither case were the k cat nor K M values significantly changed by the presence of the other enzyme (Supporting Information Fig. S1), demonstrating that PsUC and PsAH do not influence each other's enzymatic activity.
Table 1.
Kinetic Data for PsUC, PsAH, GbAH, GbAH‐UC, CaUAL, and ScUAL
| ATP cleavagea | Allophanate hydrolysisb | Urea amidolyase activitya | |||||||
|---|---|---|---|---|---|---|---|---|---|
| k cat (s−1) | K M, app urea (mM) | k cat/K M (s−1 M−1) | k cat (s−1) | (mM) | k cat/K M (s−1 M−1) | k cat (s−1) | K M, app urea (mM) | k cat/K M (s−1 M−1) | |
| PsUC | 12 ± 1c | 17 ± 3 | (7.0 ± 1.0) × 102 d | ||||||
| PsAH | 12 ± 1 | 0.22 ± 0.01 | (5.3 ± 0.3) × 104 | ||||||
| 1:1 molar ratio PsUC:PsAH | 11 ± 1 | 12 ± 1 | (9.2 ± 1.1) × 102 | 11 ± 1 | 0.15 ± 0.01 | (7.3 ± 0.8) × 104 | 1.2 ± 0.1 | 2.5 ± 0.5 | (4.7 ± 1.0) × 102 |
| GbAH | 18 ± 1 | 0.10 ± 0.01 | (1.8 ± 0.1) × 105 | ||||||
| GbAH‐UC | 29 ± 1 | 1.8 ± 0.2 | (1.6 ± 0.2) × 104 | 15 ± 1 | 0.21 ± 0.01 | (6.8 ± 0.4) × 104 | 2.7 ± 0.1 | 1.7 ± 0.1 | (1.6 ± 0.1) × 103 |
| ScUAL | 22 ± 1 | 0.7 ± 0.1 | (3.0 ± 0.3) × 104 | 32 ± 1 | 0.23 ± 0.03 | (1.4 ± 0.2) × 105 | 2.6 ± 0.1 | 0.3 ± 0.1 | (9.2 ± 3.4) × 103 |
| CaUAL | 11 ± 1 | 0.6 ± 0.1 | (1.8 ± 0.3) × 104 | 24 ± 1 | 0.23 ± 0.01 | (1.0 ± 0.1) × 105 | 0.80 ± 0.03 | 0.2 ± 0.1 | (3.6 ± 0.7) × 103 |
50 mM HEPES (pH 7.3), 50 mM NaCl, 8 mM MgSO4, 8 mM NaHCO3, 0.5‐50 mM Urea, 100 µM ATP.
50 mM HEPES (pH 7.3), 50 mM NaCl, 0.1–10 mM allophanate.
Reported errors represent the standard error from the nonlinear regression fit to the Michaelis–Menten equation. The curves were fit to the average of three independent velocity measurements at five different substrate concentrations.
Reported errors are propagated from the K M and k cat measurements in the preceding column.
Allophanate is not directly channeled from PsUC to PsAH
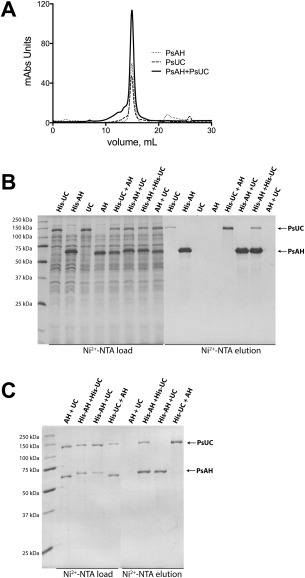
While AH and UC do not form a stable complex or influence the other's catalytic turnover kinetics, it remains possible that AH and UC transiently interact during catalytic turnover to directly channel allophanate from UC to AH. To investigate the possibility of direct substrate channeling between PsUC and PsAH, an in vitro substrate channeling assay was employed.14, 15 This assay captures even transient interactions between enzymes, offering a distinct advantage in sensitivity over more traditional methods that probe protein–protein interactions. Wild‐type PsUC (PsUCwt) and wild‐type PsAH (PsAHwt) were purified, along with an inactive variant of PsAH that was mutated at the essential nucleophilic serine 179 of PsAH (PsAHS179A).11, 22 The k cat for PsAHS179A was determined to be ∼0.03% of the wild‐type enzyme (data not shown). The rates of ammonia production from MgATP, and urea were measured using a glutamate dehydrogenase coupled assay at a 1:1 ratio of PsUCwt to PsAHwt. Subsequently, increasing ratios of PsAHS179A, were titrated into the reaction system, ranging from 0 to 100 fold molar excess over PsAHwt. If allophanate is channeled via a direct interaction between PsUC and PsAH, the addition of inactive PsAHS179A will reduce the overall catalytic activity by competitively binding to PsUC. If there is no channeling between the two enzymes, the overall activity should remain unchanged, even in the presence of a large molar excess of PsAHS179A [Fig. 2(A,B)]. No significant rate reduction was observed in the presence of increasing molar ratios of PsAHS179A [Fig. 2(C)], consistent with an absence of allophanate channeling between PsAH and PsUC.
Figure 2.
Alophanate is not channeled from PsUC to PsAH. (A) A model for the channeling or diffusion of allophanate from UC to AH as assayed by the method of Geck and Kirsch.14 The PsAHS179A inactivated enzyme interferes with substrate channeling by displacing PsAHwt and alters the overall rate. The PsAHS179A inactivated enzyme does not alter the reaction kinetics if the allophanate intermediate freely diffuses from PsUC to PsAH. The cartoon representations of UC and AH are based on published crystal structures (4GYR, 4IST, and 3VA7) with the biotin carboxylase (BC; blue), carboxyltransferase (CT; yellow), biotin carboxyl carrier protein (BCCP; red) and allophanate hydrolase (AH; purple) domains/subunits illustrated to an approximate of their relative scale. (B) Predicted plots of specific activity as a function of increasing ratios of inactivated (PsAHS179A) enzyme to wild‐type enzyme. The total enzyme concentration (PsAH179A + PsAHwt) remains constant for all ratios (C) The specific activities for the complete conversion of , MgATP and urea to NH3 and CO2 were measured at increasing ratios of inactive PsAHS179A to PsAHwt in the absence (closed circles; solid line) and presence (open triangles; dashed line) of 22% PEG4K. The total enzyme concentration (PsAH179A + PsAHwt) was kept constant at all ratios.
In vivo, both specific and nonspecific interactions in the crowded and complex cellular environment may serve to promote the direct channeling between two enzymes. Crowding agents can be added to in vitro assays to mimic a crowded environment. We repeated the substrate channeling assays in the presence of >20% (w/v) PEG4K, a commonly used crowding agent that enhances protein‐protein association rate constants in solution.23, 24 PEG4K at a concentration in excess of 25% (w/v) resulted in precipitation of the enzymes and led to a reduction of the individual enzyme catalytic activities. At 22% PEG4K, the enzymes were stable and the rates of catalysis were unaffected. At this concentration of PEG4K, the PsAH‐PsUC overall activity remained insensitive to increasing concentration of PsAHS179A [Fig. 2(C)]. While this study does not represent an exhaustive analysis of crowding agents, there is no initial evidence for substrate channeling between PsUC and PsAH under conditions that are routinely employed to mimic a crowded environment.
Two proteins encoded by proximally related genes do not alter the in vitro function of PsUC and PsAH
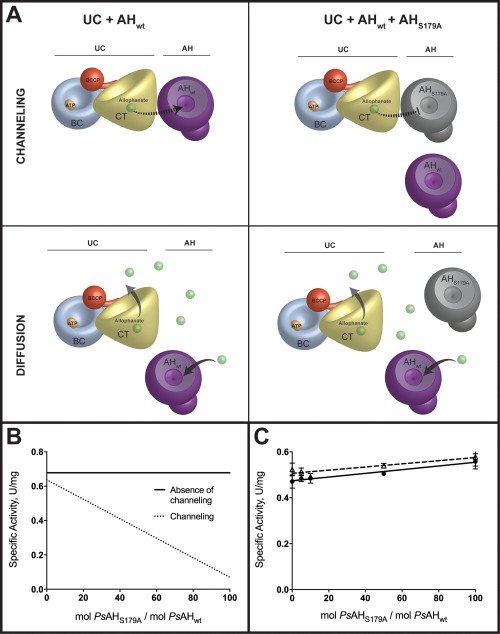
In most bacterial strains, including Wolinella succinogenes, P. syringae, and G. bethesdensis, the genes encoding two small, putatively cytoplasmic proteins are located in very close proximity to the genes encoding AH and UC [Fig. 3(A)]. In Streptomyces avermitilis, these four genes comprise a functional operon that is induced under conditions of nitrogen starvation.25 Interestingly, the genes encoding these two proteins are absent in fungi where, in most cases, UAL is fused into a single polypeptide chain. The function of these two associated proteins, typically annotated as either “hypothetical protein” or “urea amidolyase/carboxylase related protein”, is unknown. In prokaryotes, proteins encoded on the same operon have a higher tendency to exist in a complex.26 Thus, we hypothesized that these two associated proteins of unknown function may serve to mediate AH‐UC complex formation. The genes encoding the two potential chaperone proteins from P. syringae, PSPTO_4241 (encoding NP_794002.1) and PSPTO_4242 (encoding NP_794003.1), were cloned, over‐expressed and copurified. For the present study, these proteins are hereafter named “urea amidolyase associated proteins 1 and 2” (UAAP1 and UAAP2 for NP_794002.1 and NP_794003.1, respectively). Copurification of UC and AH was attempted in the presence of UAAP1 and UAAP2. The putative chaperone proteins UAAP1 and UAAP2 did not form a complex with AH or UC and did not enhance the stable complex formation between PsAH and PsUC either in the presence or absence of substrates. These results were obtained at both moderate (300 mM) and low (50 mM) salt concentrations, precluding the possibility that the buffer ionic strength interfered with protein‐protein interactions.18, 19 [Fig. 3(B)]. The presence of 10‐fold molar excess UAAP1/2 did not alter the kinetics of either PsUC or PsAH (Table 2). Substrate channeling assays between PsUC and PsAH were performed in the presence 10‐fold molar excess of UAAP1/2, with no significant decrease in overall velocity with excess PsAHS179A [Fig. 3(C)].
Figure 3.
UAAP1 and UAAP2 do not facilitate complex formation or substrate channeling between PsAH and PsUC. (A) A localized gene map of genes encoding UC, AH and UAAP1 and UAAP2 on the genome of P. syringae pv. tomato strain DC3000. The genes encoding UAAP1/2 are annotated as PSPTO_4241/2 (purple) and the genes encoding UC and AH are annotated as PSPTO_4243 (red) and PSPTO_4244 (blue). The gene annotations are indicated below the arrows that describe the relative location, size and direction of the gene. The genes annotated as PSPTO_4238‐ 4240 (green) encode a putative ABC‐type transporter and are in suitably close proximity to comprise an operon with PSPTO_4241‐4244. (B) SDS‐PAGE gel showing elution profiles for copurification of UC/AH and UAAP1/2 from a Ni2+‐NTA affinity column. Purified UC, AH and tag‐cleaved UAAP1/2 were incubated with substrate and in buffer containing either 50 or 150 mM NaCl. The samples are labeled as indicated in Figure 1(B). (C) Substrate channeling assay for PsAH and PsUC in the presence (open triangles; dashed line) or absence (closed circles; solid line) of the two hypothetical chaperone proteins. The assay was performed as described in Figure 3(C).
Table 2.
PsAH and PsUC Kinetic Parameters in the Presence and Absence of 10‐fold Molar Excess of UAAP1 and UAAP2
| Allophanate hydrolase activity a | |||
|---|---|---|---|
| k cat (s−1) | (mM) | k cat/K M (M−1s−1) | |
| PsAH | 3.64 ± 0.09c | 0.24 ± 0.02 | (1.5 ± 0.1) ×104 |
| PsAH + UAAP1/2 | 3.67 ± 0.08 | 0.23 ± 0.02 | (1.4 ± 0.1) ×104 |
| ATP hydrolysis activity b | |||
|---|---|---|---|
| k cat (s−1) | K M a TP (μM); 50 mM urea | k cat/K M (M−1s−1) | |
| PsUC | 2.21 ± 0.06 | 8.0 ± 0.8 | (2.8 ± 0.3) ×102 |
| PsUC + UAAP1/2 | 2.08 ± 0.06 | 6.5 ± 0.7 | (3.2 ± 0.4) ×102 |
| k cat (s−1) | K M, app urea (mM); 100 µM ATP | k cat/K M (M−1s−1) | |
| PsUC | 2.18 ± 0.08 | 3.8 ± 0.6 | (5.7 ± 0.9) ×102 |
| PsUC + UAAP1/2 | 2.16 ± 0.06 | 3.2 ± 0.4 | (6.8 ± 0.8) ×102 |
50 mM HEPES (pH 7.3), 50 mM NaCl, 0.1–10 mM allophanate.
Reported errors represent the standard error from the nonlinear regression fit to the Michaelis–Menten equation. The curves were fit to the average of three independent velocity measurements at five different substrate concentrations.
Reported errors are propagated from the K M and k cat measurements in the preceding column.
50 mM HEPES (pH 7.3), 50 mM NaCl, 8 mM MgSO4, 8 mM NaHCO3, 0.5–50 mM Urea, 1–100 µM ATP.
Error analysis is as described for Table I.
An artificial fusion of UC and AH from G. bethesdensis does not alter enzyme efficiency
The fusion of two related genes, whose products participate in a sequential metabolic transformation, can lead to enhanced catalytic efficiency, simply by raising the local concentration of the two reacting partners.27, 28, 29 To test if a fusion of UC and AH results in a more efficient enzyme in vitro, the UC and AH from G. bethesdensis were artificially fused into a single polypeptide, joined by the nearly identical short linker sequence that physically connects AH to UC in CaUAL. The fusion protein from G. bethesdensis, GbAH‐UC, was purified as a full‐length, homogeneous enzyme using Ni2+‐NTA and Q‐sepharose ion exchange columns [Supporting Information Fig. S2(A)] and the catalytic activity of the fused enzyme was compared to the activities of the individual components. The fusion of AH to UC did not significantly alter the k cat/K M for the allophanate hydrolase reaction, indicating that forcing UC into close physical proximity with AH does not significantly influence the rates of substrate binding, catalytic turnover or product release in AH (Table 1). Also, the k cat and K M for ATP hydrolysis, allophanate hydrolysis and urea amidolysis in the fused GbAH‐UC were very similar to the activity obtained from a (1:1) molar mixture of PsUC and PsAH (Table 1).
The inter‐domain coupling efficiency is low in both bacterial and yeast UAL
To study the efficiency of reaction coupling between UC and AH, we compared the activity of the first reaction, ATP cleavage and Pi release in the biotin‐carboxylase domain of UC, to the production of NH3 in the AH domain, all in the presence of ATP, and urea. The NH3 production/P i release ratio reflects the coupling efficiency between ATP cleavage in UC and allophanate hydrolysis in AH. When the molar ratio of UC:AH was 1:1, both P i release and NH3 production increased with increasing urea concentration [Fig. 4(A)]. However, while the coupling between the two activities increases at low urea concentrations, it drops significantly at high urea concentrations to reach a level of ∼0.2 at saturating urea concentration [Fig. 4(B)]. To investigate the reason for the low coupling efficiency, the optimal ratio of UC:AH in the overall reaction was determined. The PsAH activity was measured while titrating PsUC. In reverse, the P i release activity of PsUC was measured while titrating PsAH. Both reactions reach a maximum velocity above a 10:1 molar ratio (Supporting Information Fig. S3). Consequently, NH3 production was re‐measured as a function of urea concentration when the UC:AH ratio was 1:10. The coupling efficiency between P i release and NH3 production remained low, with a very similar coupling efficiency to that observed when the ratio was 1:1. The result suggests a highly inefficient coupling of ATP cleavage with NH3 production, consistent with a lack of intermediate channeling between the two enzyme active sites. Finally, the coupling efficiency was determined in the context of the full length ScUAL and was determined to also be very low (Fig. 5).
Figure 4.
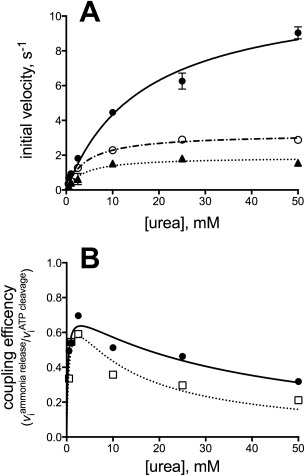
Coupling of NH3 release with ATP cleavage for the reaction catalyzed by PsUC and PsAH. (A) ATP cleavage (solid line, filled circles) and NH3 production (dashed lines) as a function of urea concentration for the combined activities of PsUC and PsAH. The rate of NH3 production was determined when PsUC and PsAH were combined at a 1:1 molar ratio (dotted line; closed triangles) and at a 1:10 molar ratio (dashed‐dotted line; open circles). The lines represent the non‐linear regression fit to the standard Michaelis‐Menten equation. (B) Replot of the ratio of NH3 production rate to ATP cleavage rate for a molar ratio of 1:1 PsUC:PsAH (dotted line; open squares) and 1:10 PsUC:PsAH (solid line; closed circles), respectively, at varying concentrations of urea. The lines represent the nonlinear regression fit to a modified form of the Michaelis–Menten equation describing classic, competitive substrate inhibition.
Figure 5.
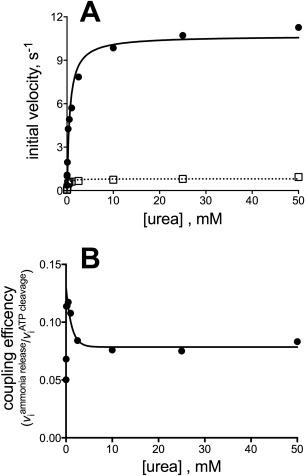
Coupling of NH3 release with ATP cleavage for the reaction catalyzed by ScUAL. (A) ATP cleavage (solid line, filled circles) and NH3 production (dashed line; open squares) as a function of urea concentration. The lines represent the nonlinear regression fit to the standard Michaelis–Menten equation. (B) Replot of the ratio of NH3 production rate to ATP cleavage rate. The lines represent the nonlinear regression fit to the equation describing exponential decay.
Discussion
Intermediate channeling facilitates efficient multi‐step enzymatic reactions and serves to prevent the loss of unstable intermediates to bulk solvent.2 Allophanate, the substrate of AH and the product of UC, is a relatively unstable intermediate that can be hydrolyzed to CO2 and urea under high buffer concentrations or acidic conditions.30, 31 The spontaneous hydrolysis of allophanate to urea contributes to an inefficient expenditure of ATP and, therefore, it is reasonable to predict that UC and AH have coevolved a mechanism to shield against this spontaneous decomposition. Furthermore, while UC and AH exist as separate enzymes in prokaryotes, the ATP cleavage reaction catalyzed by UC was reported to be tightly coupled with ammonia release from AH when the enzymes were assayed in tandem.9 This suggests that allophanate is efficiently transferred from UC to AH and leads to the hypothesis that the allophanate generated in the carboxyltransferase active site of UC is directly channeled to AH in both prokaryotic and eukaryotic UAL.
Surprisingly, our substrate channeling studies do not support allophanate channeling between UC and AH. Several additional observations are consistent with this result. First, the UC and AH coupled assays exhibit a lag phase in the coupling reactions prior to the linear phase of the reaction (Supporting Information Fig. S4), consistent with an absence of substrate channeling. In contrast, coupled reactions in substrate channeling enzymes typically do not exhibit a lag phase.32 Second, in enzymes that participate in substrate channeling, reaction coupling is typically observed between distinct domains. For example, if one active site is mutated, the activity of the other active site is affected.33, 34 Inactive mutants of full‐length UAL could not be easily expressed and purified from yeast or bacterial expression systems. However, we have shown that the presence of bacterial UC or AH does not influence the activity of the other enzyme and that physically fusing UC and AH from G. bethesdensis does not significantly alter the AH activity, consistent with a lack of coupling and physical interactions between UC and AH. Third, the efficiency of reaction coupling in UAL is very low. By contrast, in carbamoyl phosphate synthase, 100% coupling was observed, with a stoichiometry of 2 mol of MgADP and 1 mol of glutamate consumed per mol of carbamoyl phosphate synthesized.35 The coupling efficiency for AH and UC is extremely low in comparison, and decreases with increasing urea concentration (Figs. 4 and 5). Collectively, these results further support the absence of substrate channeling in both prokaryotic and fungal UAL enzymes.
Our observation of low coupling efficiency between UC and AH disagrees with an earlier study on bacterial UC and AH from Oleomonas sagaranensis, which concluded that the allophanate intermediate was efficiently transferred from UC and AH.9 A detailed comparison of this earlier study with those described herein suggests reasons for this discrepancy. First, the ATP breakdown activity for O. sagaranensis UC is 21.2 U mg−1,8 while the activity for the UC‐AH combined production of ammonia is 10.2 U mg−1,9 indicating only a 50% coupling efficiency and not a tightly coupled 1:1 stoichiometry as the authors suggest. Furthermore, in the coupling assays for ammonia‐generating activity of the combined O. sagaranesis UC and AH, the AH enzyme was present in 24‐fold excess over the concentration of UC, which was not considered in the stoichiometry calculations. In our study, a 10‐fold molar excess of AH over UC was sufficient to reflect a condition where UC‐catalyzed formation of allophanate is the rate‐limiting step and the ratio of ATP breakdown to ammonia production was ∼4:1 (Fig. 4). When UC and AH were assayed at equimolar concentrations, the ratio of ATP breakdown to ammonia production was ∼10:1 (Fig. 4). Therefore, the prior conclusions suggesting a tight coupling of UC and AH do not appear well supported. Instead, the results presented in this study offer a rigorous and detailed analysis of coupling efficiency in UAL and demonstrate a very low coupling efficiency between the two enzymatic functions.
We clearly demonstrate that UC and AH do not directly associate or channel allophanate in vitro, but we cannot exclude the possibility that they channel this substrate in a cellular milieu. It is possible, for example, that an as yet unidentified scaffolding protein(s) might be present in prokaryotic cells to assist in the formation of a complex between UC and AH. Two, relatively small candidate proteins that could serve such a function are UAAP1 and UAAP2, which are encoded by proximally associated genes in Streptomyces avermitilis and in other bacterial species.25 However, copurification and kinetic assays do not support a role for these proteins in promoting the AH and UC interaction (Fig. 3) and, consequently, the function of UAAP1/2 remains unclear. Small molecule allosteric activators are also known to increase the coupling efficiency between active sites in multifunctional enzymes. For example, in the related biotin‐dependent enzyme, pyruvate carboxylase, the absence of the allosteric activator, acetyl‐CoA, leads to an almost complete loss of coupling between catalytic domains, while the addition of acetyl‐CoA increases the coupling efficiency to ∼1:1 at saturating pyruvate concentrations.36 Given the partial homology between pyruvate carboxylase and UC, it is reasonable to speculate that an as yet unidentified activator might serve to enhance the coupling efficiency and coordination between UC and AH.
UAL is not unique in acting as a multifunctional enzyme that does not channel a substrate or intermediate: IspD/IspF in the isoprenoid biosynthetic pathway and the mitochondrial malate dehydrogenase and NADH: ubiquinone oxidoreductase (complex I) are other notable examples.37, 38 However, UAL is quite unusual in that its individual domains (UC and AH) do not associate into a stable complex in prokaryotes. It has been reported that gene fusion tends to further facilitate the formation of existing inter‐domain complexes by simplifying the protein complex topologies.39 Interestingly, UAL appears to serve as a case where gene fusion arises even in the absence of a physical interaction between the individual catalytic subunits and in the absence of intermediate channeling. This raises an interesting question: if the UC and AH activities are functionally and structurally independent, why have their genes fused into a single polypeptide in the fungal UAL enzymes?
The genes encoding UC and AH are located immediately adjacent to one another (typically separated by only a few nucleotides) in many prokaryotic genomes and they have been shown to comprise an operon in S. avermitilis.25 Bacterial operons have the advantage of coordinating multi‐gene expression but this system is not available in eukaryotes. Therefore, even though their activities are not coordinated, fusing UC and AH in fungi results in simultaneous spatiotemporal control of both enzymes. Several models have been put forward to support the evolutionary basis for gene fusion events.40 Most of them favor the idea that gene fusion facilitates intermediate transfer, increases catalytic efficiency, and facilitates regulation at the cellular level.41 Interestingly, the “selfish operon” model, has raised the hypothesis that gene organization need not necessarily favor or benefit the host.42 This model suggests that the clustering of functionally related genes serves simply to facilitate the successful inheritance of the gene cluster, both by horizontal gene transfer and by vertical transmission, because the mechanism of gene transfer is limited by the size of the mobilized DNA fragments.42 When the two genes are too far away, the gene transfer is restricted only to vertical transmission. Also, genes fuse primarily via a random process of joining and breaking, such that functionally related genes have a higher natural tendency to transition from monofunctional enzymes to multifunctional enzymes.39, 43 According to this framework and the results presented here, the combination of UC and AH into UAL in eukaryotes does not appear to arise from a beneficial linking of their enzyme activities, but rather is simply a result of their functional relatedness and the close proximity of their genes. Navarathna et al. have suggested that UAL frees the organism from reliance on urease and, by association, Ni2+, thus providing the needed evolutionary driving force for the acquisition of the biotin‐dependent UAL as a single gene.44 The present work serves as a reminder that gene fusion does not, de facto, facilitate a physical interaction between independent domains: the artificial fusion of AH and UC from G. bethesdensis does not alter the activity of either enzyme, indicating that UC and AH function as independent entities irrespective of whether or not they are linked together.
This study describes a series of in vitro experiments that do not support the association or coordination of the two individual enzymatic components of UAL. The most straightforward conclusion from these results is that there is no association or coordination between the AH and UC domains of UAL. This is a somewhat surprising and a relatively unusual finding for a multifunctional enzyme, and supports the idea that gene fusion events leading to the formation of multifunctional enzymes do not, a priori, require or result in an enhanced catalytic function.
Materials and Methods
Construction of expression vectors
Genomic DNA from Pseudomonas syringae pv. tomato str. DC3000 (strain ATCC BAA‐871D‐5) and G. bethesdensis (strain ATCC BAA‐1260/CGDN1H1) was obtained from the American Type Culture Collection (Manassas, VA). Genomic DNA from S. cerevisiae strain W303 (MATa/MATα {leu2‐3, 112 trp1‐1 can1‐100 ura3‐1 ade2‐1 his3‐11, 15} [phi+]) and C. albicans strain SC5314/ATCC MYA‐2876 was isolated using standard methods. The cloning strategies are summarized in the Supporting Information Materials and Methods and Tables SI and SII. The complete sequences of all clones were confirmed through Sanger sequencing by Functional Biosciences (Madison, WI).
Yeast strains, growth, and transformation
The haploid S. cerevisiae strain MLY40α (MATα ura3‐52)45 was grown in yeast extract/peptone/dextrose (YPD) medium. DUR1,2 is the gene encoding UAL (Dur1,2p) in S. cerevisiae. The dur1,2Δ strain was constructed using MLY40α as host, by PCR‐based chromosomal integration. The kanamycin resistance cassette (KanR) was PCR amplified from vector pFA6a‐kanMX646 using primers 25 and 26 (Supporting Information Table SII), with 50 bp flanking sequences on the 5′ and 3′ ends of DUR1,2. The PCR product (3 μg) was transformed into the MLY40α yeast cell and dur1,2Δ mutants were selected on YPD media supplemented with 20 mg L−1 G418. To confirm the DUR1,2 chromosomal gene knockout, chromosomal DNA was extracted from selected mutants and the absence of the DUR1,2 gene was confirmed by PCR using primers 27 and 28 (Supporting Information Table SII) that lie ∼100 bp upstream and downstream, respectively, of DUR1,2. The growth dependence of the dur1,2Δ mutant on urea was confirmed on glucose‐phosphate‐urea‐uracil plates, a defined minimal medium that includes urea as the sole nitrogen source. The dur1,2Δ strain was used for the expression of UAL cloned from both C. albicans and S. cerevisiae. The dur1,2Δ strains with expression plasmids harboring the URA3 marker were grown on synthetic complete medium lacking uracil (Sc‐URA), to maintain selection for the vector. To induce UAL expression, the complemented strain was grown in galactose‐phosphate urea media.47
Over‐expression of protein from Escherichia coli
E. coli HMS174 (DE3) cells were used for protein overexpression and purification of bacterial proteins. For overexpression of PsAH or PsAHS179A, HMS174 (DE3) cells were transformed with pET28a‐(His)8‐TEV vector harboring PsAH or PsAHS179A and grown in LB medium containing 25 μg/ml kanamycin. PsUC and GbAH‐UC were co‐expressed with E. coli biotin protein ligase (BirA) on vector pCY21648 in HMS174(DE3) cells. For co‐expression of UAAP1 and UAAP2, pKLD66nCBP‐UAAP1 and pET28a‐(His)8‐TEV‐UAAP2 were cotransformed into E. coli HMS174 (DE3) expression cells. For PsAH, PsAHS179A, PsUC and GbAH‐UC, 1 L overnight LB cultures were used to inoculate 12 L of LB medium, containing the previously described antibiotics. For co‐expression of UAAP1 and UAAP2, a 1 L LB overnight culture was used to inoculate 12 L of M9 minimal media containing 200 μg mL−1 ampicillin and 25 μg mL−1 kanamycin. All cultures were grown at 37°C to an OD600 of ∼0.8–1, subsequently chilled in an ice/water bath for ∼15–20 min. For PsAH, PsAHS179A, and UAAP1/UAAP2 co‐expression, cultures were induced with 1 mM IPTG. For PsUC and GbAH‐UC, cultures were induced with a final concentration of 21 mM arabinose and 1 mM IPTG. The cultures were transferred to a 16°C shaking incubator for 24 h prior to cell harvesting.
Purification of bacterial proteins
PsAH, PsUC, PsAHS179A, UAAP1/UAAP2, and GbAH‐UC were purified using Ni2+‐affinity and ion‐exchange chromatography. Cells were sonicated in buffer A (50 mM HEPES pH 8.0, 300 mM NaCl and 0.1 mM EGTA, with 10 mM β‐mercaptoethanol, 1 mM PMSF, 5 μM E‐64 and 1 mM pepstatin), centrifuged at 12,000 rpm, 4°C for 30 min. The supernatants were loaded onto Ni2+‐Profinity IMAC resin and then washed with buffer A containing 20 mM imidazole. The proteins were eluted from the column using a gradient of 20–300 mM imidazole, dialyzed overnight at 4°C against buffer B (20 mM HEPES pH 8.0, 50 mM NaCl, 1 mM EGTA, and 2 mM DTT) prior to loading on Q‐sepharose fast flow resin. The proteins were eluted using buffer B with a gradient from 100 to 750 mM NaCl. The proteins were concentrated using a 30 kDa‐molecular weight cutoff centrifugal filter to final concentrations ranging from 6 to 16.5 mg mL−1, and drop frozen in liquid nitrogen. The purity of the protein samples were confirmed by SDS‐PAGE. The N‐terminal poly(His) tags were removed from PsAH and PsUC with the tobacco etch virus (TEV) protease in vitro. The proteins were dialyzed overnight at 4°C against Buffer C (50 mM HEPES pH 8.0, 300 mM NaCl, and 1 mM DTT). Purified His‐tagged TEV protease was combined with His‐tagged PsAH and PsUC at a molar ratio of 1:50 and incubated overnight at 4°C. To separate TEV protease and uncleaved enzyme from the N‐terminally cleaved enzymes, the proteins were loaded on a Ni2+‐NTA column. The resulting column flow through contained enzymes with the poly(His) tag removed. The removal of the poly(His) tag was confirmed by western blotting with rabbit anti‐6‐His Antibody (Bethyl Laboratories, Montgomery TX; data not shown).
Copurification of P. syringae AH and UC
Both (His)8‐tagged and non (His)8‐tagged PsAH were over‐expressed in HMS174 (DE3) in the presence of biotin protein ligase, as described above. Cells were sonicated in buffer A and centrifuged at 12,000 rpm, 4°C. Supernatant were mixed and shaked gently at 4°C for 30 min in the following combinations: His‐PsAH with untagged PsUC, His‐PsUC with untagged PsAH, untagged PsAH with untagged PsUC, His‐tagged PsAH with His‐tagged PsUC. The combined cell lysate was purified through a Ni2+‐Profinity column, washed three times with buffer A containing 20 mM imidazole and the protein was eluted with 1 ml Buffer A containing 300 mM imidazole. The loaded and eluted proteins were analyzed by SDS‐PAGE.
Over‐expression and purification of UAL from S. cerevisiae
The dur1,2Δ mutant was transformed with pESC‐URA‐(His)8‐ScUAL. Galactose‐phosphate‐urea medium was used for large scale growth of yeast cells. Sc‐URA media was inoculated with a single colony, growing at 30°C overnight. Galactose‐phosphate‐urea media (12 L) was inoculated with starting culture of OD600 at 0.2. Cells were cultured at 30°C for ∼12–16 h, shaking at 225 rpm, until the OD600 reached ∼1.5. The yeast cells were subsequently harvested by centrifugation at 6,000 rpm for 10 min, resuspended in Buffer A, and lysed using a bead beater (Hamilton Beach Model No. HBB909) for 10 min. The cell debris was centrifuged at 12,000 rpm, 4°C for 30 min, followed by ultracentrifugation at 100,000g for 1 h. The supernatant was loaded onto a Ni2+‐NTA affinity column, washed (with 20 mM imidazole) and eluted with a gradient of 20–300 mM imidazole.
Size exclusion chromatography
Analytical size exclusion chromatography was performed using an ÄktaTM FPLCTM system (Amersham Phamacia Biotech Inc, Piscataway, NJ) with a Superose 6 HR 10/30 (GE Healthcare) column in buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, and 1 mM DTT. The Superose 6 HR 10/30 column was equilibrated with degased and filtered buffer at 0.3 mL min−1 flow rate for a total of two column volumes (60 mL). To prepare the protein sample for loading, the protein sample at 1 mg mL−1 was centrifuged at 15,871g for 10 min at 4°C to clarify the sample. The supernatant was also filtered through a 0.2‐µm syringe filter (NALGEN, Thermo Scientific, Waltham, MA) and was directly injected using a 200 µL sample loop. The samples were run at a flow rate of 0.30 mL min−1 with continuous detection of absorbance at 280 nm.
Enzyme assays
The initial rate of ATP cleavage was measured at varying concentrations of urea (0–50 mM) using a standard coupled ATPase assay. Assays were performed in 50 mM HEPES (pH 7.3), 50 mM NaCl, 8 mM MgSO4, 8 mM NaHCO3, 0‐50 mM Urea, 1.5 mM phosphoenolpyruvate, 0.15 mM NADH, 100 µM ATP, 5 U mL−1 of pyruvate kinase and 12.5 U mL−1 of lactate dehydrogenase. The reaction was initiated by the addition of enzyme (UC or UAL).
Allophanate hydrolase activity was assayed using a glutamate dehydrogenase coupled assay as previously described.49 The assay was performed in 50 mM HEPES (pH 7.3) and 50 mM NaCl, in the presence of 0.1–10 mM allophanate, 20 mM 2‐oxoglutarate, 0.15 mM NADH, and 20 U mL−1 glutamate dehydrogenase. Reactions were initiated by the addition of AH (∼2.5 μg mL−1).
The UC‐AH coupling activity and substrate channeling activity of UC and AH was assayed using the glutamate dehydrogenase coupled assay in a buffer containing 50 mM HEPES (pH 7.3) and 50 mM NaCl and in the presence of 0.15 mM NADH, 8 mM MgSO4, 50 mM Urea, 100 µM ATP, 8 mM NaHCO3, 10 mM 2‐oxoglutarate and 20 U mL−1 of glutamate dehydrogenase. For the substrate channeling assays, the overall activity of UC and AH was measured as described above, with increasing concentrations of PsAHS179A titrated into a 1:1 molar ratio mixture of UC (90 μg mL−1) and AH (45 μg mL−1). To simulate a crowded cellular environment, 22% (w/v) PEG4000 was added to the assay buffer.
Supporting information
Supporting Information
References
- 1. Khosla C, Harbury PB (2001) Modular enzymes. Nature 409:247–252. [DOI] [PubMed] [Google Scholar]
- 2. Spivey HO, Ovadi J (1999) Substrate channeling. Methods 19:306–321. [DOI] [PubMed] [Google Scholar]
- 3. Ghosh S, Navarathna DH, Roberts DD, Cooper JT, Atkin AL, Petro TM, Nickerson KW (2009) Arginine‐induced germ tube formation in Candida albicans is essential for escape from murine macrophage line RAW 264.7. Infect Immun 77:1596–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Navarathna DH, Lionakis MS, Lizak MJ, Munasinghe J, Nickerson KW, Roberts DD (2012) Urea amidolyase (DUR1,2) contributes to virulence and kidney pathogenesis of Candida albicans . PLoS One 7:e48475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Greenberg DE, Porcella SF, Zelazny AM, Virtaneva K, Sturdevant DE, Kupko JJ, III , Barbian KD, Babar A, Dorward DW, Holland SM (2007) Genome sequence analysis of the emerging human pathogenic acetic acid bacterium Granulibacter bethesdensis . J Bacteriol 189:8727–8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Whitney PA, Cooper TG (1972) Urea carboxylase and allophanate hydrolase: two components of a multienzyme complex in Saccharomyces cerevisiae . Biochem Biophys Res Commun 49:45–51. [DOI] [PubMed] [Google Scholar]
- 7. Strope PK, Nickerson KW, Harris SD, Moriyama EN (2011) Molecular evolution of urea amidolyase and urea carboxylase in fungi. BMC Evol Biol 11:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kanamori T, Kanou N, Atomi H, Imanaka T (2004) Enzymatic characterization of a prokaryotic urea carboxylase. J Bacteriol 186:2532–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kanamori T, Kanou N, Kusakabe S, Atomi H, Imanaka T (2005) Allophanate hydrolase of Oleomonas sagaranensis involved in an ATP‐dependent degradation pathway specific to urea. FEMS Microbiol Lett 245:61–65. [DOI] [PubMed] [Google Scholar]
- 10. Balotra S, Newman J, Cowieson NP, French NG, Campbell PM, Briggs LJ, Warden AC, Easton CJ, Peat TS, Scott C (2015) X‐ray structure of the amidase domain of AtzF, the allophanate hydrolase from the cyanuric acid‐mineralizing multienzyme complex. Appl Environ Microbiol 81:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin Y, St Maurice M (2013) The structure of allophanate hydrolase from Granulibacter bethesdensis provides insights into substrate specificity in the amidase signature family. Biochemistry 52:690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fan C, Li Z, Yin H, Xiang S (2013) Structure and function of allophanate hydrolase. J Biol Chem 288:21422–21432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fan C, Chou CY, Tong L, Xiang S (2012) Crystal structure of urea carboxylase provides insights into the carboxyltransfer reaction. J Biol Chem 287:9389–9398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Geck MK, Kirsch JF (1999) A novel, definitive test for substrate channeling illustrated with the aspartate aminotransferase/malate dehydrogenase system. Biochemistry 38:8032–8037. [DOI] [PubMed] [Google Scholar]
- 15. James CL, Viola RE (2002) Production and characterization of bifunctional enzymes. Substrate channeling in the aspartate pathway. Biochemistry 41:3726–3731. [DOI] [PubMed] [Google Scholar]
- 16. Lietzan AD, Menefee AL, Zeczycki TN, Kumar S, Attwood PV, Wallace JC, Cleland WW, St Maurice M (2011) Interaction between the biotin carboxyl carrier domain and the biotin carboxylase domain in pyruvate carboxylase from Rhizobium etli . Biochemistry 50:9708–9723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thanabalu T, Koronakis E, Hughes C, Koronakis V (1998) Substrate‐induced assembly of a contiguous channel for protein export from E. coli: reversible bridging of an inner‐membrane translocase to an outer membrane exit pore. EMBO J 17:6487–6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ianeselli L, Zhang F, Skoda MW, Jacobs RM, Martin RA, Callow S, Prevost S, Schreiber F (2010) Protein–protein interactions in ovalbumin solutions studied by small‐angle scattering: effect of ionic strength and the chemical nature of cations. J Phys Chem B 114:3776–3783. [DOI] [PubMed] [Google Scholar]
- 19. Dumetz AC, Snellinger‐O'Brien AM, Kaler EW, Lenhoff AM (2007) Patterns of protein–protein interactions in salt solutions and implications for protein crystallization. Protein Sci 16:1867–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hettwer S, Sterner R (2002) A novel tryptophan synthase beta‐subunit from the hyperthermophile Thermotoga maritima. Quaternary structure, steady‐state kinetics, and putative physiological role. J Biol Chem 277:8194–8201. [DOI] [PubMed] [Google Scholar]
- 21. Baker P, Hillis C, Carere J, Seah SY (2012) Protein–protein interactions and substrate channeling in orthologous and chimeric aldolase‐dehydrogenase complexes. Biochemistry 51:1942–1952. [DOI] [PubMed] [Google Scholar]
- 22. Shapir N, Sadowsky MJ, Wackett LP (2005) Purification and characterization of allophanate hydrolase (AtzF) from Pseudomonas sp. strain ADP. J Bacteriol 187:3731–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kozer N, Kuttner YY, Haran G, Schreiber G (2007) Protein–protein association in polymer solutions: from dilute to semidilute to concentrated. Biophys J 92:2139–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kulkarni AM, Chatterjee AP, Schweizer KS, Zukoski CF (2000) Effects of polyethylene glycol on protein interactions. J Chem Phys 113:9863. [Google Scholar]
- 25. Chen Y, Zhu H, Zheng G, Jiang W, Lu Y (2013) Functional analysis of TetR‐family regulator AmtRsav in Streptomyces avermitilis . Microbiology 159:2571–2583. [DOI] [PubMed] [Google Scholar]
- 26. Bratlie MS, Johansen J, Drablos F (2010) Relationship between operon preference and functional properties of persistent genes in bacterial genomes. BMC Genom 11:71‐2164‐11‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kuriyan J, Eisenberg D (2007) The origin of protein interactions and allostery in colocalization. Nature 450:983–990. [DOI] [PubMed] [Google Scholar]
- 28. Ribeiro LF, Furtado GP, Lourenzoni MR, Costa‐Filho AJ, Santos CR, Nogueira SC, Betini JA, Polizeli Mde L, Murakami MT, Ward RJ (2011) Engineering bifunctional laccase‐xylanase chimeras for improved catalytic performance. J Biol Chem 286:43026–43038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen B, Markillie LM, Xiong Y, Mayer MU, Squier TC (2007) Increased catalytic efficiency following gene fusion of bifunctional methionine sulfoxide reductase enzymes from Shewanella oneidensis . Biochemistry 46:14153–14161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shapir N, Cheng G, Sadowsky MJ, Wackett LP (2006) Purification and characterization of TrzF: biuret hydrolysis by allophanate hydrolase supports growth. Appl Environ Microbiol 72:2491–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thompson JF, Muenster AM (1971) Separation of the Chlorella ATP:Urea amido‐lyase into two components. Biochem Biophys Res Commun 43:1049–1055. [DOI] [PubMed] [Google Scholar]
- 32. Singh H, Arentson BW, Becker DF, Tanner JJ (2014) Structures of the PutA peripheral membrane flavoenzyme reveal a dynamic substrate‐channeling tunnel and the quinone‐binding site. Proc Natl Acad Sci USA 111:3389–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thoden JB, Holden HM, Wesenberg G, Raushel FM, Rayment I (1997) Structure of carbamoyl phosphate synthetase: a journey of 96 A from substrate to product. Biochemistry 36:6305–6316. [DOI] [PubMed] [Google Scholar]
- 34. Moxley MA, Sanyal N, Krishnan N, Tanner JJ, Becker DF (2014) Evidence for hysteretic substrate channeling in the proline dehydrogenase and Delta1‐pyrroline‐5‐carboxylate dehydrogenase coupled reaction of proline utilization A (PutA). J Biol Chem 289:3639–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang X, Raushel FM (2000) An engineered blockage within the ammonia tunnel of carbamoyl phosphate synthetase prevents the use of glutamine as a substrate but not ammonia. Biochemistry 39:3240–3247. [DOI] [PubMed] [Google Scholar]
- 36. Zeczycki TN, Menefee AL, Jitrapakdee S, Wallace JC, Attwood PV, St. Maurice M, Cleland WW (2011) Activation and inhibition of pyruvate carboxylase from Rhizobium etli . Biochemistry 50:9694–9707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lherbet C, Pojer F, Richard SB, Noel JP, Poulter CD (2006) Absence of substrate channeling between active sites in the Agrobacterium tumefaciens IspDF and IspE enzymes of the methyl erythritol phosphate pathway. Biochemistry 45:3548–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kotlyar AB, Maklashina E, Cecchini G (2004) Absence of NADH channeling in coupled reaction of mitochondrial malate dehydrogenase and complex I in alamethicin‐permeabilized rat liver mitochondria. Biochem Biophys Res Commun 318:987–991. [DOI] [PubMed] [Google Scholar]
- 39. Marsh JA, Hernandez H, Hall Z, Ahnert SE, Perica T, Robinson CV, Teichmann SA (2013) Protein complexes are under evolutionary selection to assemble via ordered pathways. Cell 153:461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cheng XY, Huang WJ, Hu SC, Zhang HL, Wang H, Zhang JX, Lin HH, Chen YZ, Zou Q, Ji ZL (2012) A global characterization and identification of multifunctional enzymes. PLoS One 7:e38979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kaessmann H (2010) Origins, evolution, and phenotypic impact of new genes. Genome Res 20:1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lawrence J (1999) Selfish operons: the evolutionary impact of gene clustering in prokaryotes and eukaryotes. Curr Opin Genet Dev 9:642–648. [DOI] [PubMed] [Google Scholar]
- 43. Koonin EV, Wolf YI, Karev GP (2002) The structure of the protein universe and genome evolution. Nature 420:218–223. [DOI] [PubMed] [Google Scholar]
- 44. Navarathna DH, Harris SD, Roberts DD, Nickerson KW (2010) Evolutionary aspects of urea utilization by fungi. FEMS Yeast Res 10:209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lorenz MC, Heitman J (1997) Yeast pseudohyphal growth is regulated by GPA2, a G protein alpha homolog. EMBO J 16:7008–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, III , Steever AB, Wach A, Philippsen P, Pringle JR (1998) Heterologous modules for efficient and versatile PCR‐based gene targeting in Schizosaccharomyces pombe . Yeast 14:943–951. [DOI] [PubMed] [Google Scholar]
- 47. Kulkarni RK, Nickerson KW (1981) Nutritional control of dimorphism in Ceratocystis ulmi . Exp Mycol 5:148–154. [Google Scholar]
- 48. Chapman‐Smith A, Turner DL, Cronan JE Jr, Morris TW, Wallace JC (1994) Expression, biotinylation and purification of a biotin‐domain peptide from the biotin carboxy carrier protein of Escherichia coli acetyl‐CoA carboxylase. Biochem J 302:881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kimura S, Yamanishi H, Iyama S, Yamaguchi Y, Kanakura Y (2003) Enzymatic assay for determination of bicarbonate ion in plasma using urea amidolyase. Clin Chim Acta 328:179–184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information