

Abstract
Purification of milligram quantities of target proteins is required for structural and biophysical studies. However, mammalian membrane proteins, many of which are important therapeutic targets, are too unstable to be expressed in heterologous hosts and to be solubilized by detergents. One of the most promising ways to overcome these limitations is to stabilize the membrane proteins by generating variants via introduction of truncated flexible regions, fusion partners, and site‐directed mutagenesis. Therefore, an effective screening strategy is a key to obtaining successful protein stabilization. Herein, we report the micro‐scale and high‐throughput screening of stabilized membrane protein variants using Saccharomyces cerevisiae as a host. All steps of the screening, including cultivation and disruption of cells, solubilization of the target protein, and the pretreatment for fluorescence‐detected size exclusion chromatography (FSEC), could be performed in a 96‐well microplate format. We demonstrated that the dispersion among wells was small, enabling detection of a small but important improvement in the protein stability. We also demonstrated that the thermally stable mutants of a human G protein‐coupled receptor could be distinguished based on an increase of the peak height in the FSEC profile, which was well correlated with increased ligand binding activity of the protein. This strategy represents a significant platform for handling numerous mutants, similar to alanine scanning.
Keywords: unstable membrane protein, expression, stabilization, screening, 96‐well microplate, Saccharomyces cerevisiae, fluorescence‐detection size‐exclusion chromatography (FSEC)
Abbreviations
- CHS
cholesteryl hemisuccinate
- DDM
n‐dodecyl‐β‐d‐maltoside
- FSEC
fluorescence‐detection size exclusion chromatography
- GFP
green fluorescent protein.
Introduction
Membrane proteins are involved in many important physiological functions, and abnormalities of these proteins are related to various diseases. Based on computational analysis, about 5500 human genes encode membrane proteins, and these genes account for approximately 26% of the protein‐coding genes.1 Membrane proteins are also important drug targets and nearly 60% of the currently utilized drugs target membrane proteins.2 High‐resolution structural analysis of target membrane proteins is a key approach for successful structure‐based drug design (SBDD). However, the number of atomic‐level structures of mammalian membrane proteins that have been elucidated is still low. At present, only around 90 unique mammalian membrane protein structures have been solved by X‐ray crystallography (from the list by the Stephen White Lab at UC Irvine; http://blanco.biomol.uci.edu/mpstruc/).
Because membrane proteins are embedded in the cell membrane, their solubilization by a detergent is required for structural and biophysical studies. However, eukaryotic membrane proteins are generally unstable in detergents compared with bacterial membrane proteins.3 In addition, although milligram quantities of purified proteins are required for these studies, the expression level of many eukaryotic membrane proteins is very low in heterologous hosts, which may be related to the instability of these proteins in different lipid conditions. Therefore, membrane proteins must be sufficiently stabilized to achieve high expression, solubilization and purification. Stabilization of membrane proteins via protein engineering has been applied mainly to G protein‐coupled receptors (GPCRs). GPCRs are the largest family of integral membrane proteins4 and are the largest drug target protein family; more than 30% of currently used drugs target GPCRs.2 For atomic‐resolution structural analysis, introduction of mutations is required to achieve stabilization of many GPCRs to overcome difficulties in expression, purification, and crystallization.5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 Furthermore, generating thermally stable membrane proteins may facilitate fragment‐based drug discovery by biophysical approaches20, 21 as well as production of therapeutic antibodies.22
During screening of membrane protein mutants for acquisition of stability and higher expression, it is important to monitor their functional activity. For this purpose, it is ideal to use a radioisotope (RI)‐labeled ligand for the protein of interest. At present, the most successful screening strategy involves the combination of alanine scanning and a thermal stability assay using RI‐labeled ligands.23, 24, 25, 26 This technique has been applied to the determination of the crystal structures of six GPCRs.5, 8, 9, 12, 13, 14, 19 However, RI‐labeled high‐affinity ligands are always not available, and in most cases, special facilities are required to use RIs. Another successful strategy is directed evolution using a fluorescently labeled ligand.27, 28 This approach has enabled determination of the crystal structure of the neurotensin receptor type 1 (NTSR1) by the vapor diffusion method after purification in nonyl‐β‐d‐glucoside.15 However, introducing a fluorescent label into small ligands often causes steric hindrance in binding to the receptor. Furthermore, the cell sorter essential for this method is too costly for many researchers. Considering these facts, a simpler and low‐cost strategy is required.
Fluorescence‐detection size‐exclusion chromatography (FSEC), in which the target protein is fused to green fluorescent protein (GFP) and analyzed by size‐exclusion chromatography (SEC) via fluorescence detection, has become a popular strategy for evaluating the stability of solubilized membrane proteins.29 In particular, the combination of FSEC and Saccharomyces cerevisiae as an expression host is suggested to be an advantageous screening system for the ensuing reasons:30, 31 First, S. cerevisiae is as easy to handle as Escherichia coli and grows fast. Second, S. cerevisiae is more advantageous for the expression of eukaryotic membrane proteins because it has the eukaryotic folding mechanism and post‐transcriptional modification system. Third, the acyl chains of yeast membrane lipids are longer than those of prokaryotes on average, which should create more suitable lipid conditions for the expression of mammalian membrane proteins.32, 33, 34 Finally, the rapid cloning and expression are possible without vector construction as in E. coli, by virtue of the high homologous recombination activity of S. cerevisiae. We previously demonstrated that a stable and highly expressed variant of the histamine H1 receptor, one of the important GPCR drug‐targets the crystal structure of which has been determined,35 could be selected using this system.36 However, thus far, the method did not adequately satisfy the requirements for high‐throughput screening from the large pool of variants. We therefore miniaturized the screening system using S. cerevisiae in a 96‐well microplate format, and significantly enhanced the throughput for screening the membrane protein variants with improved expression and/or stability.
Results
Overview of the high‐throughput screening of membrane protein variants in a 96‐well microplate format
The strategy for the miniaturization of screening of membrane protein variants in S. cerevisiae is shown in Figure 1. The transformants of S. cerevisiae harboring the expression vector of the membrane protein variant were generated by transformation of the linearized pDDGFP‐2 plasmid along with the PCR fragments of the protein of interest.36, 37 Due to the high recombination activity, a multiple variant could be constructed in S. cerevisiae in one step. The present strategy enabled the performance of all procedures in a 96‐well microplate format. The colonies were inoculated into the −Ura + Ade medium with 2% glucose in a 96‐deep‐well plate and cultured overnight. The cultures were then diluted into the −Ura + Ade medium with 0.1% glucose and cultured for 7 h. Expression of the target membrane proteins was induced by adding 2% galactose and culturing for a further 22 h. After harvesting, the cells were resuspended in the resuspension buffer, and the whole‐cell fluorescence was measured, indicating the total expression of the target membrane protein. Cells were disrupted using beads on the same deep‐well plate. The membrane proteins were solubilized by adding the detergent mixture (the final concentration was 0.5% (w/v) n‐dodecyl‐β‐d‐maltoside (DDM) and 0.1% (w/v) cholesteryl hemisuccinate (CHS)) directly to the disrupted cells. The insoluble materials were able to separate by ultracentrifugation or by passing through a 0.22 μm filter plate. FSEC analysis was performed using a conventional high‐performance liquid chromatography (HPLC) system equipped with a fluorescence detector and a UV–Vis detector. Monodispersity and sharpness of the peak in FSEC chromatogram is the indicator for stability of the membrane protein. The expression level of the membrane protein that gives a monodisperse peak in a detergent can be estimated by comparing the peak height of the monodisperse peak with that of the purified yeast‐enhanced GFP standard (Supporting Information Fig. S1). For example, if the target protein with a molecular weight of 50 kDa shows a peak height of 10,000 AU in FSEC, the yield is estimated to be ∼0.59 mg per 1 L of culture as a solubilized and monodisperse form.
Figure 1.

High‐throughput strategy of the construction and evaluation of membrane protein variants in S. cerevisiae in the 96‐well microplate format.
Optimization of the screening system to improve reliability and rapidity
We developed the efficient screening system in the 96‐well microplate format by using the S. cerevisiae strain harboring the expression vector of human adenosine A2A receptor (A2AR) [Fig. 2(A)], which is a therapeutically important GPCR, as a model mammalian membrane protein. Using a filter plate to separate insoluble materials, the throughput was largely enhanced. The void peak that disappeared after ultracentrifugation could be observed [Fig. 2(B)]. However, there was little difference in the main peak heights between the two methods of separating insoluble materials. During FSEC analysis, we also checked the chromatograms derived from the total protein detected by the UV–Vis detector at 280 nm [Fig. 2(C–F), inset], to check whether the sample concentrations are constant. Then, we compared the expression in the 96‐well plate to that of the 50 mL aerated tube. Although the FSEC peak heights of the cells cultured in the 96‐well plates were slightly higher than those in the 50 mL tube, their peak patterns were similar to each other (Supporting Information Fig. S2).
Figure 2.
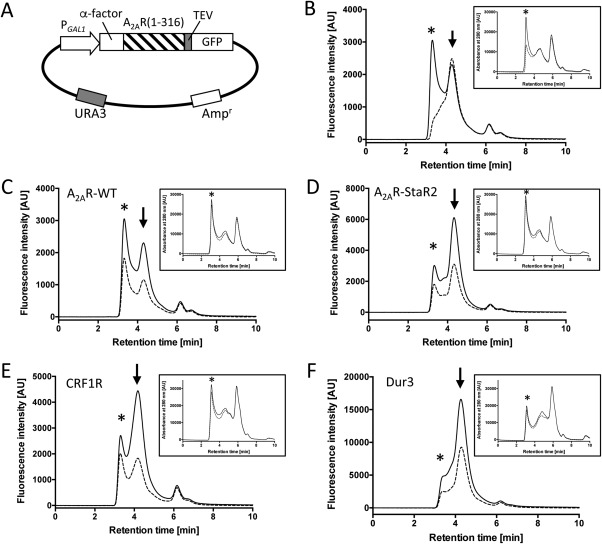
Validation of the expression of membrane protein in S. cerevisiae in the micro‐plate format. (A) Illustration of the expression vector of the adenosine A2A receptor, pDDGFP2_A2AR. Yeast α‐factor secretion signal sequence (α‐factor) was needed for higher expression of A2AR. (B) FSEC chromatogram of A2AR‐WT. The insoluble materials were separated by the 0.22 μm filter (solid line) or by ultracentrifugation (dashed line). (C–F) The FSEC chromatogram of A2AR‐WT; (C), A2AR‐StaR2; (D), corticotropin‐releasing factor 1 receptor (CRF1R); (E), S. cerevisiae urea transporter (Dur3); (F) solubilized with (solid line) and without (dashed line) cell debris. The chromatograms detected by the UV–Vis detector at 280 nm are shown in the inset. Asterisks indicate the void peak. Arrows indicate the peak of the monodisperse membrane proteins.
Mechanical disruption is required for isolation of their cell membrane, since yeast have a rigid cell wall. We examined the efficiency of cell disruption on the 96‐well microplate with glass beads or zirconia–silica beads using a conventional microplate mixer. Ninety percent disruption was achieved in 40 min with the glass beads, whereas the same degree of disruption required only 8 min with the zirconia–silica beads. The FSEC profile obtained with both methods was similar, indicating that strong breakage did not affect the condition of the membrane protein (data not shown).
We examined the solubilization process to increase the throughput in a microplate format. In our previous protocol, cell debris is removed after disruption and yeast membranes are collected by ultracentrifugation before solubilization.36, 38 In this study, the membrane suspensions with cell debris were solubilized, analyzed by FSEC and compared with those without debris, for class A GPCRs (A2AR and protease‐activated receptor 1; PAR1), class B GPCR (corticotropin‐releasing factor 1 receptor; CRF1R), and S. cerevisiae transporters (urea transporter; Dur3, glucose transporter; Hxt3 and GDP‐mannose transporter; Vgr4). Although the peak height was larger than that without cell debris, the shape of chromatogram was similar to each other [Fig. 2(C–F), Supporting Information Fig. S3], indicating that there was no adverse effect by the cell debris. This permitted omission of the most laborious step in the screening.
We examined whether the differences in the amount of inoculated cells from colonies could cause the dispersion of the expression level of the same protein among wells. A large dispersion of the OD was observed among wells up to 13 h after inoculation, which was probably due to the differences in the amount of the cells used for inoculation [Fig. 3(A)]. However, the dispersion was not observed at 19 h after inoculation. Therefore, the OD before the second dilution was similar in all wells. We grouped the eight wells with higher OD (designated “high”) and the eight wells with lower OD (designated “low”) for A2AR‐WT and A2AR‐StaR2 at 9 h after inoculation [Fig. 3(B)]. The cells were cultured for a further 13 h (total 22 h after inoculation), then diluted, subcultured, induced, and analyzed by FSEC. Our results demonstrated that, at harvest, there were little differences in the ODs, whole‐cell fluorescence (data not shown) and the peak height of FSEC between the high OD group and the low OD group [Fig. 3(C)]. These results indicated that the amount of inoculated cells hardly affected the final cell density and the expression level. The standard deviation of the peak height in each well was within ±5%, showing the reliability of the screening system.
Figure 3.
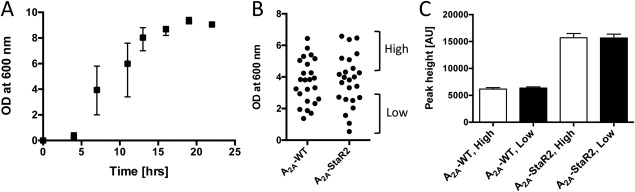
Cell growth of the preculture and expression level. (A) Cell growth of FGY217 yeast harboring the A2A‐WT plasmid in −Ura + Ade medium with 2% glucose. The means and errors for the optical densities (ODs) at 600 nm from eight randomly selected wells were plotted at the indicated time after inoculation. (B) The cell density (optical density) of 24 colonies of A2A‐WT and A2A‐StaR2 after 9 h of first inoculation. The culture wells were divided into two groups: a high‐cell density group (OD600 = 4.0–6.4, average 5.2) and a low‐cell density group (OD600 = 0.6–2.9, average 2.2). (C) The peak heights of FSEC chromatograms of A2A‐WT and A2A‐StaR2 for the group of high cell density (white bar) and low cell density (black bar). Mean values ± the standard deviations (SD) are calculated from the eight wells of each group. The ODs of the culture in all wells were similar to each other at harvest.
Selection of stabilized membrane protein variants in S. cerevisiae in the 96‐well microplate format
Substantial stabilization of unstable membrane proteins could be achieved by combining several single‐point mutations.23, 24, 25, 26, 27 Although each mutation was important, the stabilization effect was not large in many cases. Therefore, the screening system must enable secure selection of an effective single mutation. We validated the availability of this 96‐well plate format screening system by using an A2A mutant designated StaR2 as a model case.21, 39 StaR2 is the A2AR octa‐mutant containing the following mutations: A54L, T88A, R107A, K122A, L202A, L235A, V239A, and S277A, and offers improved thermal stability in the detergent n‐decyl‐β‐d‐maltoside (DM) by more than 27°C compared with the wild type. Although the experimental data for each single mutant is not available, that is, for R107A, L202A, and S277A,39 each single mutation (A54L, T88A, K122A, L235A, or V239A) contributed to the improvement of the thermal stability of A2AR.24
We evaluated each individual mutant in S. cerevisiae by whole‐cell fluorescence, FSEC, and a ligand binding assay using our microplate format screening system. There were no large differences in the OD at harvest between the WT and the mutants (data not shown). The whole‐cell fluorescence and the peak height in the FSEC profile of mutants except for K122A were larger than those of the wild type [Fig. 4(A,C)]. The functional expression was also measured by a ligand binding assay using 3H‐ZM241385. The correlation between the FSEC peak height and functional expression (R 2 = 0.95) was stronger than that between FSEC and the whole‐cell fluorescence (R 2 = 0.76) [Fig. 4(B,D)]. Especially, a much better correlation was observed in the case without StaR2 (R 2 = 0.88). It is likely that the FSEC peak height is a better indicator for selection of mutants with improved stability. In E. coli expression of A2AR, the improvement in stability was not linked to an improvement of the expression level.24 In contrast, in S. cerevisiae, the increase in stabilization was linked to an increase in the functional expression for most of the mutations, which could be judged by the increase in FSEC peak height. For the K122A mutant, decreases in the whole‐cell fluorescence, FSEC peak height, and ligand binding activity were observed. However, this mutant has been reported to improve the stability of the antagonist‐binding form.24 The present data suggest that the K122A mutant might be unstable in the apo state and exhibit improved stability only in the presence of an antagonist.
Figure 4.
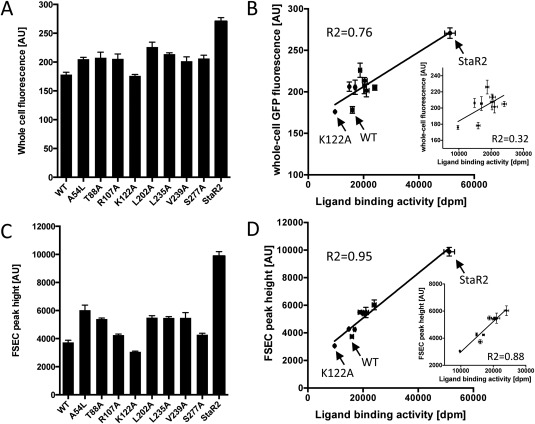
Whole‐cell fluorescence, FSEC peak height, and ligand binding activity. (A) Whole‐cell fluorescence of A2A‐WT, the single mutants, and StaR2. (B) Correlation between the whole‐cell fluorescence and ligand binding activity. Inset is the same plot without StaR2. Correlation coefficients (R 2) are shown. (C) Peak height of FSEC chromatograms of A2A‐WT, the single mutants, and StaR2. The chromatograms are shown in Supporting Information Figure S4. (D) Correlation between the peak height and ligand binding activity. Inset is the same plot without StaR2. Correlation coefficients (R 2) are shown. Mean values ± the standard deviations (SD) of four wells are shown.
A secondary screening should be needed to eliminate the mutants that lost ligand binding activities. We previously demonstrated that a higher amount of histamine H1 receptor was obtained when the cells were cultured in the presence of a ligand.40 We examined whether inactivated mutants could be distinguished from those that retained the ligand binding activity by FSEC using a non‐labeled ligand. As an example, we evaluated the L48A mutant of A2AR, which stabilizes the agonist‐binding form but largely decreases the affinity to the antagonist ZM241385.25 An increase in the FSEC peak height was observed for L48A and A54L mutants (that retain the binding activity) compared with wild type [Fig. 5(A,C)]. The cells were cultured in the presence of the antagonist ZM241385, and analyzed by FSEC. No clear differences were observed for L48A, whereas a clear increment in the peak height was observed for the A54L mutant in the presence of the ligand [Fig. 5(B,D)]. These results indicate that the inactivated receptor (L48A) could be distinguished in this high‐throughput screening manner without the use of RI‐ or fluorescence‐labeled ligands.
Figure 5.

Assessment of the ligand binding activity of the receptor by FSEC. (A) (left panel) FSEC chromatogram of A2A‐WT and the L48A mutant. (right panel) FSEC chromatogram of the L48A mutant with or without 10 μM ZM241385 during induction. (B) (left panel) FSEC chromatogram of A2A‐WT and the A54L mutant. (right panel) FSEC chromatogram of the A54L mutant with or without 10 μM ZM241385 during induction. Arrows indicate the peak of A2AR‐GFP. The mean values of the peak values derived from four colonies are indicated by horizontal lines. Each sample was centrifuged at 50,000 rpm for 30 min to eliminate insoluble materials before FSEC analysis.
Discussion
In this study, we developed a 96‐well microplate format screening method for S. cerevisiae expression and sample preparation, which enabled the high‐throughput construction and evaluation of membrane protein mutants at a low cost with simple equipment. Using the adenosine A2A receptor as a model membrane protein, we demonstrated that the increases in the thermal stability induced by single mutations could be detected as increases in the peak heights in the FSEC chromatogram. Furthermore, the peak height correlated well to the functional expression level based on the ligand binding activity. Namely, the increase in stability was related to the functional expression in S. cerevisiae. Tate and co‐workers recently reported that only correctly folded mammalian membrane proteins could be solubilized by mild detergents like DDM.41 They also pointed out that a considerable proportion of mammalian membrane proteins were misfolded even in insect cells. The present results showed that, in contrast to the 1.4‐fold increase in the whole‐cell GFP fluorescence intensity, the ligand binding activity was increased by 3.2‐fold compared with A2A‐WT and StaR2. This result indicated that the proportion of unfolded protein in A2A‐WT was much larger than that in StaR2. Conversely, this suggests that the proportion of properly folded protein can increase if the protein is thermally stable.
The most reliable way to screen the stabilized membrane protein while monitoring its activity is by combining the solubilization of the expressed membrane protein by detergent and use of an RI‐labeled or fluorescently labeled high‐affinity ligand. However, for many membrane proteins, it is not easy to obtain suitable labeled ligands. Screening of stabilized GPCR mutants using the size‐exclusion chromatography/liquid chromatography mass spectroscopy (SEC/LC‐MS)‐based ligand binding assay was recently reported; this protocol enabled a label‐free ligand binding assay.42 Since LC‐MS requires relatively costly instrumentation, it might be difficult for most researchers to utilize this technique for routine screening. In contrast, in the screening system developed herein, the stability of the membrane protein could be evaluated by FSEC, and the inactivated receptor could be distinguished also by FSEC; therefore, this method can be implemented by using conventional HPLC. The stability and ligand binding activity can be evaluated more precisely by the FSEC‐based thermostability assay (FSEC‐TS).43
The composition of lipids in S. cerevisiae is different from that in mammalian cells,44 which might result in instability and lower expression of many mammalian membrane proteins. In this study, an increase in the ligand binding activity was observed for the stabilized, single substitution A2AR mutants expressed in S. cerevisiae. These mutations might confer the stability to the protein to persist in different lipid environments, resulting in a decrease in the denaturation and degradation during expression. The same concept has been demonstrated in the stabilization of neurotensin receptor type 1 (NTSR1) by directed evolution in E. coli.27 S. cerevisiae would be an appropriate screening host for stabilizing mammalian membrane proteins given that it might provide harsher lipid conditions for the target proteins of stabilization, requiring them to become further stable. This strategy represents a significant platform for handling numerous mutants, similar to alanine scanning. The developed screening strategy is also expected to provide an avenue for confirmation of the stabilization derived from a predicted mutations via computational design, as recently proposed.45
Materials and Methods
Transformation of S. cerevisiae
The expression vector pDDGFP‐230 and the S. cerevisiae FGY217 strain (MATa, ura3‐52, lysΔ201, and pep4Δ)46 were used herein. The cDNA of adenosine A2A receptor (A2AR) with the N154Q mutation on the putative glycosylation site was provided by Prof. S. Iwata at Kyoto University, Japan. The A2AR mutants were generated with the SmaI cut pDDGFP‐2 and the PCR products of A2AR with overlapping 30 bp sequence at both ends of the fragment for homologous recombination.36 Transformation of S. cerevisiae was performed by the lithium acetate method.38 Cells were plated on the yeast synthetic complete medium without uracil plate (−Ura plate), and incubated at 30°C for 2–3 days. The DNA sequences of the A2AR mutants were confirmed with the plasmids isolated from S. cerevisiae.
Expression, measurement of whole‐cell fluorescence, and cell disruption in the 96‐well plate
Colonies of transformants were transferred into 0.5 mL of −Ura medium with 0.2 mg/mL adenine sulfate (designated as −Ura + Ade medium) with 2% (w/v) glucose in the 2 mL, 96‐well, round‐bottom, deep‐well plate (Ritter, Germany). The plate was sealed with AeraSealTM sealing film (Excel Scientific, CA) and shaken overnight (20–22 h) at 30°C at 1400 rpm on a Maximizer MBR‐022UP (TAITEC, Tokyo, Japan) plate shaker. The cells were diluted 65‐fold by introduction into 1 mL of −Ura + Ade medium with 0.1% (w/v) glucose and sub‐cultured for 7 h at 30°C with agitation at 1400 rpm. For induction, 2% (w/v) galactose was added and the cells were cultured for a further 22 h.
Cells were harvested by using a swing‐bucket rotor with microplate buckets at 3000g over the course of 30 min. The supernatant was removed by suction. The cell pellet was resuspended in 160 μL of the suspension buffer (50 mM Tris‐HCl, pH 7.5, 5 mM EDTA, 10% (v/v) glycerol, 0.12 M sorbitol, and complete protease inhibitor cocktail). The cell suspension (20 μL) was transferred into a low‐volume, 384‐well black plate (Greiner Bio‐One, Germany), and the whole‐cell GFP fluorescence was measured using the SpectraMax® Gemini plate reader (Molecular Devices, CA) at an emission wavelength of 525 nm using a 515 nm cutoff filter; the sample was excited at 490 nm.
The cells were disrupted on the same deep‐well plate by adding 100 μL of 0.5 mm glass beads or 0.5 mm zirconia–silica beads and shaking at 2500 rpm on a MicroMixer E‐36 (TAITEC, Tokyo, Japan) microplate mixer at 4°C. The suspensions with cell debris were transferred to 8‐strip PCR tubes or a 96‐well PCR plate and stored at −80°C until further use.
Solubilization and fluorescence size exclusion chromatography (FSEC) analysis
The suspensions of disrupted cells were thawed, and an equal volume of the solution containing 1% (w/v) DDM/0.2% (w/v) CHS was added (the final concentration was 0.5% DDM/0.1% CHS), mixed gently by pipetting on ice for solubilization, and allowed to stand on ice for 1 h. The samples were transferred into 0.5 mL polycarbonate tubes (Hitachi‐Koki, Tokyo, Japan) and ultracentrifuged at 100,000g for 30 min. Alternatively, the samples were transferred into a MultiScreen® HTS filtration plate (Merck‐Millipore, MA), and filtered by centrifugation at 2500g for 10 min. For FSEC analysis, an SRT SEC‐300 column (Sepax Technologies, DE,) was equilibrated with SEC buffer (20 mM Tris‐HCl pH 7.5, 150 mM NaCl, 0.03% DDM/0.006% CHS). FSEC analysis was performed using a high‐performance liquid chromatography (HPLC) system equipped with a fluorescence detector, RF‐20A (Shimadzu, Kyoto, Japan), a UV–Vis detector, SPD‐20A (Shimadzu, Kyoto, Japan), and an autosampler, SIL‐20AC (Shimadzu, Kyoto, Japan). The sample (5 μL) was injected and the GFP fluorescence was detected at an emission wavelength of 525 nm after excitation at 490 nm.
Ligand binding assay
A 40 μL suspension of the cells disrupted on the deep‐well plate (containing approximately 4 × 107 cells, as estimated from the OD at 600 nm) was incubated with 15 nM 3H‐ZM241385 in 150 μL of the assay buffer (50 mM HEPES pH 7.5, 150 mM NaCl) at 20°C for 1 h. Nonspecific binding was determined in the presence of a 1000‐fold excess of the unlabeled ligand. Membranes and cell debris were harvested on a UniFilter®‐96 GF/B microplate (PerkinElmer, MA) presoaked in 0.3% polyethylenimine using a FilterMateTM cell harvester (PerkinElmer, MA). The free ligands were separated by washing five times with 200 μL of the assay buffer. The radioactivity was measured on a MicroBeta2® (PerkinElmer, MA) instrument by adding 70 μL of MicroScintTM−20 (PerkinElmer, MA) to each well.
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Acknowledgment
We appreciate the technical assistance from The Research Support Center, Research Center for Human Disease Modeling, Kyushu University Graduate School of Medical Sciences. We appreciate Prof. S. Iwata and Dr T. Kobayashi for providing the A2A receptor DNA.
References
- 1. Fagerberg L, Jonasson K, von Heijne G, Uhlen M, Berglund L (2010) Prediction of the human membrane proteome. Proteomics 10:1141–1149. [DOI] [PubMed] [Google Scholar]
- 2. Rask‐Andersen M, Almen MS, Schioth HB (2011) Trends in the exploitation of novel drug targets. Nat Rev Drug Discov 10:579–590. [DOI] [PubMed] [Google Scholar]
- 3. Sonoda Y, Newstead S, Hu NJ, Alguel Y, Nji E, Beis K, Yashiro S, Lee C, Leung J, Cameron AD, Byrne B, Iwata S, Drew D (2011) Benchmarking membrane protein detergent stability for improving throughput of high‐resolution X‐ray structures. Structure 19:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB (2003) The G‐protein‐coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63:1256–1272. [DOI] [PubMed] [Google Scholar]
- 5. Warne T, Serrano‐Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF (2008) Structure of a beta1‐adrenergic G‐protein‐coupled receptor. Nature 454:486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330:1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC (2010) Structures of the CXCR4 chemokine GPCR with small‐molecule and cyclic peptide antagonists. Science 330:1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG (2011) Agonist‐bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474:521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. White JF, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, Gvozdenovic‐Jeremic J, Shah P, Shiloach J, Tate CG, Grisshammer R (2012) Structure of the agonist‐bound neurotensin receptor. Nature 490:508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC (2012) Structure of the human kappa‐opioid receptor in complex with JDTic. Nature 485:327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, Li T, Ma L, Fenalti G, Li J, Zhang W, Xie X, Yang H, Jiang H, Cherezov V, Liu H, Stevens RC, Zhao Q, Wu B (2013) Structure of the CCR5 chemokine receptor‐HIV entry inhibitor maraviroc complex. Science 341:1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hollenstein K, Kean J, Bortolato A, Cheng RK, Dore AS, Jazayeri A, Cooke RM, Weir M, Marshall FH (2013) Structure of class B GPCR corticotropin‐releasing factor receptor 1. Nature 499:438–443. [DOI] [PubMed] [Google Scholar]
- 13. Srivastava A, Yano J, Hirozane Y, Kefala G, Gruswitz F, Snell G, Lane W, Ivetac A, Aertgeerts K, Nguyen J, Jennings A, Okada K (2014) High‐resolution structure of the human GPR40 receptor bound to allosteric agonist TAK‐875. Nature 513:124–127. [DOI] [PubMed] [Google Scholar]
- 14. Dore AS, Okrasa K, Patel JC, Serrano‐Vega M, Bennett K, Cooke RM, Errey JC, Jazayeri A, Khan S, Tehan B, Weir M, Wiggin GR, Marshall FH (2014) Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511:557–562. [DOI] [PubMed] [Google Scholar]
- 15. Egloff P, Hillenbrand M, Klenk C, Batyuk A, Heine P, Balada S, Schlinkmann KM, Scott DJ, Schutz M, Pluckthun A (2014) Structure of signaling‐competent neurotensin receptor 1 obtained by directed evolution in Escherichia coli . Proc Natl Acad Sci USA 111:E655–E662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang K, Zhang J, Gao ZG, Zhang D, Zhu L, Han GW, Moss SM, Paoletta S, Kiselev E, Lu W, Fenalti G, Zhang W, Muller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q (2014) Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature 509:115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang D, Gao ZG, Zhang K, Kiselev E, Crane S, Wang J, Paoletta S, Yi C, Ma L, Zhang W, Han GW, Liu H, Cherezov V, Katritch V, Jiang H, Stevens RC, Jacobson KA, Zhao Q, Wu B (2015) Two disparate ligand‐binding sites in the human P2Y1 receptor. Nature 520:317–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chrencik JE, Roth CB, Terakado M, Kurata H, Omi R, Kihara Y, Warshaviak D, Nakade S, Asmar‐Rovira G, Mileni M, Mizuno H, Griffith MT, Rodgers C, Han GW, Velasquez J, Chun J, Stevens RC, Hanson MA (2015) Crystal structure of antagonist bound human lysophosphatidic acid receptor 1. Cell 161:1633–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, Schertler GF, Tate CG (2011) The structural basis for agonist and partial agonist action on a beta(1)‐adrenergic receptor. Nature 469:241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Congreve M, Rich RL, Myszka DG, Figaroa F, Siegal G, Marshall FH (2011) Fragment screening of stabilized G‐protein‐coupled receptors using biophysical methods. Methods Enzymol 493:115–136. [DOI] [PubMed] [Google Scholar]
- 21. Robertson N, Jazayeri A, Errey J, Baig A, Hurrell E, Zhukov A, Langmead CJ, Weir M, Marshall FH (2011) The properties of thermostabilised G protein‐coupled receptors (StaRs) and their use in drug discovery. Neuropharmacology 60:36–44. [DOI] [PubMed] [Google Scholar]
- 22. Hutchings CJ, Koglin M, Marshall FH (2010) Therapeutic antibodies directed at G protein‐coupled receptors. MAbs 2:594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Serrano‐Vega MJ, Magnani F, Shibata Y, Tate CG (2008) Conformational thermostabilization of the beta1‐adrenergic receptor in a detergent‐resistant form. Proc Natl Acad Sci USA 105:877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Magnani F, Shibata Y, Serrano‐Vega MJ, Tate CG (2008) Co‐evolving stability and conformational homogeneity of the human adenosine A2a receptor. Proc Natl Acad Sci USA 105:10744–10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lebon G, Bennett K, Jazayeri A, Tate CG (2011) Thermostabilisation of an agonist‐bound conformation of the human adenosine A(2A) receptor. J Mol Biol 409:298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shibata Y, White JF, Serrano‐Vega MJ, Magnani F, Aloia AL, Grisshammer R, Tate CG (2009) Thermostabilization of the neurotensin receptor NTS1. J Mol Biol 390:262–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sarkar CA, Dodevski I, Kenig M, Dudli S, Mohr A, Hermans E, Pluckthun A (2008) Directed evolution of a G protein‐coupled receptor for expression, stability, and binding selectivity. Proc Natl Acad Sci USA 105:14808–14813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dodevski I, Pluckthun A (2011) Evolution of three human GPCRs for higher expression and stability. J Mol Biol 408:599–615. [DOI] [PubMed] [Google Scholar]
- 29. Kawate T, Gouaux E (2006) Fluorescence‐detection size‐exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14:673–681. [DOI] [PubMed] [Google Scholar]
- 30. Newstead S, Kim H, von Heijne G, Iwata S, Drew D (2007) High‐throughput fluorescent‐based optimization of eukaryotic membrane protein overexpression and purification in Saccharomyces cerevisiae . Proc Natl Acad Sci USA 104:13936–13941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Drew D, Newstead S, Sonoda Y, Kim H, von Heijne G, Iwata S (2008) GFP‐based optimization scheme for the overexpression and purification of eukaryotic membrane proteins in Saccharomyces cerevisiae . Nat Protoc 3:784–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van der Rest ME, Kamminga AH, Nakano A, Anraku Y, Poolman B, Konings WN (1995) The plasma membrane of Saccharomyces cerevisiae: structure, function, and biogenesis. Microbiol Rev 59:304–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Spector AA, Yorek MA (1985) Membrane lipid composition and cellular function. J Lipid Res 26:1015–1035. [PubMed] [Google Scholar]
- 34. Morein S, Andersson A, Rilfors L, Lindblom G (1996) Wild‐type Escherichia coli cells regulate the membrane lipid composition in a “window” between gel and non‐lamellar structures. J Biol Chem 271:6801–6809. [DOI] [PubMed] [Google Scholar]
- 35. Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S (2011) Structure of the human histamine H1 receptor complex with doxepin. Nature 475:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shiroishi M, Tsujimoto H, Makyio H, Asada H, Yurugi‐Kobayashi T, Shimamura T, Murata T, Nomura N, Haga T, Iwata S, Kobayashi T (2012) Platform for the rapid construction and evaluation of GPCRs for crystallography in Saccharomyces cerevisiae . Microb Cell Fact 11:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ito K, Sugawara T, Shiroishi M, Tokuda N, Kurokawa A, Misaka T, Makyio H, Yurugi‐Kobayashi T, Shimamura T, Nomura N, Murata T, Abe K, Iwata S, Kobayashi T (2008) Advanced method for high‐throughput expression of mutated eukaryotic membrane proteins in Saccharomyces cerevisiae . Biochem Biophys Res Commun 371:841–845. [DOI] [PubMed] [Google Scholar]
- 38. Shiroishi M, Kobayashi T (2015) Screening of stable G‐protein‐coupled receptor variants in Saccharomyces cerevisiae . Methods Mol Biol 1261:159–170. [DOI] [PubMed] [Google Scholar]
- 39. Dore AS, Robertson N, Errey JC, Ng I, Hollenstein K, Tehan B, Hurrell E, Bennett K, Congreve M, Magnani F, Tate CG, Weir M, Marshall FH (2011) Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 19:1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shiroishi M, Kobayashi T, Ogasawara S, Tsujimoto H, Ikeda‐Suno C, Iwata S, Shimamura T (2011) Production of the stable human histamine H(1) receptor in Pichia pastoris for structural determination. Methods 55:281–286. [DOI] [PubMed] [Google Scholar]
- 41. Thomas J, Tate CG (2014) Quality control in eukaryotic membrane protein overproduction. J Mol Biol 426:4139–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hirozane Y, Motoyaji T, Maru T, Okada K, Tarui N (2014) Generating thermostabilized agonist‐bound GPR40/FFAR1 using virus‐like particles and a label‐free binding assay. Mol Membr Biol 31:168–175. [DOI] [PubMed] [Google Scholar]
- 43. Hattori M, Hibbs RE, Gouaux E (2012) A fluorescence‐detection size‐exclusion chromatography‐based thermostability assay for membrane protein precrystallization screening. Structure 20:1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9:112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen KY, Zhou F, Fryszczyn BG, Barth P (2012) Naturally evolved G protein‐coupled receptors adopt metastable conformations. Proc Natl Acad Sci USA 109:13284–13289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kota J, Gilstring CF, Ljungdahl PO (2007) Membrane chaperone Shr3 assists in folding amino acid permeases preventing precocious ERAD. J Cell Biol 176:617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.