

Introduction
Since the discovery of cell‐free fetal DNA (cffDNA) in maternal plasma in 19971 there has been rapid progress in harnessing this as a source of fetal genetic material for prenatal diagnosis. The majority of cell‐free DNA (cfDNA) is maternal in origin2, with the fetal proportion emanating from the placenta3, detectable in the maternal circulation from around 5 weeks' gestation2 and constituting around only 10% of cfDNA in early pregnancy4. However, as cffDNA is cleared rapidly from the maternal circulation after delivery, it offers great potential as a source of fetal genetic material for prenatal diagnosis5. Initially, in view of the high background of maternal cfDNA, technological restrictions only enabled the detection or exclusion of alleles that were not present in the mother but were present in the fetus because they were paternally inherited or arose de novo at conception. Thus, early indications were for fetal sex determination using Y‐chromosome alleles6, fetal Rhesus D (RhD) genotyping in RhD‐negative mothers7, 8 or for the diagnosis of certain genetic conditions, such as achondroplasia9, in which the majority of cases arise as a result of a new mutation. Technological advances associated with the development of next‐generation sequencing (NGS) have enabled accurate counting of DNA sequences that are associated with specific chromosomes10, 11 present in maternal blood, which has allowed very rapid development of non‐invasive prenatal testing (NIPT) for aneuploidy12. Furthermore, quantification of cffDNA may also be useful in the early identification of pregnancies at risk of other adverse outcomes, such as pre‐eclampsia and fetal growth restriction (FGR)13, 14.
These developments are delivering the biggest change seen in antenatal care over the last few decades, as the need for invasive diagnostic testing reduces dramatically. It is also likely that they will impact on the need for some therapeutic interventions, such as in‐utero fetal transfusion, as well as offering a new diagnostic tool in fetal medicine for diagnosis of a dysmorphic fetus and earlier diagnoses in pregnancies at prior risk of a genetic disorder. Here, we review the potential of cffDNA, highlighting its use in fetal medicine, and discuss how it is impacting on the practice of fetal medicine.
X‐linked disorders and disorders of genital ambiguity
The earliest clinical use of cffDNA was for the determination of fetal sex1. This relies on the detection of sequences, SRY or DYS14, in the maternal plasma that derive from the Y‐chromosome. The technique has already become incorporated into standard care in several European countries, including the UK, for management of pregnancies at risk of severe X‐linked genetic disorders15, 16, such as Duchenne muscular dystrophy. It has the potential to reduce the incidence of invasive testing for such conditions by up to 50% by allowing targeted testing in male‐bearing pregnancies16. In pregnancies at risk of congenital adrenal hyperplasia, determining fetal sex can enable early cessation of steroid treatment in male‐bearing pregnancies or, as occurs in several centers, steroid administration could be delayed until fetal sex is determined through early cffDNA testing and only offered in pregnancies in which the fetus is known to be female17. However, close co‐ordination with fetal‐medicine services is required as testing is only reliable after 7 weeks' gestation, with false‐positive results possible in twin pregnancies or in those with early fetal demise of a cotwin16.
Ambiguity of the genitalia is a rare finding on ultrasound, and even with the advent and improvement of three‐dimensional (3D) imaging techniques, differentiation between clitoromegaly in the female fetus and hypospadias in the male remains difficult (Figure 1). In cases in which genital ambiguity is isolated and cffDNA testing indicates that the fetus is male, the most likely diagnosis is hypospadias, although some rare endocrine disorders cannot be excluded completely without sequencing of the androgen receptor gene (Table 1). If, however, cffDNA testing indicates that the fetus is female genetically, referral to a team specialized in disorders of sexual development is advised as abnormalities in SRY can cause disorders of sexual differentiation and multiple markers should be assessed for determination of fetal sex in these cases (Table 1)15. Another relatively common association with hypospadias is FGR; therefore, maternal uterine artery Doppler examinations should be performed and, if Down syndrome screening is performed, maternal serum biomarker results should be reviewed for low levels of pregnancy‐associated plasma protein A (PAPP‐A) and high levels of human chorionic gonadotropin (hCG) or α‐fetoprotein levels (Table 1).
Figure 1.
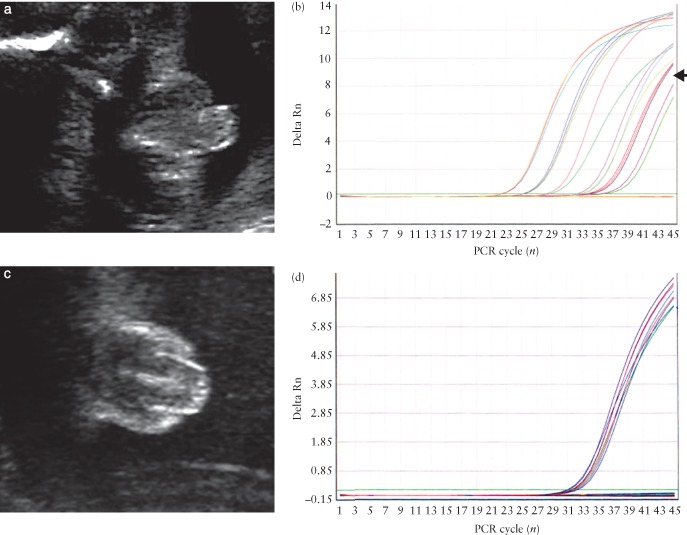
Genital ambiguity in a male fetus (a), as evidenced by amplification (arrow) of SRY sequences in cell‐free fetal DNA (cffDNA) (b), and in a female fetus (c), in which there is amplification only of the control DNA sequences (d). PCR, polymerase chain reaction; Rn, normalized reporter.
Table 1.
Clinical management of fetuses with isolated genital ambiguity based on the cell‐free fetal DNA (cffDNA) result
| cffDNA result | Differential diagnosis | Other aids for management |
|---|---|---|
| Male |
Isolated hypospadias FGR Inadequate production of testosterone because of Leydigcell hypoplasia or rare abnormalities of steroidmetabolic pathway Partial androgen insensitivity syndrome 5α‐reductase deficiency True hermaphrodite |
Look for markers of FGR: review PAPP‐A, hCG and MSAFP results if available Review maternal Doppler Consider referral to DSD team for sequencing of androgen receptor gene |
| Female |
Congenital adrenal hyperplasia 21‐hydroxylase deficiency 11‐hydroxylase deficiency 3β‐hydroxysteroid dehydrogenase deficiency True hermaphrodite Maternally derived androgens (e.g. luteoma of pregnancy) Placental aromatase deficiency |
Refer to DSD team for further investigations Amniotic steroid levels Maternal serum androgen levels Maternal urinary estrogen levels Maternal ovarian scan for multicystic change |
DSD, disorders of sexual development; FGR, fetal growth restriction; hCG, human chorionic gonadotropin; MSAFP, maternal serum alpha‐fetoprotein; PAPP‐A, pregnancy‐associated plasma protein A.
The use of cffDNA for sex determination can be an extremely useful aid to sonographic diagnoses of a number of genetic syndromes that present with multiple abnormalities, usually genital ambiguity in which determination of genetic sex can be diagnostic when combined with the presence of other relevant sonographic findings (Table 2). For example, in cases of campomelic dysplasia, which presents with varying degrees of lower limb shortening and bowing, talipes and micrognathia, at least 50% of affected male fetuses have genital ambiguity or complete sex reversal. Therefore, if a fetus has ambiguous or female genitalia with these sonographic features and cffDNA testing indicates a genetic female, the diagnosis can be made (Table 2 and Figure 2). Non‐invasive fetal sex determination using cffDNA can also be very useful in the presence of some urogenital anomalies, such as bladder cloacal exstrophy, as knowledge of the genetic sex can aid counseling with regard to long‐term outcome18.
Table 2.
Examples of genetic syndromes for which sex determination using cell‐free fetal DNA (cffDNA) or targeted non‐invasive prenatal testing for single‐gene disorders may aid ultrasound diagnosis
| Syndrome | Genitalia | Other sonographic findings | Differential diagnosis | cffDNA result | Other aids for diagnosis |
|---|---|---|---|---|---|
| Bardet–Biedel | Ambiguous/hypospadias | Large echogenic kidneys, polydactyly | Trisomy 13 | Male | Family history, consanguinity |
| Smith‐Lemli–Opitz | Ambiguous | Polysyndactyly, cardiac and CNS anomalies, FGR, cleft lip, microcephaly | Other genetic syndromes, trisomy 13 | Male | Family history, consanguinity, maternal urinary steroids |
| Malpeuch syndrome | Ambiguous | Cleft lip, FGR, renal anomalies | Trisomy 18 | Male | Family history, consanguinity |
| Campomelic dysplasia | Ambiguous/female | Short bowed lower limbs, talipes, cardiac anomalies, micrognathia | Osteogenesis imperfecta Type III/IV | Male | |
| Achondroplasia* | Normal | Rhizomelic shortening of long bones at > 24 weeks, bowed femora, frontal bossing, relative macrocephaly, trident hands, polyhydramnios (small chest) |
Down syndrome, Acromesomelic dysplasia, Hypochondroplasia |
FGFR3 mutn | Normal limb length at < 24 weeks' gestation |
| Thanatophoric dysplasia | Normal | Early‐onset shortened long bones, bowed femora, short ribs, small chest, trident hands, frontal bossing, relative macrocephaly, clover‐leaf skull, CNS anomalies, polyhydramnios |
Short‐ribbed polydactyly syndromes, Jeunes asphyxiating thoracic dystrophy, Spondyloepiphyseal dysplasia congenita |
FGFR3 mutn | |
| Apert syndrome | Normal | Abnormal skull shape, mitten hands and feet | Other craniosynostosis syndromes | FGFR2 mutn |
Shortened long bones may be the only presenting feature as this condition often presents in the third trimester when good visualization of other features can be difficult. CNS, central nervous system; FGFR2/3, fibroblast growth factor receptor 2/3 gene; FGR, fetal growth restriction; mutn, mutation.
Figure 2.
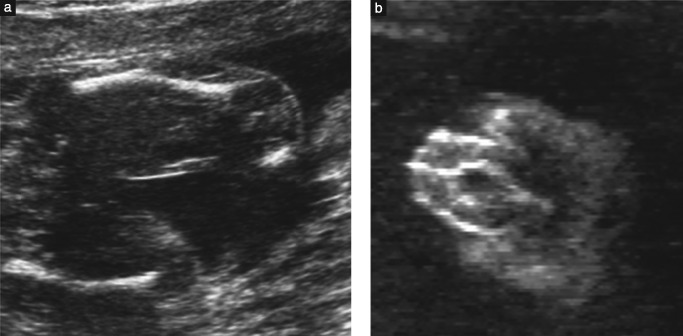
Features of campomelic dysplasia detectable on ultrasound include shortened ‘bowed’ limbs (a) and ambiguous genitalia (b).
Cell‐free fetal DNA and management of complications arising from blood‐group antigens
The second clinical application of cffDNA testing was for the determination of fetal RhD status in pregnant RhD‐negative mothers19. As with fetal sex determination, this is possible because an RhD‐negative mother does not produce any copies of the RhD gene (RHD), and thus the RHD identified in maternal blood originates from the fetus who has inherited the gene from the father. For the past decade, fetal RHD genotyping in RhD‐negative women with significant titers of anti‐RhD immunoglobulin has been possible using labor‐intensive polymerase chain reaction (PCR)‐based methods19, 20. This approach to management of these high‐risk pregnancies has avoided the need for invasive testing that was required previously. In addition to avoiding the associated risks of miscarriage, NIPT circumvents the need for assessment of paternal phenotype, which may not be known or available. If the fetus is RhD positive, increased surveillance in a tertiary center to monitor for the development of fetal anemia or hydrops is required, whereas if the fetus is predicted to be RhD negative, there is no risk of hemolytic disease of the newborn (HDN) and standard antenatal care is appropriate21.
Introduction of high‐throughput technologies for mass fetal RHD genotyping22 has provided potential for routine fetal genotyping and targeted administration of anti‐D immunoglobulin, a human blood product. Currently, many countries offer routine antenatal prophylaxis to all RhD‐negative mothers. This has significantly decreased the incidence of Rhesus sensitization, and cases resulting in fetal anemia are now rare. However, this policy results in the unnecessary administration of anti‐D immunoglobulin to around 38% of RhD‐negative women who are carrying a RhD‐negative fetus23. Routine fetal RHD genotyping and targeted anti‐D prophylaxis have been introduced recently into routine obstetric care at 26–28 weeks' gestation for RhD‐negative women in The Netherlands and Denmark24, 25. However, a UK study has shown that high‐throughput RHD genotyping is highly accurate from 11 weeks' gestation26. Introduction at this earlier stage in pregnancy would result in further avoidance of administration of anti‐D immunoglobulin for sensitizing events that occur in early pregnancy. Subsequently, there have been calls for implementation of high‐throughput RHD genotyping into routine antenatal care in the UK27. Current routine immunoprophylaxis programs do not achieve complete uptake as some women decline anti‐D immunoglobulin treatment. Routine fetal RHD genotyping is likely to be very acceptable to women28 and may improve immunoprophylaxis uptake, thereby targeting women at highest risk, causing a further decline in rates of alloimmunization and, subsequently, the need for in‐utero transfusion.
Although anti‐RhD is the most common cause of HDN, other antibodies, in particular anti‐c and anti‐K (and less commonly anti‐C and anti‐E), are responsible for an increasing proportion of cases. Unlike anti‐D, there is no prophylaxis, so the use of cffDNA as routine screening is unlikely; however, it remains an important investigation in sensitized pregnant women. The accuracy of fetal genotyping in these cases approaches 100%, thereby obviating the need for invasive testing in these pregnancies21, 29, 30.
Fetal or neonatal alloimmune thrombocytopenia (FNAIT) is caused by production of maternal alloantibodies directed against paternally inherited antigens present on fetal platelets21. Complications include intracranial hemorrhage, occurring in up to 20% of cases, which may cause severe long‐term consequences to the child. Until recently, in cases with a heterozygous father, invasive testing was required to determine whether the fetus was affected by FNAIT, which can occur only if they are positive for human platelet antigen‐1a (HPA‐1a). Analysis of cffDNA in maternal blood can detect the HPA‐1a gene31, 32, which again avoids the need for invasive testing in women with a heterozygous partner.
Non‐invasive prenatal testing for monogenic disorders
The use of cffDNA in the detection of monogenic disorders is considerably more challenging technically than fetal RHD genotyping or sex determination and is currently in clinical use only for the detection of alleles that have arisen de novo at conception, for example, achondroplasia9, or that are inherited from the father33. In recessive or X‐linked conditions for which the mother also carries the mutant allele, the high background of maternal mutation present in her plasma outweighs any fetal mutation and thus methods reliant on the detection of small differences in the ratio of mutant‐to‐wild‐type alleles are required. Accurate estimation of this ratio is also dependent on the proportion of fetal DNA in the maternal plasma, the fetal fraction, and this can only be assessed consistently in male‐bearing pregnancies as it requires measurement of an allele not present in the mother34, 35 or by use of the differential methylation in fetal and maternal DNA36; as yet, the reliability of these methods for use in routine clinical practice requires further development and evaluation. It is likely that this will become possible universally in the future, perhaps by exploiting the fact that fetal DNA is shorter than maternal cfDNA37. In recessive conditions, whereby the parents carry different allele mutations, exclusion or detection of the paternal allele can be used to refine the risk to the fetus, excluding risk if the paternal allele is not detected in maternal plasma and increasing risk if the paternal mutation is identified. In the latter situation, an invasive test is required to determine whether or not the fetus has inherited the maternal allele. This has recently been reported in conditions such as thalassemia38 and cystic fibrosis39.
Definitive diagnosis of achondroplasia9 and thanatophoric dysplasia40 by NIPT has been available on a research basis since 2007 and was approved for use in routine clinical practice in the UK in 2012. The early tests were based on restriction‐enzyme‐digest‐PCR methodology, whereby mutations had to be tested for individually. This is labor intensive, slow and costly, and the results can be difficult to interpret (Figure 3). The advent of NGS has allowed the development of gene panels for use in NIPT33 that allow all possible disease‐causing mutations to be tested for simultaneously and a digital result enables easier interpretation. This has greatly enhanced the utility of NIPT for conditions such as thanatophoric dysplasia, of which there are multiple possible mutations and all cases arise de novo as this is a lethal dominant condition. NIPT can be a very useful aid to clinical management of skeletal dysplasias. Thanatophoric dysplasia is detected increasingly in early pregnancy when the differential diagnosis includes the short‐ribbed polydactyly syndromes and other autosomal recessively inherited conditions associated with a high recurrence risk41 (Table 2), whilst thanatophoric dysplasia is a new dominant mutation with a low recurrence risk. If positive for thanatophoric dysplasia, NIPT can deliver a definitive diagnosis without the need for invasive testing and allows the option of a surgical termination, as a postmortem will not be required. Additionally, NIPT can allow the safe differentiation of thanatophoric dysplasia from achondroplasia, both of which arise from mutations in the fibroblast growth factor receptor 3 gene (FGFR3), but the former is lethal and the latter is the most common viable short‐stature syndrome. Distinguishing between these two conditions can be challenging as they have many common features, such as frontal bossing, relative macrocephaly, short limbs, bowing of the femora, small chest and short fingers giving rise to the ‘trident’ hand appearance; these features are all more extreme in thanatophoric dysplasia. NIPT can also offer safer diagnostic testing in twin pregnancies with fetuses discordant for the abnormalities as it permits a safe definitive diagnosis whilst avoiding the risk of miscarriage of the normal fetus. In addition, it allows for conservative management of pregnancy in lethal conditions with no requirement for termination of pregnancy. In cases presenting at risk of achondroplasia late in pregnancy, NIPT allows for definitive diagnosis and accurate parental counseling without the risk of precipitating preterm labor. In all cases, it also allows for an early, safe, non‐invasive test to exclude recurrence or inheritance of a paternal mutant allele in future pregnancies. This can be performed from 9 weeks' gestation, earlier than invasive testing, which cannot be performed safely until 11 weeks42, 43, and much earlier than an ultrasound scan for women not wanting to put the pregnancy at risk by an invasive test.
Figure 3.
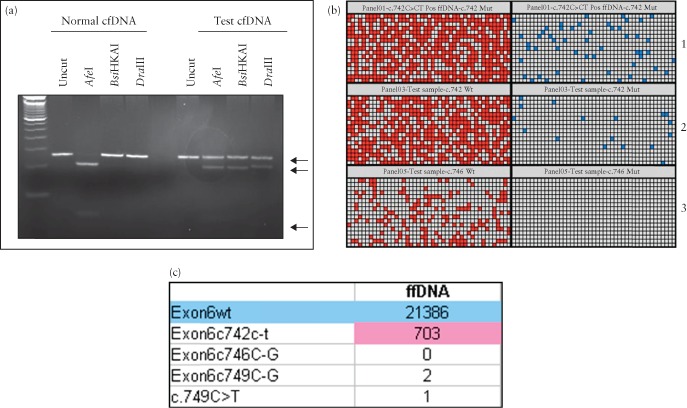
Detection of a mutation in the fibroblast growth factor receptor 3 (FGFR3) gene causing thanatophoric dysplasia, showing the increasing ease of interpretation between polymerase chain reaction (PCR)‐based method (a), digital PCR (b) and digital readout obtained from sequencing (c). PCR‐based method (a) relies on subjective interpretation; very faint bands for mutant alleles in affected cell‐free (cf) DNA can be seen (bottom arrows). The wild‐type (normal) allele is strongly present in all samples (upper arrow). This compares with digital PCR (b) for detection of the mutant allele c.742 C > T (blue dot) and wild‐type alleles (red dot). Each row represents one sample. Wild‐type signals are present in all samples but the mutant allele is only present in the positive control (panel 1) and test sample (panel 2). Panel 3 is the result obtained from a normal pregnancy and shows only wild‐type alleles present. The digital readout obtained from sequencing (c) reveals a very high wild‐type allele count (blue), as this represents both maternal and fetal alleles, and a lower mutant allele (pink) count, but is still very high compared with the counts for other disease‐causing mutations, indicating that the fetus has thanatophoric dysplasia as a result of the c.742 C > T mutation.
The use of cffDNA for the diagnosis of monogenic disorders has great potential. In the UK it has been approved for use in clinical National Health Service (NHS) practice to screen for mutations in the FGFR3 (achondroplasia and thanatophoric dysplasia) and FGFR2 (Apert syndrome) genes and for paternal exclusion of common cystic fibrosis mutations. With the introduction of these safer tests we are seeing a dramatic decrease in the use of invasive testing for monogenic disorders (Table 3), with a further decline in the likely need for invasive testing as more non‐invasive tests are developed and validated.
Table 3.
Shift from invasive to non‐invasive prenatal testing (NIPT) for achondroplasia and thanatophoric dysplasia in the UK from 2009 to 2013, as tests became validated and approved for use in the North East Thames Regional National Health Service genetics laboratory
| Achondroplasia | Thanatophoric dysplasia | |||
|---|---|---|---|---|
| Year | Invasive | NIPT | Invasive | NIPT |
| 2009–2010 | 28 | 0 | 16 | 0 |
| 2010–2011 | 27 | 13 | 21 | 0 |
| 2011–2012 | 28 | 14 | 25 | 2 |
| 2012–2013 | 20 | 22 | 17 | 11 |
| 2013– | 10 | 14 | 7 | 18 |
Data are given as n. Other conditions for which NIPT has been performed in high‐risk families include Apert syndrome (n = 7), Crouzon syndrome (n = 2), Fraser's syndrome (n = 4), autosomal polycystic kidney disease, osteogenesis imperfecta (n = 2) and cystic fibrosis.
Cell‐free DNA testing for aneuploidy
The use of cfDNA testing for aneuploidy is bringing the most radical change to the practice of fetal medicine. Early attempts at providing NIPT for Down syndrome relied on quantifying the amount of placenta‐specific 4 (PLAC4) in the maternal plasma44. This gene originates from chromosome 21, is expressed only in the placenta and is therefore fetal in origin. By detection of two separate alleles in a 1 : 1 ratio, the fetus can be assumed to be euploid. If there is duplication of an allele (because of an extra copy of chromosome 21), the ratio will be 2 : 1 and the fetus can be assumed to be trisomic for chromosome 21. Although groundbreaking, this initial approach was flawed as it required the identification of genetic differences in the parents and was therefore only applicable in around 40% of pregnancies. The advent of NGS brought a new approach that could be applied universally. Rather than detection of a gene product derived from chromosome 21, all DNA circulating in maternal plasma is sequenced. This, by definition, includes both maternal and fetal DNA, and sequencing will analyze the entire fetal and maternal genome. There are three NGS‐based approaches to NIPT for aneuploidy: whole‐genome NGS; targeted NGS; and single nucleotide polymorphisms (SNPs). The whole‐genome approach10 requires sequencing of cfDNA from maternal plasma to generate millions of short sequence reads from the whole genome. These are then mapped to a reference human genome sequence to determine from which chromosome the fragment is derived; the number of fragments mapped uniquely to the chromosome of interest are then counted and compared with the number of counts obtained from other chromosomes. A variety of bioinformatics algorithms have been developed10, 45, 46 to determine whether there is an increase or decrease in the expected number of counts around a set threshold which is suggestive of aneuploidy; for example, if the fetus has trisomy 21, more fragments from chromosome 21 will be present than expected in maternal plasma. Alternative NGS approaches involve the selective amplification of specific genomic loci on the chromosome of interest followed by sequencing. This approach may be more economical as the amount of sequencing required is reduced but has the limitation that only the preselected regions of interest can be studied and the development of these tests is potentially more labor intensive12, although it does allow estimation of fetal fraction. Early studies validated this approach for the detection of trisomies 21 and 18 in high‐risk pregnancies47 but, more recently, the results of a large general‐population study have shown similar high performance in pregnancies at low prior risk48. The third approach, a variation of the targeted approach, is based on the amplification of large numbers of polymorphic loci (SNPs) on the chromosome of interest49. Sensitivity and specificity for the detection of common aneuploidies are high for whole‐genome, targeted and SNP approaches, irrespective of the sequencing platform or bioinformatic algorithms used12.
Since the first reports describing the use of NGS for NIPT to detect trisomy 2110, 11, the pace of development has been extremely rapid and entirely commercially driven. NIPT is now available in the private sector in more than 50 countries, with detection rates in excess of 99% for trisomy 21 and slightly lower for trisomies 18 and 13, at around 96% and 92%, respectively50, 51. Sensitivity for detection of sex‐chromosome aneuploidy remains lower, at 88.6%, with a false‐positive rate of 0.12% for monosomy X50, 51. NIPT for the detection of aneuploidy has been shown to be a highly effective test in both high‐risk12 and low‐risk48 pregnancies.
Although more accurate than conventional combined screening (using nuchal translucency, PAPP‐A and hCG) in the first trimester48, NIPT should still be regarded as a highly sensitive screening test, rather than a diagnostic one52, 53, 54, and any positive result should be confirmed by invasive testing52, 53, 54, ideally by amniocentesis or, at minimum, karyotyping on cultured chorionic villi to avoid confined placental mosaicism. This is because of increasing, well‐documented evidence of discordant results (mostly false positive, but false‐negative results have also been reported)55 between NIPT using cfDNA and conventional karyotyping following invasive testing56. cffDNA is derived placentally and discordant results can therefore be attributed to confined placental mosaicism57, 58 or to early demise of an aneuploid cotwin (vanishing twin)55 as the placenta can continue to shed cffDNA after death of the fetus3. Other causes are derived maternally, including the detection of maternal chromosomal rearrangements59, 60, 61 or mosaicism62 as sequencing analyzes maternal as well as fetal cfDNA. Finally, maternal malignancy secondary to a tumor secreting abnormal cell lines has also been reported as the cause of a discordant NIPT result63.
Failure of NIPT or inconclusive results occur in up to 5% of cases. These are usually caused by a low fetal fraction of cffDNA50, 51, most commonly because of either testing at an early gestation, as the quantity of cffDNA increases with placental mass and hence gestation64, 65, or high maternal body mass index64, 65, in which low fetal fraction has been attributed to high levels of maternal cfDNA derived from adipose tissue. In situations associated with a small placental volume, for example, trisomies 13 and 1866, 67, fetal fraction has been reported to be lower.
NIPT for aneuploidy is now available widely across the globe, albeit only in the private sector at present. The cost of testing remains high but it has already decreased significantly and there is considerable debate as to how it might be implemented into public‐sector healthcare. It is likely that different approaches will be taken depending on local care pathways already in place and the local economy68. However, at the moment, most commentators advocate introduction of NIPT as a contingent test after traditional Down syndrome screening69 and there are at least two national studies ongoing in Europe evaluating different approaches70, 71. Although NIPT for aneuploidy is not yet available outside the context of a research study in public health services, widespread availability in the private sector is having a significant effect on the practice of fetal medicine by decreasing the need for invasive diagnostic testing.
Currently, NIPT can only be used reliably for detection of the major trisomies and sex chromosome abnormalities and, as such, will fail to detect the majority of other chromosomal rearrangements that are the underlying pathology in a significant proportion of structurally abnormal fetuses. However, how long this remains the case is in question as there are already reports of NGS detecting other chromosomal rearrangements. Initial studies reported using very high depths of sequencing72 but, more recently, rearrangements detectable from the karyotype have been detected using sequencing required for standard aneuploidy detection (Figure 4)73, 74, 75. The main drawback with this approach is that the false‐positive rates and limits of detection are as yet unknown, thus limiting its value at present. Despite this, a number of companies have launched commercial tests for a limited range of microdeletion syndromes, including Di George (22q‐), Wolf–Hirschhorn (4p‐), Cri‐du‐Chat (5p‐), Prader–Willi, Angelman and 1p36‐. These tests have largely been developed using artificially produced samples, and reasonable validation data describing sensitivity, specificity and the positive predictive value in maternal plasma samples are yet to be published. Whilst this approach might increase the detection of pathogenic mutations, this targeted approach will only detect around 25% of pathogenic rearrangements as these occur across all chromosomes. There is also concern that using extended NIPT may increase the false‐positive rate, potentially reversing the downward trend seen in invasive testing subsequent to the introduction of NIPT for aneuploidy. If this approach is to be used, it would seem sensible to confine its use to cases in which there is an increased incidence of pathogenic rearrangements, for example in euploid fetuses with multiple ultrasound anomalies76, 77, 78.
Figure 4.
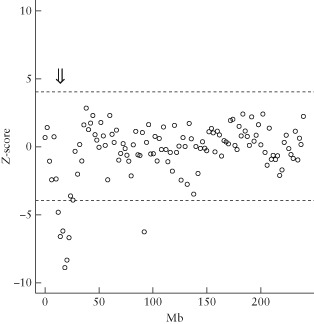
Detection of chromosomal rearrangements in cell‐free fetal DNA (cffDNA) using standard aneuploidy sequencing. A small deletion ( ) of chromosome 2, confirmed as 46,XY,del(2) (p23p25.1), is indicated when the expected number of reads falls outside a Z‐score of ± 4 (
) of chromosome 2, confirmed as 46,XY,del(2) (p23p25.1), is indicated when the expected number of reads falls outside a Z‐score of ± 4 ( ).
).
Conclusions
The use of cffDNA for the diagnosis of fetal genetic and chromosomal conditions is having a profound effect on the practice of fetal medicine worldwide. The advent of NIPT for aneuploidy is reducing the need for invasive testing, the rate of which has declined dramatically in some countries79, a fall that is met with approval from both women and health professionals alike as we move toward safer and earlier prenatal diagnosis80, 81. The use of cffDNA to direct invasive testing or treatment in sex‐linked diseases is also decreasing the need for invasive testing, whilst making prenatal diagnosis safer and more acceptable to high‐risk families82; however, this may increase the economic burden on health services as an increasing number of families elect to undergo NIPT for information83, 84. These changes will inevitably impact on care pathways and fetal medicine in general, in respect to both training and service provision, as the indications for invasive tests decrease. The pace of change has been rapid and we must urgently address how we structure our services so that we can provide safe services for those who continue to need invasive testing and treatment.
REFERENCES
- 1. Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, Wainscoat JS. Presence of fetal DNA in maternal plasma and serum. Lancet 1997. ; 350 : 485–487. [DOI] [PubMed] [Google Scholar]
- 2. Lo YM, Tein MS, Lau TK, Haines CJ, Leung TN, Poon PM, Wainscoat JS, Johnson PJ, Chang AM, Hjelm NM. Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Human Genet 1998. ; 62 : 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alberry M, Maddocks D, Jones M, Abdel Hadi M, Abdel‐Fattah S, Avent N, Soothill PW. Free fetal DNA in maternal plasma in anembryonic pregnancies: confirmation that the origin is the trophoblast. Prenat Diagn 2007. ; 27 : 415–418. [DOI] [PubMed] [Google Scholar]
- 4. Lunn FM, Chiu RW, Allen Chan KC, Yeung Leung T, Kin Lau T, Dennis Lo YM. Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin Chem 2008. ; 54 : 1664–1672. [DOI] [PubMed] [Google Scholar]
- 5. Lo YM, Zhang J, Leung TN, Lau TK, Chang AM, Hjelm NM. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet 1999. ; 64 : 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hyett JA, Gardener G, Stojilkovic‐Mikic T, Finning KM, Martin PG, Rodeck CH, Chitty LS. Reduction in diagnostic and therapeutic interventions by non‐invasive determination of fetal sex in early pregnancy. Prenat Diagn 2005. ; 25 : 1111–1116. [DOI] [PubMed] [Google Scholar]
- 7. Clausen FB. Integration of non‐invasive prenatal prediction of fetal blood group into clinical prenatal care. Prenat Diagn 2014. ; 34 : 409–415. [DOI] [PubMed] [Google Scholar]
- 8. Teitelbaum L, Metcalfe A, Clarke G, Parboosingh JS, Wilson RD, Johnson JM. Costs and benefits of non‐invasive fetal RhD determination. Ultrasound Obstet Gynecol 2015. ; 45 : 84–88. [DOI] [PubMed] [Google Scholar]
- 9. Chitty LS, Griffin DR, Meaney C, Barrett A, Khalil A, Pajkrt E, Cole TJ. New aids for the non‐invasive prenatal diagnosis of achondroplasia: dysmorphic features, charts of fetal size and molecular confirmation using cell‐free fetal DNA in maternal plasma. Ultrasound Obstet Gynecol 2011. ; 37 : 283–289. [DOI] [PubMed] [Google Scholar]
- 10. Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Non‐invasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci U S A 2008. ; 105 : 16266–16271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lo YM, Chiu RW, Lo YMD, Chiu RWK. Non‐invasive prenatal diagnosis of fetal chromosomal aneuploidies by maternal plasma nucleic acid analysis. Clin Chem 2008. ; 54 : 461–466. [DOI] [PubMed] [Google Scholar]
- 12. Boon EM, Faas BH. Benefits and limitations of whole genome versus targeted approaches for non‐invasive prenatal testing for fetal aneuploidies. Prenat Diagn 2013. ; 33 : 563–568. [DOI] [PubMed] [Google Scholar]
- 13. Martin A, Krishna I, Nartina B, Samuel A. Can the quantity of cell‐free fetal DNA predict pre‐eclampsia: a systematic review . Prenat Diagn 2014. ; 34 : 685–691. [DOI] [PubMed] [Google Scholar]
- 14. Rolnik DL, O'Gorman N, Fiolna M, van den Boom D, Nicolaides KH, Poon LC. Maternal plasma cell‐free DNA in the prediction of pre‐eclampsia. Ultrasound Obstet Gynecol 2015. ; 45 : 106–111. [DOI] [PubMed] [Google Scholar]
- 15. Chitty LS, Chatelain P, Wolffenbuttel KP, Aigrain Y. Prenatal management of disorders of sex development. J Pediatr Urol 2012. ; 8 : 576–584. [DOI] [PubMed] [Google Scholar]
- 16. Hill M, Finning K, Martin P, Hogg J, Meaney C, Norbury G, Daniels G, Chitty LS. Non‐invasive prenatal determination of fetal sex: translating research into clinical practice. Clin Genet 2011. ; 80 : 68–75. [DOI] [PubMed] [Google Scholar]
- 17. Tardy‐Guidollet V, Menassa R, Costa JM, David M, Bouvattier‐Morel C, Baumann C,Houang M, Lorenzini F, Philip N, Odent S, Guichet A, Morel Y. New management strategy of pregnancies at risk of congenital adrenal hyperplasia using fetal sex determination in maternal serum: French cohort of 258 cases (2002–2011). J Clin Endocrinol Metab 2014. ; 99 : 1180–1188. [DOI] [PubMed] [Google Scholar]
- 18. Wilcox DT, Chitty LS. Non‐visualisation of the fetal bladder: aetiology and management. Prenat Diagn 2001. ; 21 : 977–983. [PubMed] [Google Scholar]
- 19. Finning KM, Martin PG, Soothill PW, Avent ND. Prediction of fetal D status from maternal plasma: introduction of a new non‐invasive fetal RHD genotyping service. Transfusion 2002. ; 42 : 1079–1085. [DOI] [PubMed] [Google Scholar]
- 20. Finning K, Martin P, Daniels G. A clinical service in the UK to predict fetal Rh (Rhesus) D blood group using free fetal DNA in maternal plasma. Ann N Y Acad Sci 2004. ; 1022 : 119–123. [DOI] [PubMed] [Google Scholar]
- 21. Scheffer PG, van der Schoot CE, Page‐Christiaens GC, de Haas M. Non‐invasive fetal blood group genotyping of rhesus D, c, E and of K in alloimmunised pregnant women: evaluation of a 7‐year clinical experience. BJOG 2011. ; 118 : 1340–1348. [DOI] [PubMed] [Google Scholar]
- 22. Finning K, Martin P, Summers J, Massey E, Poole G, Daniels G. Effect of high throughput RHD typing of fetal DNA in maternal plasma on use of anti‐RhD immunoglobulin in RhD negative pregnant women: prospective feasibility study. BMJ 2008. ; 336 : 816–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Daniels G, Poole J, de Silva M, Callaghan T, MacLennan S, Smith N. The clinical significance of blood group antibodies. Transfus Med 2002. ; 12 : 287–295. [DOI] [PubMed] [Google Scholar]
- 24. Clausen FB, Christiansen M, Steffensen R, Jorgensen S, Nielsen C, Jakobsen MA, Madsen RD, Jensen K, Krog GR, Rieneck K, Sprogoe U, Homburg KM, Grunnet N, Dziegiel MH. Report of the first nationally implemented clinical routine screening for fetal RHD in D− pregnant women to ascertain the requirement for antenatal RhD prophylaxis. Transfusion 2012. ; 52 : 752–758. [DOI] [PubMed] [Google Scholar]
- 25. De Haas M, van der Ploeg CPB, Scheffer PG, Verlinden DA, Hirschberg H, Abbink F. A nationwide fetal RHD screening programme for targeted antenatal and postnatal anti‐D. ISBT Sci Ser 2012. ; 7 : 164–167. [Google Scholar]
- 26. Chitty LS, Finning K, Wade A, Soothill P, Martin B, Oxenford K, Daniels G, Massey E. Diagnostic accuracy of routine antenatal determination of fetal RHD status across gestation: population based cohort study. BMJ 2014. ; 349 : g5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alfirevic Z , Callaghan T. Anti‐RhD prophylaxis for RhD negative pregnant women. BMJ 2014. ; 49 : g5437. [DOI] [PubMed] [Google Scholar]
- 28. Oxenford K, Silcock C, Hill M, Chitty LS. Routine testing of fetal Rhesus D status in Rhesus D negative women using cell free fetal DNA: an investigation into the preferences and information needs of women. Prenat Diagn 2013. ; 33 : 688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daniels G, Finning K, Martin P, Massey E. Non‐invasive prenatal diagnosis of fetal blood group phenotypes: current practice and future prospects. Prenat Diagn 2009. ; 29 : 101–107. [DOI] [PubMed] [Google Scholar]
- 30. Geifman‐Holtzman O, Grotegut CA, Gaughan JP, Gaughan JP, Holtzman EJ, Floro C, Hernandez E. Non‐invasive fetal RhCE genotyping from maternal blood. BJOG 2009. ; 116 : 144–151. [DOI] [PubMed] [Google Scholar]
- 31. Scheffer PG, Ait Soussan A, Verhagen OJ, Gaughan JP, Holtzman EJ, Floro C, Hernandez E. Non‐invasive fetal genotyping of human platelet antigen‐1a. BJOG 2011. ; 118 : 1392–1395. [DOI] [PubMed] [Google Scholar]
- 32. Le Toriellec E, Chenet C, Kaplan C. Safe fetal platelet genotyping: new developments. Transfusion 2013. ; 53 : 1755–1762. [DOI] [PubMed] [Google Scholar]
- 33. Lench N, Barrett A, Fielding S, McKay F, Hill M, Jenkins L, White H, Chitty LS. The clinical implementation of non‐invasive prenatal diagnosis for single gene disorders: challenges and progress made. Prenat Diagn 2013. ; 33 : 555–562. [DOI] [PubMed] [Google Scholar]
- 34. Lun FM, Tsui NB, Chan KC, Leung TY, Lau TK, Charoenkwan P, Chow KC, Lo WY, Wanapirak C, Sanguansermsri T, Cantor CR, Chiu RW, Lo YM. Non‐invasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc Nat Acad Sci U S A 2008. ; 105 : 19920–19925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barrett AN, McDonnell TCR, Allen Chan KC, Chitty LS. Digital PCR analysis of maternal plasma for non‐invasive detection of sickle cell anemia. Clin Chem 2012. ; 58 ; 1026–1032. [DOI] [PubMed] [Google Scholar]
- 36. Nygren AO, Dean J, Jensen TJ, Kruse S, Kwong W, van den Boom D, Ehrich M. Quantification of fetal DNA by use of methylation‐based DNA discrimination. Clin Chem 2010. ; 56 : 1627–1636. [DOI] [PubMed] [Google Scholar]
- 37. Yu SCY, Allen Chan KC, Zheng YWL, Jiang P, Liao GJW, Sun H, Akolekar R, Leung TY, Go ATJI, van Vugt, JMG , Minekawa R, Oudejans CBM, Nicolaides KH, Chiu RWK, Dennis Lo YM. Size‐based molecular diagnostics using plasma DNA for non‐invasive prenatal testing. Proc Natl Acad Sci U S A 2014. ; 111 : 8583–8588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Papasavva T, van Ijcken WF, Kockx CE, van den Hout MC, Kountouris P, Kythreotis L, Kalogirou E, Grosveld FG, Kleanthous M. Next generation sequencing of SNPs for non‐invasive prenatal diagnosis: challenges and feasibility as illustrated by an application to beta‐thalassaemia. Eur J Hum Genet 2013. ; 21 : 1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Twiss P, Hill M, McKay F, Fielding S, Jenkins L, Chitty LS. Non‐invasive prenatal diagnosis for cystic fibrosis: detection of paternal mutations and exploration of patient preferences. Prenat Diagn 2014. ; 34 (Suppl 1): 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chitty LS, Khalil A, Barrett AN, Pajkrt E, Griffin DR, Cole T. Safer, accurate prenatal diagnosis of thanatophoric dysplasia using ultrasound and cell free fetal DNA. Prenat Diagn 2013. ; 33 : 416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Khalil A, Pajkrt E, Chitty LS. Early prenatal diagnosis of skeletal anomalies. Prenat Diagn 2011. ; 31 : 115–124. [DOI] [PubMed] [Google Scholar]
- 42. Tabor A, Alfirevic Z. Update on procedure‐related risks for prenatal diagnosis techniques. Fetal Diagn Ther 2010. ; 27 : 1–7. [DOI] [PubMed] [Google Scholar]
- 43. Akolekar R, Beta J, Picciarelli G, Ogilvie C, D'Antonio F. Procedure‐related risk of miscarriage following amniocentesis and chorionic villus sampling: a systematic review and meta‐analysis. Ultrasound Obstet Gynecol 2015. ; 45 : 16–26. [DOI] [PubMed] [Google Scholar]
- 44. Lo YM, Lun FM, Chan KC, Tsui NB, Chong KC, Lau TK, Leung TY, Zee BC, Cantor CR, Chiu RW. Digital PCR for the molecular detection of fetal chromosomal aneuploidy. Proc Nat Acad Sci U S A 2007. ; 104 : 13116–13121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lo KK, Boustred C, Chitty LS, Plagnol V. RAPIDR: an analysis package for non‐invasive prenatal testing of aneuploidy. Bioinformatics 2014. ; 30 : 2965–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sehnert AJ, Rhees B, Comstock D, de Feo E, Heilek G, Burke J, Rapa RP. Optimal detection of fetal chromosomal abnormalities by massively parallel DNA sequencing of cell‐free fetal DNA from maternal blood. Clin Chem 2011. ; 57 : 1042–1049. [DOI] [PubMed] [Google Scholar]
- 47. Norton ME, Brar H, Weiss J, Karimi A, Laurent LC, Caughey AB Rodriguez MH, Williams J 3rd, Mitchell ME, Adair CD, Lee H, Jacobsson B, Tomlinson MW, Oepkes D, Hollemon D, Sparks AB, Oliphant A, Song K. Non‐Invasive Chromosomal Evaluation (NICE) Study: results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. Am J Obstet Gynecol 2012. ; 207 : 137.e1–8. [DOI] [PubMed] [Google Scholar]
- 48. Norton ME, Jacobsson B, Swamy G, Laurent LC, Ranzini A, Brar H, Tomlinson M, Pereira L, Spitz J, Holleman D, Cuckle H, Musci T, Wapner R. Non‐invasive examination of Trisomy using directed cell‐free DNA analysis: The NEXT study. Prenat Diagn 2014. ; 34 (Suppl 1): e2. [Google Scholar]
- 49. Nicolaides KH, Syngelaki A, Gil M, Atanasova V, Markova D. Validation of targeted sequencing of single‐nucleotide polymorphisms for non‐invasive prenatal detection of aneuploidy of chromosomes 13, 18, 21, X, and Y. Prenat Diagn 2013. ; 33 : 575–579. [DOI] [PubMed] [Google Scholar]
- 50. Gil MM, Akolekar R, Quezada MS, Bregant B, Nicolaides KH. Analysis of cell‐free DNA in maternal blood in screening for aneuploidies: meta‐analysis. Fetal Diagn Ther 2014. ; 35 : 156–173. [DOI] [PubMed] [Google Scholar]
- 51. Gil MM, Quezada MS, Revello R, Akolekar R, Nicolaides KH. Analysis of cell‐free DNA in maternal blood in screening for fetal aneuploidies: updated meta‐analysis. Ultrasound Obstet Gynecol 2015. ; 45 : 249–266. [DOI] [PubMed] [Google Scholar]
- 52.American College of Obstetricians and Gynecologists. Non‐invasive prenatal testing for fetal aneuploidy. Committee Opinion No. 545. Obstet Gynecol 2012. ; 120 : 1532–1534. [DOI] [PubMed] [Google Scholar]
- 53. Salomon LJ, Alfirevic Z, Audibert F, Kagan KO, Yeo G, Raine‐Fenning N; ISUOG Clinical Standards Committee. ISUOG consensus statement on the impact of non‐invasive prenatal testing (NIPT) on prenatal ultrasound practice. Ultrasound Obstet Gynecol 2014. ; 44 : 122–123. [DOI] [PubMed] [Google Scholar]
- 54. Benn P, Borrell A, Cuckle H, Dugoff L, Gross S, Johnson JA, Maymon R, Odibo A, Schielen P, Spencer K, Wright D, Yaron Y. Prenatal detection of Down Syndrome using massively parallel sequencing (MPS): a rapid response statement from a committee on behalf of the Board of the International Society for Prenatal Diagnosis, 24 October 2011. Prenat Diagn 2011. ; 32 : 1–2. [DOI] [PubMed] [Google Scholar]
- 55. Futch T, Spinosa J, Bhatt S, de Feo E, Rava RP, Sehnert AJ. Initial clinical laboratory experience in non‐invasive prenatal testing for fetal aneuploidy from maternal plasma DNA samples. Prenat Diagn 2013. ; 33 : 569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang JC, Sahoo T, Schonberg S, Kopita KA, Ross L, Patek K, Strom CM. Discordant noninvasive prenatal testing and cytogenetic results: a study of 109 consecutive cases. Genet Med 2015. ; 17 : 234–236. [DOI] [PubMed] [Google Scholar]
- 57. Pan M, Li FT, Li Y, Jiang FM, Li DZ, Lau TK, Liao C. Discordant results between fetal karyotyping and non‐invasive prenatal testing by maternal plasma sequencing in a case of uniparental disomy 21 due to trisomic rescue. Prenat Diagn 2013. ; 33 : 598–601. [DOI] [PubMed] [Google Scholar]
- 58. Verweij EJJ, de Boer MA, Oepkes D. Non‐invasive prenatal testing for trisomy 13; more harm than good? Ultrasound Obstet Gynecol 2014. ; 44 : 112–114. [DOI] [PubMed] [Google Scholar]
- 59. Searle CJ, Smith K, Daniels G, Maher EJ, Quarrell O. Cell‐free fetal DNA sex determination identified a maternal SRY gene with a known X chromosome deletion. Prenat Diagn 2013. ; 33 : 612–613. [DOI] [PubMed] [Google Scholar]
- 60. Yao H, Jiang F, Hu H, Gao Y, Zhu Z, Zhang H, Wang Y, Guo Y, Liu L, Yuan Y, Zhou L, Wang J, Du B, Qu N, Zhang R, Dong Y, Xu H, Chen F, Jiang H, Liu Y, Zhang L, Tian Z, Liu Q, Zhang C, Pan X, Yang S, Zhao L, Wang W, Liang Z. Detection of fetal sex chromosome aneuploidy by massively parallel sequencing of maternal plasma DNA: initial experience in a Chinese hospital. Ultrasound Obstet Gynecol 2014. ; 44 : 17–24. [DOI] [PubMed] [Google Scholar]
- 61. Lau TK, Jiang FM, Stevenson RJ, Lo TK, Chan LW, Chan MK, Lo PS, Wang W, Zhang HY, Chen F, Choy KW. Secondary findings from non‐invasive prenatal testing for common fetal aneuploidies by whole genome sequencing as a clinical service. Prenat Diagn 2013. ; 33 : 602–608. [DOI] [PubMed] [Google Scholar]
- 62. Wang Y, Chen Y, Tian F, Zhang J, Song Z, Wu Y, Han X, Hu W, Ma D, Cram D, Cheng W. Maternal mosaicism is a significant contributor to discordant sex chromosomal aneuploidies associated with non‐invasive prenatal testing. Clin Chem 2014. ; 60 : 251–259. [DOI] [PubMed] [Google Scholar]
- 63. Osborne M, Hardisty E, Devers P, Kaiser‐Rogers K, Hayden MA, Goodnight W, Vora NL. Discordant non‐invasive testing results in a patient subsequently diagnosed with metastatic disease. Prenat Diagn 2013. ; 33 : 609–611. [DOI] [PubMed] [Google Scholar]
- 64. Wang E, Batey A, Struble C, Musci T, Song K, Oliphant A. Gestational age and maternal weight effects on fetal cell‐free DNA in maternal plasma. Prenat Diagn 2013. ; 33 : 662–666. [DOI] [PubMed] [Google Scholar]
- 65. Ashoor G, Syngelaki A, Poon LC, Rezende JC, Nicolaides KH. Fetal fraction in maternal plasma cell‐free DNA at 11–13 weeks' gestation: relation to maternal and fetal characteristics. Ultrasound Obstet Gynecol 2013. ; 41 : 26–32. [DOI] [PubMed] [Google Scholar]
- 66. Wegrzyn P, Faro C, Falcon O, Peralta CF, Nicolaides KH. Placental volume measured by three‐dimensional ultrasound at 11 to 13 + 6 weeks of gestation: relation to chromosomal defects. Ultrasound Obstet Gynecol 2005. ; 26 : 28–32. [DOI] [PubMed] [Google Scholar]
- 67. Rava RP, Srinivasan A, Sehnert AJ, Bianchi DW. Circulating fetal cell‐free DNA fractions differ in autosomal aneuploidies and monosomy X. Clin Chem 2014. ; 60 : 243–250. [DOI] [PubMed] [Google Scholar]
- 68. Chitty LS, Hill M, White H, Wright D, Morris S. Non‐invasive prenatal testing for aneuploidy – ready for prime time? Am J Obstet Gynecol 2012. ; 206 : 269–275. [DOI] [PubMed] [Google Scholar]
- 69. Benn P, Cuckle H, Pergament E. Non‐invasive prenatal testing for aneuploidy: current status and future prospects. Ultrasound Obstet Gynecol 2013. ; 42 : 15–33. (http://www.ncbi.nlm.nih.gov/pubmed/?term=cuckle+h+and+NIPT) [DOI] [PubMed] [Google Scholar]
- 70.Hill M, Wright D, Daley R, Lewis C, McKay F, Mason S, Lench N, Howarth A, Boustred C, Lo K, Plagnol V, Spencer K, Fisher J, Kroese M, Morris S, Chitty LS . BMC Pregnancy Childbirth 2014. ; 14 : 229 (http://www.ncbi.nlm.nih.gov/pubmed/25027965) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Verweij EJ, Jacobsson B, van Scheltema PA, de Boer MA, Hoffer MJ, Hollemon D, Westgren M, Song K, Oepkes D . European non‐invasive trisomy evaluation (EU‐NITE) study: a multicenter prospective cohort study for non‐invasive fetal trisomy 21 testing. Prenat Diagn 2013. ; 33 : 996–1001. [DOI] [PubMed] [Google Scholar]
- 72. Srinivasan A, Bianchi DW, Huang H, Sehnert AJ, Rava RP. Non‐invasive detection of fetal subchromosome abnormalities via deep sequencing of maternal plasma. Am J Hum Genet 2013. ; 92 : 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen S, Lau TK, Zhang C, Xu C, Xu Z, Hu P, Xu J, Huang H, Pan L, Jiang F, Chen F, Pan X, Xie W, Liu P, Li X, Zhang L, Li S, Li Y, Xu X, Wang W, Wang J, Jiang H, Zhang X. A method for non‐invasive detection of fetal large deletions/duplications by low coverage massively parallel sequencing. Prenat Diagn 2013. ; 33 : 584–590. [DOI] [PubMed] [Google Scholar]
- 74. Lau TK, Cheung SW, Lo PS, Pursley AN, Chan MK, Jiang F, Zhang H, Wang W, Jong LF, Yuen OK, Chan HY, Chan WS, Choy KW. Non‐invasive prenatal testing for fetal chromosomal abnormalities by low‐coverage whole‐genome sequencing of maternal plasma DNA: review of 1982 consecutive cases in a single center. Ultrasound Obstet Gynecol 2014. ; 43 : 254–264. [DOI] [PubMed] [Google Scholar]
- 75. Lo K, Boustred C, McKay F, Fielding S, Plagnol V, Chitty L. Detection of “sub‐chromosomal” pathogenic changes by sequencing cfDNA in maternal plasma: feasibility and implementation strategies. Prenat Diagn 2014. ; 34 (Suppl 1): 10. [Google Scholar]
- 76. Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, Savage M, Platt LD, Saltzman D, Grobman WA, Klugman S, Scholl T, Simpson JL, McCall K, Aggarwal VS, Bunke B, Nahum O, Patel A, Lamb AN, Thom EA, Beaudet AL, Ledbetter DH, Shaffer LG, Jackson L. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012. ; 367 : 2175–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Callaway JL, Shaffer LG, Chitty LS , Rosenfeld JA, Crolla JA . The clinical utility of microarray technologies applied to prenatal cytogenetics in the presence of a normal conventional karyotype: a review of the literature. Prenat Diagn 2013. ; 33 : 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. de Wit MC, Srebniak MI, Govaerts LC, Van Opstal D, Galjaard RJ, Go AT. Additional value of prenatal genomic array testing in fetuses with isolated structural ultrasound abnormalities and a normal karyotype: a systematic review of the literature. Ultrasound Obstet Gynecol 2014. ; 43 : 139–146. [DOI] [PubMed] [Google Scholar]
- 79. Ferres MA, Lichten L, Sachs A, Lau K, Bianchi D. Rate of diagnostic procedures for aneuploidy in the post non‐invasive DNA testing (NIDT) era. Prenat Diagn 2014. ; 34 (Suppl 1): 53. [Google Scholar]
- 80. Lewis C, Hill M, Silcock C, Daley R, Chitty L. Non‐invasive prenatal testing for trisomy 21: a cross‐sectional survey of service users' views and likely uptake. BJOG 2014. ; 121 : 582–594. [DOI] [PubMed] [Google Scholar]
- 81. Allyse M, Sayres LC, Goodspeed TA, Cho MK. Attitudes towards non‐invasive prenatal testing for aneuploidy among US adults of reproductive age. J Perinatol 2014. ; 34 : 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lewis C, Hill M, Skirton H, Chitty LS. Non‐invasive prenatal diagnosis for fetal sex determination: benefits and disadvantages from the service users' perspective. Eur J Hum Genet 2012. ; 20 : 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lewis C, Hill M, Chitty LS. Non‐invasive prenatal diagnosis for single gene disorders: experience of patients. Clin Genet 2014. ; 85 : 336–342. [DOI] [PubMed] [Google Scholar]
- 84. Oepkes D, Yaron Y, Kozlowski P, de Sousa MJ Rego, Bartha JL, van den Akker ES, Dornan SM, Krampl‐Bettelheim E, Schmid M, Wielgos M, Cirigliano V, Di Renzo GC, Cameron A, Calda P, Tabor A. Counseling for non‐invasive prenatal testing (NIPT): what pregnant women may want to know. Ultrasound Obstet Gynecol 2014. ; 44 : 1–5. [DOI] [PubMed] [Google Scholar]