

Abstract
The formation of endohedral metallofullerenes (EMFs) in an electric arc is reported for the mixed‐metal Sc–Ti system utilizing methane as a reactive gas. Comparison of these results with those from the Sc/CH4 and Ti/CH4 systems as well as syntheses without methane revealed a strong mutual influence of all key components on the product distribution. Whereas a methane atmosphere alone suppresses the formation of empty cage fullerenes, the Ti/CH4 system forms mainly empty cage fullerenes. In contrast, the main fullerene products in the Sc/CH4 system are Sc4C2@C80 (the most abundant EMF from this synthesis), Sc3C2@C80, isomers of Sc2C2@C82, and the family Sc2C2 n (2 n=74, 76, 82, 86, 90, etc.), as well as Sc3CH@C80. The Sc–Ti/CH4 system produces the mixed‐metal Sc2TiC@C2 n (2 n=68, 78, 80) and Sc2TiC2@C2 n (2 n=80) clusterfullerene families. The molecular structures of the new, transition‐metal‐containing endohedral fullerenes, Sc2TiC@Ih‐C80, Sc2TiC@D 5h‐C80, and Sc2TiC2@Ih‐C80, were characterized by NMR spectroscopy. The structure of Sc2TiC@Ih‐C80 was also determined by single‐crystal X‐ray diffraction, which demonstrated the presence of a short Ti=C double bond. Both Sc2TiC‐ and Sc2TiC2‐containing clusterfullerenes have Ti‐localized LUMOs. Encapsulation of the redox‐active Ti ion inside the fullerene cage enables analysis of the cluster–cage strain in the endohedral fullerenes through electrochemical measurements.
Keywords: fullerenes, methane, mixed-metal compounds, scandium, titanium
Introduction
Creating molecules with unprecedented structural, chemical, electronic, and magnetic properties is the main motivation behind the developments in the field of endohedral fullerenes (EMFs). In this class of molecules, carbon cages of various sizes can encapsulate one, two, or three metal atoms as well as complex clusters comprising of up to seven atoms.1 The vast majority of endohedral clusterfullerenes is based on Group III metals, including Sc, Y, and lanthanides. In particular, Sc provides the largest variety of clusterfullerenes, such as nitrides (Sc3N@C2 n (2n=68, 70, 78–82)2), carbides with one or two interior carbon atoms (Sc4C@C2 n (2n=80, 82),3 Sc2C2@C2 n (2n=72–88),4 Sc3C2@C80,5 Sc4C2@C80 6), carbohydrides (Sc3CH@C80,7 Sc4C2H@C80 8), oxides (Sc2O@C2 n (2n=70–82),9 Sc4O2@C80,10 Sc4O3@C80 11), sulfides (Sc2S@C2 n (2n=70–82)12), cyanide (Sc3CN@C2 n (2n=78, 80)13), and even mixed carbidocyanide (Sc3C2CN@C80).14
Fullerenes are electro‐active and form a special type of non‐innocent π ligand for the metals inside. Yet, there are certain endohedral species, which can exhibit their own redox activity in the potential window of the carbon cage.15 In this situation, the fullerene cage plays the role of an “electron‐transparent” container that protects the endohedral charge states from the environment. Thus, a combination of the stable carbon cage shielding the endohedral species from the environment with the encapsulation of the redox pair (be it a particular metal or part of the cluster) presents the opportunity to change the properties of the EMF of interest. Although some Group III metal clusterfullerenes exhibit an endohedral redox activity due to the complex electronic structure of the cluster (e.g., Sc3N@Ih‐C80 and its derivatives,16 Sc4O2@Ih‐C80,17 or Sc3CN@Ih‐C80 18), encapsulation of redox‐active metal ions is an appealing approach to create endohedrals with electrochemical activity. Transition metals, with their rich electrochemistry and variety of spin states, would be ideal for the formation of new endohedral species for electronically and magnetically tunable EMFs.
The number of structurally characterized EMFs that involve transition metals is quite limited. Group IV metals have been found to form EMFs in early laser ablation experiments (Ti@Td‐C28 being most famous,19 and its formation mechanism has been recently disclosed20). However, the bulk synthesis of MIV‐EMFs was delayed for almost a decade, until the reports on the arc‐discharge synthesis and isolation of Ti2C80 and three isomers of Ti2C84 by Shinohara et al.21 It was later determined that Ti2C80 was actually a carbide clusterfullerene, that is, Ti2C2@D 3h‐C78.22 Another Ti‐based clusterfullerene, namely, Ti2S@D 3h‐C78, was obtained by Echegoyen et al.23 Note that both Ti‐based EMFs utilize the D 3h‐C78 cage. Hafnium was also reported to yield small amounts of the EMFs Hf2C80 and HfC84, but their structures have not been determined yet.24
Another strategy for the encapsulation of transition metals within EMFs was first applied by Yang and coworkers. The process employed the use of Group III metals (which readily form EMFs) as “templates” to create mixed‐metal clusterfullerenes. When titanium was mixed with Sc or Y, the nitride clusterfullerenes TiM2N@Ih‐C80 (M=Sc, Y) were obtained when a nitrogen atmosphere was involved in the arc synthesis.25 The first successful synthesis of vanadium EMFs, that is, VSc2N@Ih‐C80 and V2ScN@Ih‐C80, utilized a similar strategy.26 It is noteworthy that vanadium‐containing EMFs are not formed through laser‐ablation experiments.27 By using the reactive gas atmosphere method, we have discovered that a Ti–lanthanide system formed a special type of μ3‐C carbido clusterfullerene featuring a Ti=C double bond, M2TiC@Ih‐C80 (M=Y, Nd, Gd, Dy, Er, Lu).28 Such clusterfullerenes are formally isostructural and isoelectronic with the nitride clusterfullerenes M2ScN@Ih‐C80 and can be synthesized with high selectivity when methane is added to the arc‐discharge reactor atmosphere.
In this article, we explore the formation of endohedral metallofullerenes in the mixed‐metal Sc–Ti system by using the reactive gas method with methane and study their electrochemical properties. The mutual influence of the two metals in the synthesis of clusterfullerenes is studied. Finally, electrochemical studies reveal a systematic dependence of the reduction potentials on the endohedral TiIV/TiIII redox couple in a series of Ti–lanthanide carbide clusterfullerenes. This is characteristic of the size of the lanthanide, which is interpreted as a manifestation of the inherent cage/cluster strain present in many clusterfullerenes.
Results and Discussion
Synthesis of EMFs
Methane was shown to be an efficient reactive gas in the synthesis of endohedral metallofullerenes that suppressed the yield of empty cage fullerenes and produced carbide clusterfullerenes as the main fullerene products.3, 28a To obtain a complete overview on the influence of the individual metals, a series of arc‐discharge syntheses with Sc, Ti, and CH4 was performed. The results are summarized in Figure 1 and selected high‐pressure liquid chromatography (HPLC) chromatograms of the raw CS2 extracts are shown in Figure 2.
Figure 1.
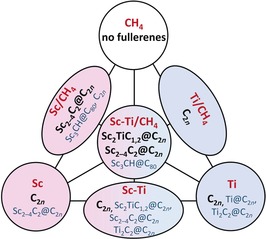
Overview of the EMF syntheses conditions and resulting fullerenes (the amount of graphite and helium gas is constant for all syntheses). Initial conditions (metals and reactive gas) are printed in red, the main fullerene products in black, and the minor fullerene products in blue.
Figure 2.
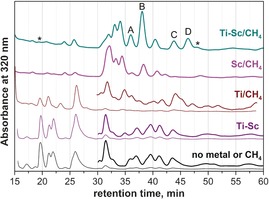
HPLC chromatograms of raw fullerene mixtures synthesized under different conditions (Buckyprep column, toluene as eluent). A–D denote the fractions with a large content of mixed Sc2Ti carbide clusterfullerenes, asterisks mark the minor Sc2Ti clusterfullerene fractions. The retention times of C60 and C70 are t R=9.2 and 14.3 min, respectively (not shown). The other empty fullerenes elute at t R=18.9 (C76), 20.5–21.2 (C78), 26.5 min (C84).
When methane was used as a reactive gas in the arc‐discharge synthesis, no empty cage fullerenes were formed. However, others have reported the isolation of C64H4 29 and C70CH2 30 in somewhat different arc‐discharge syntheses by using methane. In the Sc/CH4 system, the main fullerene products are carbide clusterfullerenes, including Sc4C2@C80 (the most abundant EMF, retention time t R=32 min), Sc3C2@C80 (t R=39 min), isomers of Sc2C2@C82 and the family Sc2C2 n (2 n=74, 76, 82, 86, 90, etc.), as well as Sc3CH@C80 (see also Refs. 3, 7b). Some amounts of Sc3N@C80 are also formed because of the presence of trace nitrogen in the generator. Surprisingly, a completely different behavior was observed in the Ti/CH4 system. Instead of producing Ti–carbide EMFs, we found that Ti has a suppressing influence of CH4 during the synthesis. As a result, the Ti/CH4 system produced only empty cage fullerenes, but with a considerably different size and isomeric distribution (compare the HPLC curves in Figure 2, see also Figure S1 in the Supporting Information). Formation of Ti EMFs in the Ti/CH4 system could not be detected even by mass spectrometry.
The mixed‐metal Sc–Ti/CH4 system was also examined. Figure 2 shows that in the mixed‐metal system methane efficiently suppresses empty cage‐fullerene formation. One of the major differences between the Sc/CH4 and Sc–Ti/CH4 systems is the decrease in the yield of Sc4C2@C80 in the presence of Ti. Sc4C2@C80 is the main EMF formed in the Sc/CH4 synthesis, but it is a minor component in the Sc–Ti/CH4 system. Another major difference involves the formation of a series of mixed‐metal Sc2TiCx clusterfullerenes with both even and odd numbers of carbon atoms.
The main fractions containing Sc2TiCx clusterfullerenes are marked with block letters in the chromatogram in Figure 2. Fraction A contains Sc2TiC79 (presumably Sc2TiC@C78) mixed with comparable amounts of Sc2C82 and Sc2C2@C84. Pure Sc2TiC@C78 was isolated by recycling HPLC on a Buckyprep column (Figure S2 in the Supporting Information). The most abundant EMF product in the Sc–Ti/CH4 system is Sc2TiC81‐I (Sc2TiC@C80‐I) eluting in the fraction B (t R=37–39 min). Unlike M2TiC@Ih‐C80 with lanthanides, which was a single compound in one fraction, Sc2TiC@C80‐I co‐elutes with Sc3C2@C80 (≈15 % of the fraction B; the corresponding peak is also seen in the chromatogram of the Sc/CH4 system). Isolation of pure Sc2TiC@C80‐I was accomplished with recycling HPLC on a Buckyprep‐M column (see Figure S3 in the Supporting Information). In Figure 2, Fractions C and D contain pure Sc2TiC@C80‐II and Sc2TiC82‐I (presumably Sc2TiC2@C80‐I), respectively. Mass spectrometry also provided evidence for the formation of Sc2TiC@C68 and Sc2TiC2@C80‐II (the corresponding fractions are marked with asterisks in Figure 2, see Figures S4 and S5 in the Supporting Information), but their low amounts and similar retention times with other EMFs made further separation impractical. To summarize, the cage‐size distribution of the Sc2TiC@C2 n clusterfullerenes is similar to that of Sc3N@C2 n (note that Sc3N is isoelectronic to Sc2TiC). However, the retention times of the Sc2Ti carbide clusterfullerenes are generally longer. In addition, the difference in the retention times of the two isomers for Sc2TiC@C80 is considerably larger than for the isomers of Sc3N@C80, a situation that facilitates separation.
Molecular structure elucidation for Sc2TiC@Ih‐C80, Sc2TiC@D 5h‐C80, and Sc2TiC2@Ih‐C80
Elucidation of the molecular structure of two isomers of Sc2TiC81 and Sc2TiC82 was accomplished by 13C NMR spectroscopy. Sc2TiC81‐I has a characteristic two‐line spectrum, which unambiguously points to the freely rotating Sc2TiC cluster encapsulated within the Ih(7)‐C80 cage. A similar spectrum with slightly different chemical shifts was observed for Sc2TiC82, which suggests that the compound can be formulated as Sc2TiC2@Ih(7)‐C80. Table 1 compares the 13C chemical shifts of the Sc clusterfullerenes with the Ih‐C80‐cage.
Table 1.
13C and 45Sc NMR chemical shifts (in [ppm]) of the Sc‐based clusterfullerenes with an Ih(7)‐C80 carbon cage
| EMF | 13C5:6:6 [a] | 13C6:6:6 [a] | 45Sc | Reference[b] |
|---|---|---|---|---|
| Sc2TiC@C80 | 143.15 | 135.86 | 277 | t.w. |
| Sc2TiC2@C80 | 143.52 | 136.24 | 316 | t.w. |
| Lu2TiC@C80 | 143.46 | 136.60 | – | 28 |
| Lu2TiC2@C80 | 143.20 | 136.05 | – | 28a |
| Sc3N@C80 | 144.76 (144.57) | 136.41 (137.24) | 191 (200) | t.w. (2b) |
| Sc3CH@C80 | 144.06 | 136.78 | 292 | 7b |
| Sc3C2@C80 − | 145.6 | 138.9 | n/a | 5c |
| Sc3CN@C80 | 144.9 | 137.7 | 280/360 | 13b |
| Sc4C2@C80 | 143.90 (144.7) | 137.14 (137.8) | 373 | t.w. (6) |
| Sc4O2@C80 | 144.82 | 137.29 | 129/292 | 17 |
[a] “C5:6:6” and “C6:6:6” denote two types of carbon atoms in the Ih‐C80 cage: C5:6:6 (60 atoms) is on a pentagon/hexagon/hexagon junction, whereas C6:6:6 (20 atoms) is on a triple hexagon junction. [b] “t.w.” stands for “this work”
The 13C NMR spectrum of Sc2TiC81‐II has six lines, characteristic of the D 5h(6)‐C80 cage, which indicates that the compound is Sc2TiC@D 5h(6)‐C80. A similar spectrum was observed for Sc3N@D 5h(6)‐C80 as shown in Figure 3. The amounts of Sc2TiC69, Sc2TiC79, and Sc2TiC82‐II we obtained were not sufficient for characterization by 13C NMR spectroscopy. However, by analogy with the well‐established structures of Sc3N@D 3(6140)‐C68,2c Sc3N@D 3h(5)‐C78,2a and Sc3N@D 5h(6)‐C80,31 it is likely that they are Sc2TiC@D 3(6140)‐C68, Sc2TiC@D 3h(5)‐C78 (see also the UV/Vis spectra in Figure S7 in the Supporting Information), and Sc2TiC2@D 5h(6)‐C80.
Figure 3.
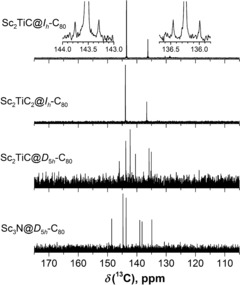
13C NMR spectra of Sc2TiC@Ih‐C80, Sc2TiC2@Ih‐C80, Sc2TiC@D 5h‐C80, and Sc3N@D 5h‐C80. The insets show 13C satellites for 13C‐enriched Sc2TiC@Ih‐C80 (1 J(C,C)=58 Hz).
Attempts to observe the 13C NMR signals of the internal carbon atoms for 13C‐enriched Sc2TiC@Ih‐C80 and Sc2TiC2@Ih‐C80 were not successful. 13C satellites of the cage signals are clearly visible for Sc2TiC@Ih‐C80 (Figure 3), which shows that the sensitivity of the measurement was sufficient for detection in cases with similar linewidths. We propose that broadening of the resonance of the internal carbon atom inhibited the detection as was observed previously for Sc2C2@C 3v‐C82.32
The 45Sc NMR chemical shifts of Sc clusterfullerenes have been found to be quite characteristic and sensitive to the EMF structure as shown in Figure 4 and Table 1. The signals of Sc2TiC@Ih‐C80 (δ=277 ppm) and Sc2TiC2@Ih‐C80 (δ=316 ppm) are shifted downfield versus Sc3N@C80; the value for the former is close to the 45Sc chemical shift in Sc3CH@C80 (δ=292 ppm), another clusterfullerene with a single internal carbon atom. For comparison, we also measured the 45Sc NMR spectrum of Sc4C2@Ih‐C80, which was not reported before. The value is more positive (δ=373 ppm) than for other clusterfullerenes with the Ih‐C80 cage. The 45Sc NMR linewidth in Sc2TiC@Ih‐C80 is similar to that in Sc3N@Ih‐C80 (≈4500 Hz and 4000 Hz, respectively), whereas in Sc2TiC2@Ih‐C80, the peak is noticeably broader (6000 Hz). The broadening may be tentatively ascribed to the internal dynamics of the cluster (such as the motion of the C2 unit).
Figure 4.
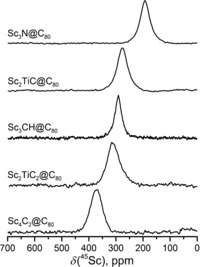
45Sc NMR spectra of Sc2TiC@Ih‐C80, Sc2TiC2@Ih‐C80, and some other clusterfullerenes with the Ih‐C80 carbon cage.
Single‐crystal X‐ray diffraction
Crystals suitable for X‐ray diffraction were grown by co‐crystallization of Sc2TiC@Ih(7)‐C80 with [Ni(OEP)]; OEP is the dianion of octaethylporphyrin. The crystals of Sc2TiC@Ih(7)‐C80 ⋅Ni(OEP)⋅2 (C7H8) are isostructural to crystals of Lu2TiC@Ih(7)‐C80 ⋅Ni(OEP)⋅2 (C7H8), which were previously characterized.28b The asymmetric unit contains one endohedral fullerene, one porphyrin, and two molecules of toluene. The endohedral fullerene consists of a nearly planar Sc2TiC unit inside an Ih‐C80 cage, with the central C81 atom adopting a μ3 configuration (Figure 5). The deviation of the C81 atom from the least‐squares plane of Ti1/Sc1/Sc2 is only 0.022(4) Å. The cage is ordered, whereas there is some disorder in the positioning of the Sc2TiC unit. However, the major site for the Sc2TiC unit has an occupancy of 0.87. For this major site, the distances to the central carbide are: Ti1−C81 1.917(4), Sc1−C81 2.102(4), and Sc2−C81, 2.104(4) Å. The trimetallic carbide is tipped in such a way that the Ti is the metal atom closest to the porphyrin (Figure 5). The shortest distance between the nearest cage carbon atom and the Ni atom is 2.834(4) Å, compared to 2.871(5) Å observed in the Lu2TiC cluster structure.
Figure 5.
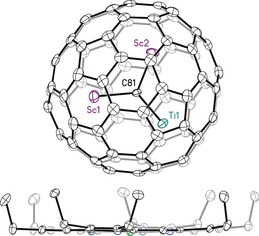
View for the structure of Sc2TiC@Ih(7)‐C80 ⋅Ni(OEP)⋅toluene with hydrogen atoms omitted for clarity. Only the predominant Sc and Ti positions (occupancy 0.87) are shown. Displacement parameters are shown at the 50 % probability level. Selected bond lengths: Ti1−C81 1.917(4), Sc1−C81 2.102(4), and Sc2−C81, 2.104(4) Å.
Inside the Ih(7)‐C80 cage, the Sc2TiC cluster is slightly shifted away from the central cage position by 0.21 Å in the direction of Ti, in keeping with the shorter contacts between Ti⋅⋅⋅C(cage) relative to Sc⋅⋅⋅C(cage). In addition, each metal is closest to a 6:6:5 junction where the carbon is highly pyramidalized at the C3 (13.6°), C58 (12.5°), and C67 (13.1°) atoms as shown in Figures 6 and 7, which show the pyramidalization of the individual carbon atoms in the cage. For comparison, the average pyramidalization angle for an Ih‐C80 endohedral is 10.1°. A similar increase in the pyramidalization of carbon atoms near the internal metal ions has been observed for a functionalized version of Sc3N@C80.33 Those carbon atoms showing unusually small POAV (π‐orbital vector analysis) angles of 8.29–8.95° are one or two bonds away from the C3, C58, and C67 atoms and their flattening compensates for the high pyramidalization of the C3, C58, and C67 atoms as shown in Figure 7. In the present case, all three metals are evidently deforming the geometry at these points. In the case of Lu2TiC@Ih‐C80 ⋅Ni(OEP)⋅2 (C7H8), only the Ti atom had such a large effect (13.7°). In sum, the positioning of the Sc2TiC unit within the Ih‐C80 cage resembles, but is not identical, to that of Lu2TiC. The Ti=C bond in the Sc2TiC cluster is 0.043 Å longer than in the Lu2TiC cluster (1.874(6) Å) but still remains in the bond length range typical for Ti=C double bonds.
Figure 6.
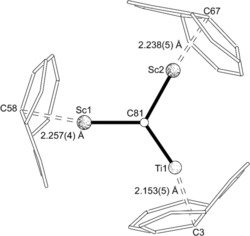
Depiction of the interaction of the metal ions with the closest portions of the cage.
Figure 7.
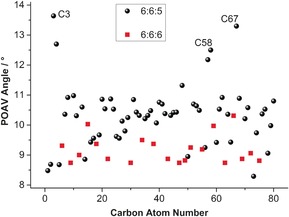
POAV angles for the various carbon atoms in Sc2TiC@Ih‐C80. The red squares denote carbon atoms at 6:6:6 ring intersections, whereas the black circles represent carbon atoms at 6:6:5 intersections. The C3, C58, and C67 atoms are the carbon atoms closest to the metal ions.
DFT calculations of the cluster geometry
In the absence of single‐crystal X‐ray diffraction data for Sc2TiC2@Ih‐C80, its molecular structure was studied computationally at the PBE/TZ2P level of theory. Optimization of multiple initial configurations showed that the structure of the Sc2TiC2 cluster resembles Sc3C2 in Sc3C2@C80.34, 35 The Sc2TiC2 unit adopts a bat ray structure with the three metal atoms forming a triangle whereas the C2 unit is tilted from the plane of the metal atoms. Two metal atoms have a η2‐coordination to the C2 unit, whereas the third atom is bonded to only one carbon atom in an acetylide fashion (η1‐coordination). Due to the fast rearrangement of the C2 unit, all Sc atoms in Sc3C2@C80 are dynamically equivalent. In the Sc2TiC2 case, such configuration of the cluster can be realized in two ways: in the lowest‐energy structure, Ti shares a η2‐coordination with the other Sc atoms, whereas the Ti of the higher‐energy configuration (14.3 kJ mol−1) has a η1‐coordination. Both Sc atoms are η2‐coordinated to the acetylide (Figure 8). For each of the cluster configurations, several near‐isoenergetic orientations (within a few kJ mol−1, see Figure S8 in the Supporting Information) with respect to the cage were found. We also located two different pathways between the two configurations, both with barriers of 80–90 kJ mol−1. Note that for the M2TiC2@Ih‐C80 clusterfullerenes with larger metals (i.e., Y, Lu), the lowest‐energy configuration corresponds to the structure with the C2 unit perpendicular to the plane of three metals (so that all metals are η2‐coordinated to the acetylide).28a The structure of Lu2TiC2@C80 with the C2 unit tilted from the plane was found to be 9 kJ mol−1 less stable. For the Sc2TiC2@Ih‐C80, the cluster configuration with a perpendicular C2 unit is 45 kJ mol−1 less stable than the conformer with the tilted arrangement. Presumably, such variations of the structure traversing from Lu to Sc are caused by the increase of the metal atom size, which forces the cluster to adopt a more compact shape.
Figure 8.
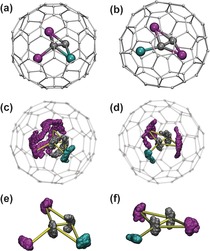
a,b) DFT‐optimized molecular structures of two low‐energy configurations of the Sc2TiC2@Ih‐C80 clusterfullerene with η2‐ and η1‐coordination of Ti (shown in a) and b), respectively). c,d) Born–Oppenheimer molecular dynamics trajectories of the two structures (PBE/DZVP, T=300 K, propagation time >50 ps). e,f) MD trajectories of the Sc2TiC2 cluster in both types of structures obtained after subtraction of the rotational and translational degrees of freedom of the cluster (the cage is not shown for clarity); in essence, e) and f) illustrate internal dynamics of the cluster. Color coding for all figures: magenta=Sc, cyan=Ti, and dark gray=endohedral carbon atoms. In c–f), the yellow lines show bonds in the starting configuration of the cluster (i.e., before molecular dynamics).
To study the dynamic behavior of the Sc2TiC2 cluster in Sc2TiC2@Ih‐C80, Born–Oppenheimer molecular dynamics simulations were performed at the PBE/DZVP level of theory. Two stable configurations were chosen as starting points and the structures were then equilibrated at 300 K during 5 ps. The trajectories were followed for 50 ps with a Nosé–Hoover thermostat. Figure 8 shows the trajectories that were obtained. The two‐line 13C NMR spectrum of Sc2TiC2@Ih‐C80 indicates that the cluster rotates freely on the NMR time scale (which is usually in the nanosecond range). The accessible time frame for our MD simulations is much shorter, approximately 50 ps. Over this period of time, the cluster cannot fully establish rotational motion, but large displacements of the Sc atoms parallel to the inner surface of the cage can be seen, especially in Figure 8 c. Interestingly, the mobility of the Ti atoms is clearly much lower, an observation that may point to stronger metal–cage bonding. Similar observations were found earlier in the MD studies of Sc2TiN@C80.36 The internal dynamics of the cluster are better seen when its external degrees of freedom (rotations and translations) are subtracted. Such trajectories are shown in Figures 8 e and f. Both cluster configurations are found to be dynamically stable (i.e., substantial changes of the cluster geometry are not observed over 50 ps). The most dynamic part of the cluster is the acetylide fragment, which is continuously changing its tilt angle with respect to the plane of the metal atoms. Note that similar motion of the C2 unit was reported in the MD studies of Sc3C2@Ih‐C80.37
The bonding situation in Sc2TiC@C80 and Sc2TiC2@C80 was studied by applying Bader's quantum theory of atoms in molecules (QTAIM).38 Table 2 lists the atomic charges q and the delocalization indices δ(A,B) (roughly equivalent to bond order between the atoms A and B).
Table 2.
QTAIM charges and delocalization indices in Sc2TiC@Ih‐C80, Sc2TiC2@Ih‐C80, and [Sc3C2@Ih‐C80]−.
| EMF | R | q | δ(R,μ3‐C) | δ(R,μ2‐C) |
|---|---|---|---|---|
| Sc2TiC@Ih‐C80 | Ti | +1.65 | 1.43 | |
| Sc | +1.71 | 0.71/0.74 | ||
| μ3‐C | −1.75 | |||
| Sc2TiC2@Ih‐C80 | Ti | +1.59 | 0.55 | 0.66 |
| η2‐Sc | +1.71 | 0.35 | 0.42 | |
| η1‐Sc | +1.65 | 0.60 | 0.08 | |
| μ3‐C | −1.13 | 1.94 | ||
| μ2‐C | −0.63 | 1.94 | ||
| [Sc3C2@Ih‐C80]− | η2‐Sc | +1.62 | 0.43/0.39 | 0.44/0.48 |
| η1‐Sc | +1.69 | 0.64 | 0.09 | |
| μ3‐C | −1.17 | 1.99 | ||
| μ2‐C | −0.63 | 1.99 |
The charge and bond distribution in Sc2TiC@C80 closely resembles that reported for Lu2TiC@Ih‐C80.28b Ti and Sc have similar charges (+1.65 and +1.71, respectively), whereas the central carbon atom bears a large negative charge (−1.75) similar to that in nitride clusterfullerenes. The delocalization index δ(Ti,C) of 1.43 is roughly two times larger than the δ(Sc,C) indices, which is in accord with the Ti=C double bond.
The charges of the metal atoms in Sc2TiC2@Ih‐C80 are similar to those in Sc2TiC@Ih‐C80. The acetylide group bears a negative charge of −1.76, which is unevenly distributed between the μ3‐ and μ2‐carbon atoms (−1.13 and −0.63, respectively). The δ(η2‐Ti,C) indices are systematically larger than the δ(η2‐Sc,C) values, and hence the total metal–acetylide delocalization index, δ(Ti,C2) of 1.21 is noticeably higher than the δ(Sc,C2) value, that is, 0.76. Analysis of the charges and delocalization indices shows that the acetylide unit in Sc2TiC2@Ih‐C80 is similar to that of the single carbon atom in Sc2TiC@Ih‐C80. The C−C bond index in Sc2TiC2 is 1.94, thus the formal charge and bond distribution in the C2 unit can be described as (C=C)4−. QTAIM analysis of the [Sc3C2@Ih‐C80]− ion, which is isostructural and isoelectronic with Sc2TiC2@Ih‐C80, gives very similar values for the atomic charges and delocalization indices (Table 2).
Electronic structure and electrochemistry
The frontier molecular orbitals of Sc2TiC@Ih‐C80 and Sc2TiC2@Ih‐C80 are visualized in Figure 9. The predominant localization of the LUMO on the Ti atom is characteristic for both of these clusterfullerenes. The HOMO of Sc2TiC@Ih‐C80 is localized on the carbon cage and resembles the HOMO of Sc3N@Ih‐C80, whereas the HOMO of Sc2TiC2@Ih‐C80 is equally delocalized between the cluster and the carbon cage. Comparison to lanthanide‐based clusterfullerenes shows that substitution by Sc does not affect the nature of the frontier MOs in M2TiC@Ih‐C80. For M2TiC2@Ih‐C80, the difference between Sc and lanthanides is more pronounced, especially for the HOMO, which has almost no cage contribution in the lanthanide‐based M2TiC2@Ih‐C80 clusterfullerenes.
Figure 9.
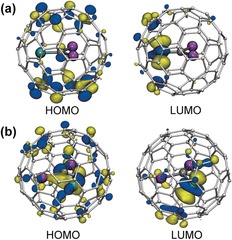
Frontier molecular orbitals of a) Sc2TiC@Ih‐C80 and b) Sc2TiC2@Ih‐C80 computed at the PBE/def2‐TZVP level of theory (isovalues: ±0.038 a.u.). Magenta=Sc, cyan=Ti, and dark gray=internal carbon atoms.
The cyclic voltammetry study shows that the first reduction and oxidation of Sc2TiC@Ih‐C80 are electrochemically reversible (Figure 10 a), whereas increasing the potential window to include the second and third reduction steps also affects the reversibility of the first reduction (observe new re‐oxidation peak at −0.34 V). The first reduction potentials of both Sc2TiC@Ih‐C80 and Sc2TiC2@Ih‐C80 (−0.67 and −0.76 V, respectively, Table 3) are substantially more positive than that of Sc3N@Ih‐C80 (−1.14 V) and can be ascribed to the endohedral TiIV/TiIII redox couple. The oxidation potential of Sc2TiC@Ih‐C80 is virtually equal to that of the nitride clusterfullerene, which is in accord with almost identical HOMOs in both molecules. The first oxidation potential of the Sc2TiC2@Ih‐C80 is shifted in the cathodic direction due to the cluster contribution to the HOMO, but the shift is not dramatic, which is consistent with the distribution of the HOMO between the cluster and the cage.
Figure 10.
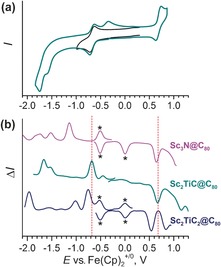
a) Cyclic voltammograms of Sc2TiC@Ih‐C80 and b) square‐wave voltammograms of Sc2TiC@Ih‐C80, Sc2TiC2@Ih‐C80, and Sc3N@Ih‐C80. Measurements are performed at room temperature in o‐dichlorobenzene/TBAPF6 (TBA=tetrabutylammonium) with a scan rate of a) 100 mV s−1 and b) 50 mV s−1. In a), the black curve shows the measurement in the limited potential range covering only the first reduction, whereas the cyan curve shows the measurement in the whole accessible range of potentials. To guide the eye, vertical bars in b) mark the first reduction and oxidation potentials of Sc2TiC@Ih‐C80. Asterisks in b) mark the redox processes of [Fe(Cp*)2] and [Fe(Cp)2] (Cp*=1,2,3,4,5‐pentamethylcyclopentadienyl, Cp=cyclopentadienyl) used as internal standards.
Table 3.
Redox potentials[a] of Sc2TiC@Ih‐C80, Sc2TiC2@Ih‐C80, and selected Ti‐based EMFs.
| EMF | Ox‐I | Red‐I | Red‐II | Red‐III | gapEC | Reference |
|---|---|---|---|---|---|---|
| Sc2TiC@Ih‐C80 | 0.66 | −0.67 | −1.51 | −1.66 | 1.33 | t.w. |
| Sc2TiC2@Ih‐C80 | 0.53 | −0.76 | −1.01 | −1.96 | 1.26 | t.w. |
| Gd2TiC@Ih‐C80 | 0.60 | −1.04 | −1.72 | −1.91 | 1.64 | 28a |
| Y2TiC@Ih‐C80 | 0.60 | −0.99 | −1.67 | −1.89 | 1.59 | 28a |
| Dy2TiC@Ih‐C80 | 0.61 | −0.97 | −1.62 | −1.87 | 1.58 | 28a |
| Dy2TiC2@Ih‐C80 | 0.47 | −1.14 | −1.58 | −2.29 | 1.61 | 28a |
| TiSc2N@Ih‐C80 | 0.16 | −0.94 | −1.58 | −2.21 | 1.10 | 36 |
| TiY2N@Ih‐C80 | 0.00 | −1.11 | −1.79 | 1.11 | 25a | |
| Sc3N@Ih‐C80 | 0.63 | −1.15 | −1.54 | −1.73 | 1.78 | t.w. |
| Ti2S@D 3h(5)‐C78 | 0.23 | −0.92 | −1.53 | −1.80 | 1.15 | 23 |
[a] Potentials are listed in Volt versus the [Fe(Cp)2]+/0 pair, “Ox” stands for oxidation, “Red” stands for reduction, gapEC is an electrochemical gap defined as the difference of the first reduction and oxidation potentials.
Comparison of the first reduction potential of Sc2TiC@Ih‐C80 with that of other Ti‐based clusterfullerenes reveals substantial variability of the endohedral TiIV/TiIII redox couple (Table 3). The variation of the redox potential in the M2TiC@Ih‐C80 series, from −0.67 V in Sc2TiC@Ih‐C80 to −1.04 V in Gd2TiC@Ih‐C80 (Table 3) is especially striking. Figure 11 shows that a linear correlation exists between the reduction potential of M2TiC@Ih‐C80 and the ionic radius of the metal, R(M3+). Such a strong variation of the Ti‐based reduction on the second metal, which is not involved in the LUMO, suggests that the energetics of the reduction is controlled by geometric factors. Earlier, we observed a similar phenomenon for the endohedral CeIV/CeIII redox couple in CeM2N@Ih‐C80 (M=Sc, Lu, Y) and in other Ce‐containing nitride clusterfullerenes.39 In these EMFs, the first oxidation step is usually described as an oxidation of the trivalent Ce. Because the ionic radius of CeIV is smaller than that of CeIII, oxidation of CeIII results in a decrease of the size of the endohedral cluster. For a metal with a large ionic radius, such as Y, the cluster in CeM2N@C80 is highly strained due to the limited room inside the cage, and the oxidation of Ce releases the strain. The variation of the oxidation potential in the CeM2N@Ih‐C80 series is therefore ascribed to the alteration of the strain energy of the nitride cluster. Similar arguments apply here for the M2TiC@Ih‐C80 family. The ionic radius of Ti is increased on going from TiIV (0.605 Å) to TiIII (0.67 Å), and therefore, the size of the M2TiC cluster is increased in the anion (we use Shannon ionic radii throughout this discussion40). When the size of the cluster is relatively small, as in Sc2TiC@Ih‐C80, the increase of the cluster size does not result in a considerable increase of the strain energy (ΔE stain is relatively small). On the contrary, in Gd2TiC@Ih‐C80 the size of the cluster is rather large resulting in a non‐negligible steric strain. A further increase of the cluster size upon reduction is, therefore, not thermodynamically favorable (ΔE stain is large) and requires additional energy input (e.g., more negative potential) to overcome the strain. Thus, the variation of the potential of the internal TiIV/TiIII redox couple can serve as a measure of the strain energy in the M2TiC@Ih‐C80 clusterfullerenes. The increment of the linear correlation in Figure 11 is −1.93 V Å−1, which can be interpreted as an increase of the strain energy by 19 kJ mol−1 with an increase of the ionic radius by 0.1 Å. Although the inner strain in EMFs is an intuitively clear concept, it is not straightforward to give a numerical estimation of the strain energy, especially within experimental studies. Implantation of redox‐active transition metals that change their sizes upon reduction or oxidation into the endohedral cluster thus provides a rare opportunity to address the strain problem in EMFs experimentally. In a similar fashion, the variation of the redox potential of transition metals has been used to analyze the ligand strain in organometallic complexes of Fe, Co, or Cu.41
Figure 11.
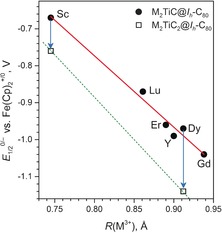
Correlation between the reduction potentials of M2TiC@Ih‐C80 (dots) and M2TiC2@Ih‐C80 (squares) and the Shannon ionic radii of the metals, R(M3+). The arrows denote negative shifts of the reduction potentials when going from M2TiC@C80 to M2TiC2@C80.
This line of argument can be used for other types of Ti‐based clusterfullerenes. Thus, the reduction potentials of Sc2TiC2@Ih‐C80 and Dy2TiC2@Ih‐C80 are shifted negatively versus Sc2TiC@Ih‐C80 and Dy2TiC@Ih‐C80, and the shift is larger for Dy2TiC2@C80 due to the larger ionic radius of Dy3+ (and hence larger ΔE stain value). In a similar fashion, in nitride clusterfullerenes TiM2N@C80, the first reduction step is also a Ti‐based process, and the first reduction of TiY2N@Ih‐C80 is more negative than in TiSc2N@Ih‐C80, because of the larger cluster size in the former. Furthermore, in TiM2N@C80 the valence state of titanium is TiIII, and it becomes TiII when the molecule is reduced. The ionic radius of TiII is 0.86 Å, and therefore, the increase of the metal size in the TiIII/TiII couple (ΔR(M3+/2+)=0.19 Å) is more pronounced than in the TiIV/TiIII couple (ΔR(M4+/3+)=0.06 Å). This observation explains why the reduction potentials of the TiM2N@Ih‐C80 nitride clusterfullerene are more negative than their carbide counterparts M2TiC@Ih‐C80.
Conclusion
The synthesis of the mixed Ti–Sc system in a He/CH4 atmosphere affords a series of new EMFs with mixed‐metal Sc2TiC and Sc2TiC2 clusters. Both clusters have a formal charge of 6+ and thus, prefer the Ih‐C80 cage as disclosed by 13C NMR spectroscopy. Clusterfullerenes with the D 5h‐C80 cage isomer and other cage sizes (i.e., Sc2TiC@C68, Sc2TiC@C78) are also formed in smaller amounts. A single‐crystal X‐ray diffraction study of Sc2TiC@Ih‐C80 suggested the presence of the Ti=C double bond. Sc2TiC@Ih‐C80 and Lu2TiC@Ih‐C80 are the only single carbide‐containing EMFs that have been crystallographically characterized.28b They serve as potential models for the series of EMFs with odd numbers of carbon atoms such as Y3C107 to Y3C125 and Lu3C107 to Lu3C115, which have been discovered recently.42
The presence of methane in the reactor atmosphere is crucial for the high selectivity of the synthesis of carbide clusterfullerenes. In particular, it dramatically reduces the yield of empty fullerenes. Interestingly, this function of CH4 is efficient in the presence of Sc (either Sc/CH4 or Sc–Ti/CH4 systems), whereas Ti blocks the reactive atmosphere effect with the predominant formation of empty fullerenes in the Ti/CH4 system. The presence of Ti also affects the distribution of Sc–carbide clusterfullerenes. The most striking effect is a dramatic decrease of the yield of Sc4C2@C80, the most abundantly formed clusterfullerene in the Sc/CH4 system. Finally, the distribution of empty fullerenes formed in the Ti/CH4 system is noticeably different from that of standard empty fullerene synthesis. Thus, Ti is actively involved in the fullerene formation in the arc discharge even when Ti EMFs are not formed.
Electrochemical and frontier orbital studies of Sc2TiC@Ih‐C80 and Sc2TiC2@Ih‐C80 revealed a Ti‐based reduction in both types of clusterfullerenes. Reduction potentials of endohedral TiIV/TiIII redox couples showed dramatic variation with the size of the endohedral clusters. In particular, the reduction potential in the M2TiC@Ih‐C80 series scales linearly with the ionic radius of the electrochemically inert metals. This phenomenon is explained by consideration of the inner strain in clusterfullerenes emerging when the size of the cluster is non‐commensurable with that of the carbon cage. The increase of the ionic radius of Ti upon reduction increases the strain energy and pushes the reduction potentials of the EMFs with larger (and hence more strained) clusters to more negative values.
Experimental Section
Arc‐discharge synthesis: The arc‐discharge synthesis was performed in a static 250 mbar helium atmosphere in the presence of a several mbar of methane. Graphite rods were drill‐holed and packed with a mixture of Sc/graphite, Ti/graphite, or Sc–Ti/graphite powder (Sc and Ti were used as metals, the molar ratio in the mixed‐metal system was 1:1); 13C enrichment (≈5 %) was achieved by adding amorphous 13C powder. According to our recent study of Sc3CH@C80, 13C enrichment through carbon powder gives a comparable 13C content for the cage and endohedral carbon atoms.7b Note that reducing the amount of residual nitrogen in the generator is crucial for the successful synthesis because nitride clusterfullerenes are readily formed in the presence of nitrogen.
Spectroscopic measurements: MALDI mass spectra were measured with a Bruker autoflex mass spectrometer by using sulfur as a matrix. NMR measurements were performed on a Bruker Avance 500 spectrometer equipped with the multiprobe head 1152Z. The measurements were performed for compounds dissolved in CS2 with [D6]acetone placed in a coaxial tube as a lock; relaxation agents were not used in the NMR measurements. Electrochemical measurements were performed in a glovebox with a three‐electrode cell (Pt wire as working and counter electrodes, Ag wire as a pseudo‐reference electrode; the potentials were calibrated versus [Fe(Cp)2] and [Fe(Cp*)2] as internal standards).
Crystal‐structure determination of Sc2TiC@I h (7)‐C80⋅Ni(OEP)⋅2 (C7H8): Crystals suitable for X‐ray diffraction studies were obtained by layering a toluene solution of [Ni(OEP)] over a toluene solution of Sc2TiC@Ih(7)‐C80 in a 5 mm outside diameter glass tube approximately 18 cm in length. The crystal selected for data collection was a black plate of the dimensions 0.005×0.090×0.140 mm3. The crystal was mounted in the 100 K cold nitrogen stream provided by an Oxford Cryostream low‐temperature apparatus on the goniometer head of a Bruker D8 diffractometer equipped with a Bruker Photon 100 CMOS detector. Data were collected with the use of synchrotron radiation (λ=0.7749 Å) at the Beamline 11.3.1 at the Advanced Light Source, Lawrence Berkeley Laboratory. The structure was solved by a dual space method, (SHELXT)43 and refined by full‐matrix least‐squares on F 2 (SHELXL‐2014).44 The structure is a pseudo‐merohedral twin with twin law (0 −1 0 −1 0 0 0 0 −1) and refined twin parameter of 0.2410(9). The Sc2TiC cluster is disordered over three orientations. There is only one position for the central carbide carbon atom. The three clusters are comprised of Ti1/Sc1/Sc2, Ti2/Sc3/Sc4, and Ti3/Sc1/Sc5 with relative occupancies of 0.87:0.05:0.08. The atoms of the minor orientations were refined with isotropic thermal parameters. All other non‐hydrogen atoms were refined with anisotropic displacement parameters. One of the two toluene molecules is disordered over two orientations with relative occupancies of 0.88:0.12. The minor component of this second toluene site was restrained to have the same geometry as the major component. CCDC 1472263 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Crystal data: C131H60N4NiSc2Ti; M w=1886.36 g mol−1; triclinic; P ; a=14.6160(6), b=14.6376(7), c=20.4430(9) Å; α=83.135(2), β=83.596(2), γ=60.686(2)°; V=3779.1(3) Å3; Z=2; R 1 [15 380 reflections with I>2 σ(I)]=0.0631; wR 2 (all 17 305 unique data)=0.1726, 1309 parameters; 15 restraints; largest diff. peak and hole 1.488 and −1.547 e Å−3.
Computational studies: Density functional theory computations were carried out within the generalized gradient approximation (GGA) PBE45 for the exchange‐correlation term and the original TZ2P‐quality basis set as implemented in the PRIRODA package.46 Wavefunctions for QTAIM analysis were obtained in single‐point‐energy calculations at the PBE/def2‐TZVP level with full‐electron basis sets and scalar‐relativistic DKH correction as implemented in the Orca suite.47 QTAIM analysis was performed with the AIMAll code.48 Born–Oppenheimer molecular dynamics (MD) calculations performed in the CP2K code49 and employed the velocity Verlet algorithm with a time step of 0.5 fs and the Nosé–Hoover thermostat set at 300 K. Before a production run, the systems were first thermostated for 5 ps. MD calculations were performed with the PBE functional and employed Gaussian and plane wave GPW scheme with Goedecker–Teter–Hutter pseudopotentials and DZVP basis set.49a, 50 Molecular structures, orbitals, and MD trajectories were visualized by using the VMD package.51
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors acknowledge funding by the DFG (grant PO 1602/1‐2) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No 648295 “GraM3”) and the U. S. NSF Grant CHE‐1305125 to A.L.B. and M.M.O. Computational resources were provided by the Center for Information Services and High‐Performance Computing (ZIH) at the TU Dresden. The authors thank Ulrike Nitzsche for technical assistance with computational resources at the IFW Dresden, Sandra Schiemenz for the measurements of the absorption spectra, and Christin Scheunert and Pauline Voigt for their help with the synthesis of the fullerenes. We thank the Advanced Light Source, supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U. S. Department of Energy under Contract No. DE‐AC02‐05CH11231 for synchrotron beam time and a fellowship to K.B.G.
K. Junghans, K. B. Ghiassi, N. A. Samoylova, Q. Deng, M. Rosenkranz, M. M. Olmstead, A. L. Balch, A. A. Popov, Chem. Eur. J. 2016, 22, 13098.
Contributor Information
Prof. Marilyn M. Olmstead, Email: mmolmstead@ucdavis.edu.
Prof. Alan L. Balch, Email: albalch@ucdavis.edu
Dr. Alexey A. Popov, Email: a.popov@ifw-dresden.de.
References
- 1.
- 1a. Popov A. A., Yang S., Dunsch L., Chem. Rev. 2013, 113, 5989–6113; [DOI] [PubMed] [Google Scholar]
- 1b. Lu X., Feng L., Akasaka T., Nagase S., Chem. Soc. Rev. 2012, 41, 7723–7760; [DOI] [PubMed] [Google Scholar]
- 1c. Rodríguez-Fortea A., Balch A. L., Poblet J. M., Chem. Soc. Rev. 2011, 40, 3551–3563; [DOI] [PubMed] [Google Scholar]
- 1d. Wang T., Wang C., Acc. Chem. Res. 2014, 47, 450–458; [DOI] [PubMed] [Google Scholar]
- 1e. Yang S., Liu F., Chen C., Jiao M., Wei T., Chem. Commun. 2011, 47, 11822–11839. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Olmstead M. M., de Bettencourt-Dias A., Duchamp J. C., Stevenson S., Marciu D., Dorn H. C., Balch A. L., Angew. Chem. Int. Ed. 2001, 40, 1223–1225; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 1263–1265; [Google Scholar]
- 2b. Stevenson S., Rice G., Glass T., Harich K., Cromer F., Jordan M. R., Craft J., Hadju E., Bible R., Olmstead M. M., Maitra K., Fisher A. J., Balch A. L., Dorn H. C., Nature 1999, 401, 55–57; [Google Scholar]
- 2c. Olmstead M. M., Lee H. M., Duchamp J. C., Stevenson S., Marciu D., Dorn H. C., Balch A. L., Angew. Chem. Int. Ed. 2003, 42, 900–903; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 928–931; [Google Scholar]
- 2d. Wei T., Wang S., Liu F., Tan Y., Zhu X., Xie S., Yang S., J. Am. Chem. Soc. 2015, 137, 3119–3123; [DOI] [PubMed] [Google Scholar]
- 2e. Yang S. F., Popov A. A., Dunsch L., Angew. Chem. Int. Ed. 2007, 46, 1256–1259; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 1278–1281. [Google Scholar]
- 3. Deng Q., Junghans K., Popov A. A., Theor. Chem. Acc. 2015, 134, 10. [Google Scholar]
- 4.
- 4a. Chen C.-H., Ghiassi K. B., Cerón M. R., Guerrero-Ayala M. A., Echegoyen L., Olmstead M. M., Balch A. L., J. Am. Chem. Soc. 2015, 137, 10116–10119; [DOI] [PubMed] [Google Scholar]
- 4b. Feng Y., Wang T., Wu J., Feng L., Xiang J., Ma Y., Zhang Z., Jiang L., Shu C., Wang C., Nanoscale 2013, 5, 6704–6707; [DOI] [PubMed] [Google Scholar]
- 4c. Kurihara H., Lu X., Iiduka Y., Mizorogi N., Slanina Z., Tsuchiya T., Akasaka T., Nagase S., J. Am. Chem. Soc. 2011, 133, 2382–2385; [DOI] [PubMed] [Google Scholar]
- 4d. Iiduka Y., Wakahara T., Nakajima K., Tsuchiya T., Nakahodo T., Maeda Y., Akasaka T., Mizorogi N., Nagase S., Chem. Commun. 2006, 2057–2059; [DOI] [PubMed] [Google Scholar]
- 4e. Wang C. R., Kai T., Tomiyama T., Yoshida T., Kobayashi Y., Nishibori E., Takata M., Sakata M., Shinohara H., Angew. Chem. Int. Ed. 2001, 40, 397–399; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 411–413. [Google Scholar]
- 5.
- 5a. Yannoni C. S., Hoinkis M., Devries M. S., Bethune D. S., Salem J. R., Crowder M. S., Johnson R. D., Science 1992, 256, 1191–1192; [DOI] [PubMed] [Google Scholar]
- 5b. Shinohara H., Sato H., Ohkohchi M., Ando Y., Kodama T., Shida T., Kato T., Saito Y., Nature 1992, 357, 52–54; [Google Scholar]
- 5c. Iiduka Y., Wakahara T., Nakahodo T., Tsuchiya T., Sakuraba A., Maeda Y., Akasaka T., Yoza K., Horn E., Kato T., Liu M. T. H., Mizorogi N., Kobayashi K., Nagase S., J. Am. Chem. Soc. 2005, 127, 12500–12501. [DOI] [PubMed] [Google Scholar]
- 6. Wang T.-S., Chen N., Xiang J.-F., Li B., Wu J.-Y., Xu W., Jiang L., Tan K., Shu C.-Y., Lu X., Wang C.-R., J. Am. Chem. Soc. 2009, 131, 16646–16647. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Krause M., Ziegs F., Popov A. A., Dunsch L., ChemPhysChem 2007, 8, 537–540; [DOI] [PubMed] [Google Scholar]
- 7b. Junghans K., Rosenkranz M., Popov A. A., Chem. Commun. 2016, 52, 6561–6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feng Y., Wang T., Wu J., Zhang Z., Jiang L., Han H., Wang C., Chem. Commun. 2014, 50, 12166–12168. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Tang Q., Abella L., Hao Y., Li X., Wan Y., Rodríguez-Fortea A., Poblet J. M., Feng L., Chen N., Inorg. Chem. 2016, 55, 1926–1933; [DOI] [PubMed] [Google Scholar]
- 9b. Yang T., Hao Y., Abella L., Tang Q., Li X., Wan Y., Rodríguez-Fortea A., Poblet J. M., Feng L., Chen N., Chem. Eur. J. 2015, 21, 11110–11117; [DOI] [PubMed] [Google Scholar]
- 9c. Tang Q., Abella L., Hao Y., Li X., Wan Y., Rodríguez-Fortea A., Poblet J. M., Feng L., Chen N., Inorg. Chem. 2015, 54, 9845–9852; [DOI] [PubMed] [Google Scholar]
- 9d. Zhang M., Hao Y., Li X., Feng L., Yang T., Wan Y., Chen N., Slanina Z., Uhlik F., Cong H., J. Phys. Chem. C 2014, 118, 28883–28889; [Google Scholar]
- 9e. Mercado B. Q., Stuart M. A., Mackey M. A., Pickens J. E., Confait B. S., Stevenson S., Easterling M. L., Valencia R., Rodriguez-Fortea A., Poblet J. M., Olmstead M. M., Balch A. L., J. Am. Chem. Soc. 2010, 132, 12098–12105. [DOI] [PubMed] [Google Scholar]
- 10. Stevenson S., Mackey M. A., Stuart M. A., Phillips J. P., Easterling M. L., Chancellor C. J., Olmstead M. M., Balch A. L., J. Am. Chem. Soc. 2008, 130, 11844–11845. [DOI] [PubMed] [Google Scholar]
- 11. Mercado B. Q., Olmstead M. M., Beavers C. M., Easterling M. L., Stevenson S., Mackey M. A., Coumbe C. E., Phillips J. D., Phillips J. P., Poblet J. M., Balch A. L., Chem. Commun. 2010, 46, 279–281. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Chen N., Mulet-Gas M., Li Y.-Y., Stene R. E., Atherton C. W., Rodriguez-Fortea A., Poblet J. M., Echegoyen L., Chem. Sci. 2013, 4, 180–186; [Google Scholar]
- 12b. Chen N., Beavers C. M., Mulet-Gas M., Rodriguez-Fortea A., Munoz E. J., Li Y.-Y., Olmstead M. M., Balch A. L., Poblet J. M., Echegoyen L., J. Am. Chem. Soc. 2012, 134, 7851–7860; [DOI] [PubMed] [Google Scholar]
- 12c. Dunsch L., Yang S., Zhang L., Svitova A., Oswald S., Popov A. A., J. Am. Chem. Soc. 2010, 132, 5413–5421; [DOI] [PubMed] [Google Scholar]
- 12d. Chen N., Chaur M. N., Moore C., Pinzon J. R., Valencia R., Rodriguez-Fortea A., Poblet J. M., Echegoyen L., Chem. Commun. 2010, 46, 4818–4820; [DOI] [PubMed] [Google Scholar]
- 12e. Mercado B. Q., Chen N., Rodriguez-Fortea A., Mackey M. A., Stevenson S., Echegoyen L., Poblet J. M., Olmstead M. M., Balch A. L., J. Am. Chem. Soc. 2011, 133, 6752–6760. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Wu J., Wang T., Ma Y., Jiang L., Shu C., Wang C., J. Phys. Chem. C 2011, 115, 23755–23759; [Google Scholar]
- 13b. Wang T.-S., Feng L., Wu J.-Y., Xu W., Xiang J.-F., Tan K., Ma Y.-H., Zheng J.-P., Jiang L., Lu X., Shu C.-Y., Wang C.-R., J. Am. Chem. Soc. 2010, 132, 16362–16364. [DOI] [PubMed] [Google Scholar]
- 14. Wang T., Wu J., Feng Y., Dalton Trans. 2014, 43, 16270–16274. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Zhang Y., Popov A. A., Organometallics 2014, 33, 4537–4549; [Google Scholar]
- 15b. Popov A. A., Dunsch L., J. Phys. Chem. Lett. 2011, 2, 786–794. [Google Scholar]
- 16.
- 16a. Jakes P., Dinse K. P., J. Am. Chem. Soc. 2001, 123, 8854–8855; [DOI] [PubMed] [Google Scholar]
- 16b. Popov A. A., Pykhova A. D., Ioffe I. N., Li F.-F., Echegoyen L., J. Am. Chem. Soc. 2014, 136, 13436–13441; [DOI] [PubMed] [Google Scholar]
- 16c. Elliott B., Pykhova A. D., Rivera J., Cardona C. M., Dunsch L., Popov A. A., Echegoyen L., J. Phys. Chem. C 2013, 117, 2344–2348; [Google Scholar]
- 16d. Shustova N. B., Peryshkov D. V., Kuvychko I. V., Chen Y.-S., Mackey M. A., Coumbe C. E., Heaps D. T., Confait B. S., Heine T., Phillips J. P., Stevenson S., Dunsch L., Popov A. A., Strauss S. H., Boltalina O. V., J. Am. Chem. Soc. 2011, 133, 2672–2690; [DOI] [PubMed] [Google Scholar]
- 16e. Popov A. A., Shustova N. B., Svitova A. L., Mackey M. A., Coumbe C. E., Phillips J. P., Stevenson S., Strauss S. H., Boltalina O. V., Dunsch L., Chem. Eur. J. 2010, 16, 4721–4724. [DOI] [PubMed] [Google Scholar]
- 17. Popov A. A., Chen N., Pinzón J. R., Stevenson S., Echegoyen L. A., Dunsch L., J. Am. Chem. Soc. 2012, 134, 19607–19618. [DOI] [PubMed] [Google Scholar]
- 18. Feng Y., Wang T., Wu J.-Y., Ma Y., Zhang Z., Jiang L., Ge C., Shu C.-Y., Wang C.-R., Chem. Commun. 2013, 49, 2148–2150. [DOI] [PubMed] [Google Scholar]
- 19. Dunk P. W., Kaiser N. K., Mulet-Gas M., Rodríguez-Fortea A., Poblet J. M., Shinohara H., Hendrickson C. L., Marshall A. G., Kroto H. W., J. Am. Chem. Soc. 2012, 134, 9380–9389. [DOI] [PubMed] [Google Scholar]
- 20. Deng Q., Heine T., Irle S., Popov A. A., Nanoscale 2016, 8, 3796–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Cao B. P., Suenaga K., Okazaki T., Shinohara H., J. Phys. Chem. B 2002, 106, 9295–9298; [Google Scholar]
- 21b. Cao B. P., Hasegawa M., Okada K., Tomiyama T., Okazaki T., Suenaga K., Shinohara H., J. Am. Chem. Soc. 2001, 123, 9679–9680. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Sato Y., Yumura T., Suenaga K., Moribe H., Nishide D., Ishida M., Shinohara H., Iijima S., Phys. Rev. B 2006, 73, 193 401; [Google Scholar]
- 22b. Yumura T., Sato Y., Suenaga K., Iijima S., J. Phys. Chem. B 2005, 109, 20251–20255; [DOI] [PubMed] [Google Scholar]
- 22c. Tan K., Lu X., Chem. Commun. 2005, 4444–4446. [DOI] [PubMed] [Google Scholar]
- 23. Li F.-F., Chen N., Mulet-Gas M., Triana V., Murillo J., Rodriguez-Fortea A., Poblet J. M., Echegoyen L., Chem. Sci. 2013, 4, 3404–3410. [Google Scholar]
- 24. Akiyama K., Sueki K., Kodama T., Kikuchi K., Takigawa Y., Nakahara H., Ikemoto I., Katada M., Chem. Phys. Lett. 2000, 317, 490–496. [Google Scholar]
- 25.
- 25a. Chen C., Liu F., Li S., Wang N., Popov A. A., Jiao M., Wei T., Li Q., Dunsch L., Yang S., Inorg. Chem. 2012, 51, 3039–3045; [DOI] [PubMed] [Google Scholar]
- 25b. Yang S., Chen C., Popov A., Zhang W., Liu F., Dunsch L., Chem. Commun. 2009, 6391–6393. [DOI] [PubMed] [Google Scholar]
- 26. Wei T., Wang S., Lu X., Tan Y., Huang J., Liu F., Li Q., Xie S., Yang S., J. Am. Chem. Soc. 2016, 138, 207–214. [DOI] [PubMed] [Google Scholar]
- 27. Dunk P. W., Mulet-Gas M., Nakanishi Y., Kaiser N. K., Rodríguez-Fortea A., Shinohara H., Poblet J. M., Marshall A. G., Kroto H. W., Nat. Commun. 2014, 5, 5844. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Junghans K., Schlesier C., Kostanyan A., Samoylova N. A., Deng Q., Rosenkranz M., Schiemenz S., Westerström R., Greber T., Büchner B., Popov A. A., Angew. Chem. Int. Ed. 2015, 54, 13411–13415; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13 609–13 613; [Google Scholar]
- 28b. Svitova A. L., Ghiassi K. B., Schlesier C., Junghans K., Zhang Y., Olmstead M. M., Balch A. L., Dunsch L., Popov A. A., Nat. Commun. 2014, 5, 3568. [DOI] [PubMed] [Google Scholar]
- 29. Wang C. R., Shi Z. Q., Wan L. J., Lu X., Dunsch L., Shu C. Y., Tang Y. L., Shinohara H., J. Am. Chem. Soc. 2006, 128, 6605–6610. [DOI] [PubMed] [Google Scholar]
- 30. Li B., Shu C., Lu X., Dunsch L., Chen Z., Dennis T. J. S., Shi Z., Jiang L., Wang T., Xu W., Wang C., Angew. Chem. Int. Ed. 2010, 49, 962–966; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 974–978. [Google Scholar]
- 31. Cai T., Xu L. S., Anderson M. R., Ge Z. X., Zuo T. M., Wang X. L., Olmstead M. M., Balch A. L., Gibson H. W., Dorn H. C., J. Am. Chem. Soc. 2006, 128, 8581–8589. [DOI] [PubMed] [Google Scholar]
- 32. Yamazaki Y., Nakajima K., Wakahara T., Tsuchiya T., Ishitsuka M. O., Maeda Y., Akasaka T., Waelchli M., Mizorogi N., Nagase H., Angew. Chem. Int. Ed. 2008, 47, 7905–7908; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 8023–8026. [Google Scholar]
- 33. Iezzi E. B., Duchamp J. C., Harich K., Glass T. E., Lee H. M., Olmstead M. M., Balch A. L., Dorn H. C., J. Am. Chem. Soc. 2002, 124, 524–525. [DOI] [PubMed] [Google Scholar]
- 34. Tan K., Lu X., J. Phys. Chem. A 2006, 110, 1171–1176. [DOI] [PubMed] [Google Scholar]
- 35. Fang H., Cong H., Suzuki M., Bao L., Yu B., Xie Y., Mizorogi N., Olmstead M. M., Balch A. L., Nagase S., Akasaka T., Lu X., J. Am. Chem. Soc. 2014, 136, 10534–10540. [DOI] [PubMed] [Google Scholar]
- 36. Popov A. A., Chen C., Yang S., Lipps F., Dunsch L., ACS Nano 2010, 4, 4857–4871. [DOI] [PubMed] [Google Scholar]
- 37. Taubert S., Straka M., Pennanen T. O., Sundholm D., Vaara J., Phys. Chem. Chem. Phys. 2008, 10, 7158–7168. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Popov A. A., Dunsch L., Chem. Eur. J. 2009, 15, 9707–9729; [DOI] [PubMed] [Google Scholar]
- 38b. Bader R. F. W., Atoms in Molecules—A Quantum Theory, Oxford University Press, Oxford, 1990; [Google Scholar]
- 38c. Matta C. F., Boyd R. J., The Quantum Theory of Atoms in Molecules. From Solid State to DNA and Drug Design, Wiley-VCH, Weinheim, 2007. [Google Scholar]
- 39. Zhang Y., Schiemenz S., Popov A. A., Dunsch L., J. Phys. Chem. Lett. 2013, 4, 2404–2409. [Google Scholar]
- 40. Shannon R., Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar]
- 41.
- 41a. Geselowitz D., Inorg. Chem. 1981, 20, 4457–4459; [Google Scholar]
- 41b. Comba P., Jakob H., Helv. Chim. Acta 1997, 80, 1983–1991; [Google Scholar]
- 41c. Comba P., Coord. Chem. Rev. 1999, 182, 343–371; [Google Scholar]
- 41d. Comba P., Coord. Chem. Rev. 1993, 123, 1–48; [Google Scholar]
- 41e. Hambley T. W., Inorg. Chem. 1988, 27, 2496–2501. [Google Scholar]
- 42. Sarina E. A., Mercado B. Q., Franco J. U., Thompson C. J., Easterling M. L., Olmstead M. M., Balch A. L., Chem. Eur. J. 2015, 21, 17035–17043. [DOI] [PubMed] [Google Scholar]
- 43. Sheldrick G., Acta Crystallogr. Sect. A 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sheldrick G., Acta Crystallogr. Sect. C 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865–3868. [DOI] [PubMed] [Google Scholar]
- 46.
- 46a. Laikov D. N., Ustynuk Y. A., Russ. Chem. Bull. 2005, 54, 820–826; [Google Scholar]
- 46b. Laikov D. N., Chem. Phys. Lett. 2005, 416, 116–120; [Google Scholar]
- 46c. Laikov D. N., Chem. Phys. Lett. 1997, 281, 151–156. [Google Scholar]
- 47.
- 47a. Neese F., WIREs Comput. Mol. Sci. 2012, 2, 73–78; [Google Scholar]
- 47b. Pantazis D. A., Chen X.-Y., Landis C. R., Neese F., J. Chem. Theory Comput. 2008, 4, 908–919. [DOI] [PubMed] [Google Scholar]
- 48.T. A. Keith, in AIMAll (Version 14.04.17), http://aim.tkgristmill.com, 2014.
- 49.
- 49a. VandeVondele J., Krack M., Mohamed F., Parrinello M., Chassaing T., Hutter J., Comput. Phys. Commun. 2005, 167, 103–128; [Google Scholar]
- 49b. Hutter J., Iannuzzi M., Schiffmann F., VandeVondele J., WIREs Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar]
- 50.
- 50a. Lippert G., Hutter J., Parrinello M., Theor. Chem. Acc. 1999, 103, 124–140; [Google Scholar]
- 50b. Goedecker S., Teter M., Hutter J., Phys. Rev. B 1996, 54, 1703–1710. [DOI] [PubMed] [Google Scholar]
- 51. Humphrey W., Dalke A., Schulten K., J. Molec. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary