

Abstract
Organisms exhibit an incredible diversity of life history strategies as adaptive responses to environmental variation. The establishment of novel life history strategies involves multilocus polymorphisms, which will be challenging to establish in the face of gene flow and recombination. Theory predicts that adaptive allelic combinations may be maintained and spread if they occur in genomic regions of reduced recombination, such as chromosomal inversion polymorphisms, yet empirical support for this prediction is lacking. Here, we use genomic data to investigate the evolution of divergent adaptive ecotypes of the yellow monkey flower Mimulus guttatus. We show that a large chromosomal inversion polymorphism is the major region of divergence between geographically widespread annual and perennial ecotypes. In contrast, ∼40,000 single nucleotide polymorphisms in collinear regions of the genome show no signal of life history, revealing genomic patterns of diversity have been shaped by localized homogenizing gene flow and large‐scale Pleistocene range expansion. Our results provide evidence for an inversion capturing and protecting loci involved in local adaptation, while also explaining how adaptive divergence can occur with gene flow.
Keywords: Adaptation, chromosome inversion, Mimulus, phylogeography, population genomics
Life history theory predicts that natural selection operating within a given environment shapes patterns of reproduction, growth, and senescence (Roff 2002). These suites of life history traits involve multitrait phenotypes, which are likely determined by numerous genes throughout the genome. To explain how polymorphic life‐history strategies are established and maintained within a species is challenging, as adaptive allelic combinations are constantly broken down by recombination and gene flow (Slatkin 1987). An explanation relies on understanding the balance between divergent natural selection and homogenizing gene flow (Lenormand 2002), as well as the genetic architecture of adaptive divergence. These are central factors influencing the origin and maintenance of diverging taxa.
A feature that can maintain polymorphism in complex traits in the face of gene flow is if the genetic targets of selection occur in chromosomal regions with reduced recombination (Feder et al. 2012). In particular, chromosomal rearrangements may play a key role in the process of local adaptation, in addition to their role in speciation (Dobzhansky 1970; White 1978). Recent theoretical models show that chromosomal inversion polymorphisms can play a critical role in local adaptation when they capture two or more locally adapted alleles (Kirkpatrick and Barton 2006). This is because the inversion suppresses meiotic recombination in heterozygous individuals, and can prevent mixtures of adapted and maladapted alleles. The local adaptation mechanism for inversion spread has received support from studies in malaria mosquitoes (Anopheles; Coluzzi et al. 2002) and fruit flies (Drosophila; Krimbas and Powell 1992; Umina et al. 2005), where inversions are clinal and track environmental gradients. Examples from mimetic butterflies (Joron et al. 2011) suggest that the local adaptation hypothesis has great potential to explain the abundance of inversions both within and between species (Hoffmann and Rieseberg 2008), although empirical support for inversions playing a major role in adaptive divergence is limited.
The recent discovery of a widespread chromosomal inversion that differs between two adaptive life history ecotypes of the yellow monkey flower, Mimulus guttatus, provides an opportunity to investigate how an inversion polymorphism contributes to adaptive divergence (Lowry and Willis 2010). The paracentric inversion contains hundreds of genes, and is a major quantitative trait locus (QTL) (20–30% variation explained) for flowering time and growth‐related traits (Hall et al. 2006; Lowry and Willis 2010). Reciprocal transplant experiments have demonstrated strong local adaptation in traits that map to the inversion and differ between the annual and perennial ecotypes (Lowry et al. 2008a; Hall et al. 2010; Lowry et al. 2008b). Annual populations of M. guttatus grow in habitats that experience summer drought, and face strong selection to flower rapidly prior to desiccation. In contrast, perennial populations grow in permanently moist sites, and invest heavily in vegetative growth, before flowering later in the season. The phenotypic differences between ecotypes (e.g., time to flowering and senescence, potential for clonal spread, and flower size; Fig. 1A) represent exceptional diversity within a species.
Figure 1.
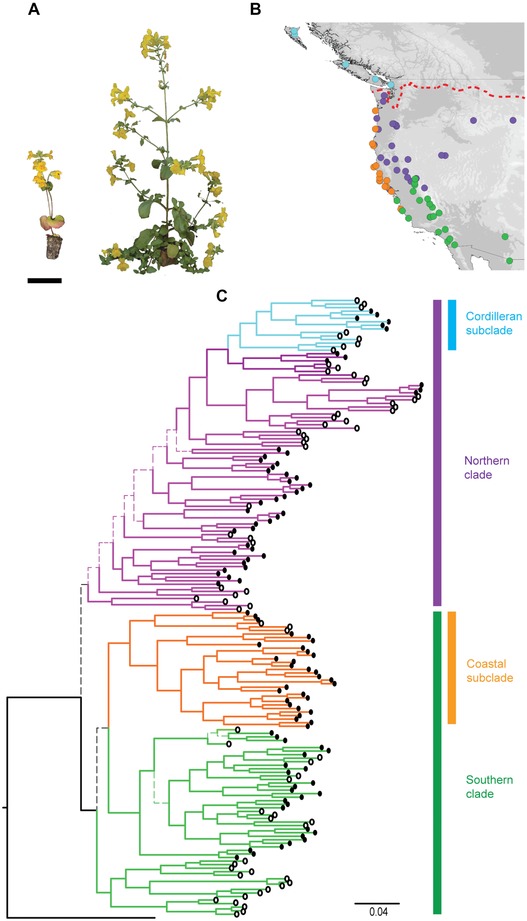
Phylogenetic relationships between annual and perennial populations of Mimulus guttatus. (A) Representative phenotypes of annual (population LMC, left) and perennial (HUM, right) ecotypes grown in a common glasshouse. Perennials are typically larger, produce stoloniferous branches, and flower later than annuals. Both photographed populations originate from California. Scale bar is 5 cm. (B) Geographic map of genotyped samples. Populations are colored according to the phylogenetic groupings in panel c. The red dashed line represents the southern extent of the Cordilleran Ice Sheet. (C) Bayesian majority rule consensus tree of 15,277 SNP loci genotyped in 174 individuals of M. guttatus. The tree is rooted with M. moniliformis as an outgroup. Branches are colored to represent major clades, with green being southern, orange coastal, purple northern, and light blue Cordilleran. The circles at tips indicate life history, with annuals as open circles, and perennials as filled circles. All nodes are supported with Bayesian posterior probabilities greater than 0.9, except those shown with thin dashed branches.
Surprisingly, given their morphological differences, the annual and perennial ecotypes of M. guttatus have overlapping distributions throughout most of their native range in Western North America. Although there is some disagreement about their taxonomic status (Nesom 2012), the two types are fully interfertile. Furthermore, studies based on microsatellite loci outside of the inversion suggest that there is no clear genetic differentiation between the annual and perennial forms, whereas inversion loci show a contrasting genetic structure associated with environmental variables (Oneal et al. 2014). Overall, the genetic relationships between annual and perennial ecotypes in close proximity, and from across their geographic distribution, are still unclear.
Here, we use population genomic approaches to sequence range‐wide populations of M. guttatus, to explain whether an inversion polymorphism contributes to adaptive divergence. Our first aim is to clarify the evolutionary relationships between annual and perennial populations of M. guttatus, as studies with fewer markers have provided limited insight into intraspecific relations in this highly diverse group (Sweigart and Willis 2003; Oneal et al. 2014). Specifically, we test whether genome‐wide SNPs reveal an annual–perennial separation, or instead phylogeographic structure reflecting the complex Pleistocene history of North America (Soltis et al. 1997; Brunsfeld et al. 2001). In light of this genome‐wide survey of genetic diversity, our second aim is to identify whether the large chromosomal inversion polymorphism, or other genomic regions, is associated with phenotypic divergence of ecotypes in M. guttatus. Finally, we seek to provide a first insight into the history of the inversion, including which orientation is derived, whether the inversion is of recent origin, and the pattern of inversion spread. Together this enables us to explain the history and maintenance of coexisting life history ecotypes across a widespread species’ native range.
Materials and Methods
POPULATION SAMPLING, SEQUENCING, AND DATA FILTERING
We genotyped representative populations from throughout the native range of M. guttatus DC. (Phrymaceae), from northern Mexico to British Columbia (Table S1). We used a total of 174 individuals from 69 populations for genotyping, as well as an individual of M. moniliformis (Mimulus section Mimulosa), which was used as an outgroup. We classified M. guttatus populations as annual or perennial in the field, and then confirmed this by phenotyping individuals in a common greenhouse environment for traits that differ between ecotypes (number of stolons, days to first flower, stem width, internode length, flower size; Friedman et al. 2015).
We extracted total genomic DNA from silica dried leaves or flash‐frozen flower buds from live plants, using the DNeasy Plant Kit (Qiagen, Germantown, MD). We used a genotyping‐by‐sequencing (GBS) approach to generate genome‐wide SNP data (Elshire et al. 2011). GBS test libraries using enzymes PstI, ApeKI, and EcoT22 showed that PstI was the most suitable for phylogeography, as it produces a few thousand cut sites with high sequence coverage, without cutting in repetitive regions of the genome. We created sequencing libraries by digesting individual Mimulus samples, technical replicates, and negative controls (blank samples) with PstI, tagging samples with a unique barcoded adapter, performing polymerase chain reaction, and pooling and diluting samples in a 96‐plex according to the protocol of Elshire et al. (2011). We sequenced multiplexed libraries on the Illumina HiSeq 2500 at the Biotechnology Resource Center, Cornell, and the University of Rochester Medical Center. A total of 531 million 100 bp single‐end reads were generated across the two libraries.
We used TASSEL‐GBS (Glaubitz et al. 2014) to de‐multiplex, remove barcodes, perform quality filtering, and call SNPs, following recommended settings for population studies of outcrossing species. We aligned the GBS tags to the M. guttatus genome version 2.0_256 (phytozome.net) using the default settings of BWA (Li and Durbin 2009). GBS data usually undercall heterozygous sites due to the relatively low sequencing coverage. Therefore, we prepared two SNP matrices, one where heterozygous sites were allowed for biallelic SNPs, and one where only homozygous SNP calls were allowed. As population genetic analyses gave identical results for both SNP matrices (results not shown), we present the results that include the heterozygous calls.
GENOME‐WIDE PATTERNS OF GENETIC STRUCTURE
Phylogenetic analyses were performed on the 15,277 SNP loci present in at least half the individuals we sequenced. Genome‐wide SNPs were analyzed using phylogenetic networks, which do not assume a tree‐like pattern between populations connected by gene flow, and using Bayesian phylogenetic inferences. We used SplitsTree4 (Huson and Bryant 2006) to perform phylogenetic network analyses, with default parameters. We used TASSEL to make neighbor‐joining trees.
We then constructed Bayesian phylogenies using MrBayes MPI version 3.2.1 (Ronquist et al. 2012) with BEAGLE version 1.0 (Ayres et al. 2012). Analyses used the Mkv model (Lewis 2001), and the gamma prior. A starting tree generated from short MrBayes runs (with three randomly introduced perturbations) was used as an input, and the mean branch length prior set to 0.01 (Thomas et al. 2012). Analyses consisted of two replicates of six chains, with 5,000,000 iterations, with the first 25% discarded as burn‐in. We confirmed convergence by inspecting trace plots and marginal probability distributions in TRACER (part of the BEAST package; Drummond and Rambaut 2007), and confirming the standard deviation of splits was <0.05. We used the CIPRES cluster (www.phylo.org) using 12 CPUs to perform phylogenetic analyses, and used FigTree version 1.4 to visualize the output trees.
We performed STRUCTURE analyses on 1400 well‐spaced SNPs (100 per linkage group, mean distance 201 Kb between loci) present across 75% of individuals. We filtered by missing data and distances between loci in PLINK (Purcell et al. 2007). This filtered SNP dataset removes adjacent SNPs that may be in linkage disequilibrium, accounts for sequencing heterogeneity by equally representing sequences on each major linkage group, and decreases the analysis time. The filtered data gave quantitatively similar results to a linkage disequilibrium filtered matrix of 10,000 SNPs (results not shown). Broad patterns of genetic clustering (K = 1–20) were first explored in fastSTRUCTURE (Raj et al. 2013), with 10 replicates of 1,000,000 iterations for each K‐value. The chooseK.py script determined K = 2 to be the model that best explained variation in the data. We then ran analyses in STRUCTURE (Pritchard et al. 2000), accounting for linkage between loci (Falush et al. 2003), for K‐values 1–5. We used ten replicate runs for each K‐value, each with 500,000 generations, with the first 250,000 generations discarded as burn‐in. The optimal number of clusters was confirmed as K = 2 using the ad hoc statistic ΔK (Evanno et al. 2005). We performed analyses using the parallel version of STRUCTURE on the iPlant Discovery Environment server. We combined multiple STRUCTURE runs in CLUMPP (Jakobsson and Rosenberg 2007).
We used analysis of molecular variance (AMOVA) to compare the portion of genome‐wide genetic diversity partitioned by life history or geography. Analyses were performed in Arlequin version 3.50 (Excoffier and Lischer 2010), using loci with SNP calls in every population. Analyses by geography tested the proportion of variance explained (VE) by two equal sized groups, north and south, based on the latitude of the collection site. We also examined the relationship between geography and life history using principal component analysis (PCA). PCA was performed in TASSEL version 4.0 (Bradbury et al. 2007), using the “collapse minor allele” function, and the correlation matrix setting. Plots were made of the first two principal components, and visually inspected for clusters corresponding to geographic groupings or life history ecotypes.
GENOME SCANS AND POPULATION GENOMICS OF INVERSION LOCI
We expect that if life history ecotypes have a common genetic basis, candidate regions underlying adaptive divergence would cluster by ecotype, rather than by geography. We looked for candidates by scanning the genome for regions enriched for allele frequency differences (FCT outliers), and regions of high nucleotide diversity (π), between annuals and perennials. The FCT scan for allele frequency differences allows us to identify regions of putative divergent selection (Gompert and Buerkle 2011; Bourret et al. 2013), while accounting for population‐level variation (FST). The analysis used the 134 individuals with less than 25% missing data. The FCT scan was performed as a locus‐by‐locus AMOVA in Arlequin, with significant outliers detected using a hierarchical island model with 10,000 simulations. We calculated regions of high nucleotide diversity within annuals (πA) and within perennials (πP) using a sliding window analysis (30‐SNP window, moving 10 SNPs per iteration) in DnaSP version 5.0 (Librado and Rozas 2009). We also performed a sliding‐window analysis of nucleotide divergence between ecotypes (DXY), as above. Nucleotide diversity (πA and πP) and divergence (DXY) were calculated as the number of fixed SNPs out of the total number of SNPs (πSNP). We plotted both FCT outliers and π against the nucleotide position of the M. guttatus version 2.0 reference genome.
The linkage group 8 chromosomal inversion was identified as a major outlier region, and given that it has previously been recognized as a major adaptive QTL (Hall et al. 2006; Lowry and Willis 2010), we further investigated patterns of genetic diversity within the inversion. There is currently some uncertainty as to the size of the LG8 inversion (Holeski et al. 2014), and the exact position of the inversion breakpoints is unknown, therefore we performed analyses on two different assemblies of the inversion. First, we extracted loci from the genome‐wide data that correspond to the inversion in the most recent M. guttatus genome build (inversion size ∼6.5 Mb, positions 1.1–7.6 Mb; phytozome.net). This assembly contains six sequence scaffolds (scaffolds 11, 59, 76, 233, 604, and 1093). We repeated all analyses on the M. guttatus version 1.0 genome assembly, where the inversion was considered smaller (∼2.2 Mb), containing only scaffold 11. Results from both SNP datasets gave highly consistent results (data not shown), therefore results are presented for the M. guttatus version 2.0 genome aligned SNPs.
We used PLINK to filter SNP loci within the inversion for linkage disequilibrium (20 kb sliding window, move five SNPs per window, remove pairwise SNPs R 2 < 0.2), and then analyzed the SNP data in STRUCTURE using K = 1–5. We conducted AMOVAs in Arlequin to test the partitioning of genetic variance by life history and geographic groups. Settings for these analyses were as before.
Results
GENOME‐WIDE PATTERNS OF GENETIC STRUCTURE
Our next‐generation sequencing approach generated a total of 531 million raw sequence reads, of which 94.1% mapped to the reference M. guttatus genome, giving a mean read count of 2.4 million reads (±95,000) per individual. In total, 38,872 genome‐wide SNPs were called across the 14 linkage groups, in 69 populations representative of the species’ native range (Fig. 1B, Table S1).
The genome‐wide phylogeny clearly resolves the relationships between populations of M. guttatus (Fig. 1C, Fig. S1). The phylogeny shows annual and perennial populations do not form reciprocally monophyletic clades, instead the two ecotypes are scattered across the phylogenetic tree. Support for the relationships between populations is generally high (mean posterior probability = 0.986 ± 0.003), with individuals from a given population always clustering together. A similar pattern is observed in neighbor‐joining and SplitsTree networks (Fig. S1), where annuals and perennials are intermixed. This pattern is also evident in PCAs (Fig. S2), and STRUCTURE analyses with the optimum K‐value (K = 2, Fig. 2A), which show the lack of association by life history. The only group in the phylogeny that comprises a single life history strategy is that of coastal perennial plants (colored orange, Fig. 1), which represent a distinct genotypic and phenotypic cluster within M. guttatus (Lowry et al. 2008b; Fig. S2).
Figure 2.
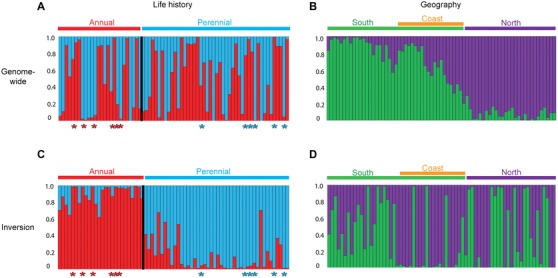
Contrasting patterns of genetic structure across 69 populations of Mimulus guttatus at genome‐wide and inversion loci. Bayesian STRUCTURE plots for each population are shown for the optimal number of genetic clusters (K = 2), with ancestry proportion (Q) on the y‐axis. (A) Plot for 1400 genome‐wide loci (100 per linkage group) grouped by life history, with the annual cluster in red and the perennial cluster in blue. (B) Plot for genome‐wide loci ordered by latitude, with the southern‐most on the left and the most northerly on the right. (C) Plot for 276 LD‐filtered loci within the inversion, ordered by life history. (D) Plot for inversion loci ordered by latitude. Colored asterisks indicate those individuals known to be of annual (red) or perennial (blue) orientation from previous experimental crosses (Lowry and Willis 2010).
Rather than divergence related to life history, our genomic data show fine‐scale geographic clustering, and reveal previously unresolved broad‐scale genetic structure. We resolve two major genetic groups, reflecting north‐south divergence across the Pacific Northwest. The southern clade is restricted to the south of the Shasta Cascades in the Sierra Nevada, but has spread more widely to the west of the Sierras, to include much of the California and Oregon coastline (Fig 1B). This coastal group forms a discrete genetic cluster in STRUCTURE with K = 3 (Fig. S2). The northern clade has similarly strong geographic substructuring, with a ladderized topology showing sequential northwards colonization (Fig. S3). A single group derived from within the northern clade includes populations inhabiting the area formerly covered by the Cordilleran Ice Sheet. Included within the northern clade is the annual population from Iron Mountain, which is used as the M. guttatus reference genome (see Hellsten et al. 2013; www.mimulusevolution.org). The topology of the phylogeny, as well as the strong correlation between latitude and Bayesian clustering scores (Fig. 2B, Fig. S4, R 2 = 0.61, P < 0.001), and latitude and principal component 1 (PC1, R 2 = 0.7, P < 0.001), support a pattern of northwards migration of M. guttatus as a result of postglacial range expansion.
The population genomic analyses show that the northern and southern groups are not discrete genetic clusters, and instead they blend into one another where they overlap in their geographic distributions. This can be observed in STRUCTURE results at K = 2, and higher K‐values (Fig. S2), where geographically intermediate populations show mixed ancestry. The nodes in the phylogeny with reduced support correspond to clades containing these admixed individuals that are least genetically distinct (Fig. S2). Incomplete lineage sorting and rapid range expansion may explain some of this pattern, with evidence from the star‐like topology of short branches in the SplitsTree network (Fig. S1). Intermediate STRUCTURE Q scores in derived populations away from the main area of geographic overlap (e.g., population OSW near the state border between Oregon and Washington) suggest that mixed ancestry may also reflect recent admixture between groups.
GENOME SCANS AND POPULATION GENOMICS OF INVERSION LOCI
We looked for candidate regions underlying annual–perennial divergence by scanning the genome for outlier loci between ecotypes. We found exceptional allelic differences between ecotypes within the linkage group 8 inversion (maximum FCT = 0.751, P < 0.001, Fig. 3A). Forty percent (12/30) of all FCT outliers colocalize with the inversion, even though it represents just 3.5% of the genome‐wide SNPs. Other outlier loci are spread across eight of the other 13 linkage groups. Bayesian clustering analyses performed with the 276 SNPs within the inversion revealed the data were optimally partitioned in to two genetic clusters, corresponding to the two life history categories based on morphology. Moreover, populations of known inversion orientation from previous crossing experiments (Lowry and Willis 2010) were placed in the correct genetic cluster based on the inversion SNPs (Fig. 2C). Of the 69 populations sampled, 67 are assigned by STRUCTURE as wholly comprised of either annuals or perennials based on inversion loci, whereas two populations are mixed. In the mixed populations, annual–perennial phenotypic differences co‐segregate with the inferred genetic assignment from STRUCTURE. AMOVA corroborate that inversion loci follow a clear annual–perennial separation (11.7% VE, P < 0.001, Table S2), in conflict with the genome‐wide data that is more strongly partitioned by geography (8.4% VE, P < 0.001) than by life history (1.3% VE, P < 0.01).
Figure 3.
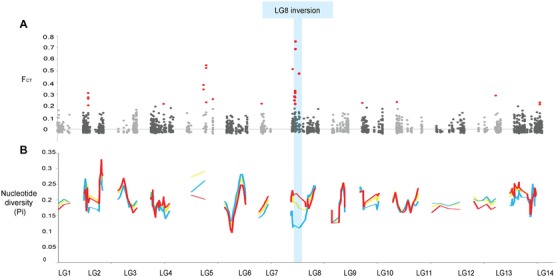
Genomic patterns of divergence across annual and perennial ecotypes of Mimulus guttatus. (A) Genome scan for allele frequency differences between ecotypes (FCT outlier scan). The x‐axis represents physical position relative to the M. guttatus reference genome. Significant outliers with FCT greater than 0.2 are highlighted in red. The linkage group 8 inversion is shaded in blue. (B) Sliding window analysis of genetic diversity (π) within and between ecotypes. Pairwise nucleotide divergence between ecotypes (DXY) is shown in yellow, nucleotide diversity in annuals (πA) in red, and in perennials (πP) in blue.
Although the inversion loci most strongly cluster by life history, geographic clustering can be observed in STRUCTURE analyses at higher K‐values (K = 3–5). These plots show a signal of geography within the inversion in both annual and perennial ecotypes, which is similar to the geographic clusters from the genome‐wide data (Fig. S5). Broadly, annuals can be separated in to north and south genetic clusters, whereas perennials comprise north, south, and coastal genetic clusters. To further understand the origin and history of the inversion spread, we investigated patterns of nucleotide diversity within the inversion for each ecotype, and divergence between ecotypes at inversion loci. We observed that the level of divergence between ecotypes (DXY = 0.311 ± 0.001; Fig. 3B) was similar to pairwise nucleotide diversity in the annuals (πA = 0.306 ± 0.001); whereas diversity was lower in the perennials (πP = 0.270 ± 0.001). Together the nucleotide diversity results and the STRUCTURE patterns can be used to distinguish between a scenario of recent inversion spread, and an ancient origin of the inversion (see below).
Discussion
Our results provide important insights into the evolution of adaptive life history ecotypes in a widespread species, M. guttatus. Across all populations, loci within a large chromosomal inversion are divergent between annuals and perennials. This pattern of life history divergence at inversion loci contrasts with the strong pattern of geographic structure at genome‐wide loci, suggesting the inversion is maintaining adaptive divergence despite populations experiencing strong homogenizing gene flow. Overall, these genomic data implicate a key role for a polymorphic chromosomal inversion protecting multiple adaptive loci from recombination, and thus facilitating adaptive divergence across the species’ range.
PHYLOGEOGRAPHIC PATTERNS IN WIDELY CO‐OCCURRING LIFE HISTORY ECOTYPES
Here, we use genomic data to generate the first well‐resolved intraspecific phylogeny of the widely studied plant species M. guttatus. Consistent with previous studies using microsatellite markers (Oneal et al. 2014) or genome resequencing of a few inbred lines (Brandvain et al. 2014; Puzey and Vallejo‐Marín 2014), this phylogeny reveals neighboring populations are always most closely genetically related, regardless of life history. As such, there are numerous examples of closely related annual and perennial populations that exhibit strongly contrasting morphologies. The lack of phylogenetic signal for life history demonstrates that reproductive isolation between ecotypes, due to flowering time differences and immigrant inviability (Lowry et al. 2008a, Lowry and Willis 2010), is not strong enough to prevent homogenizing gene flow between neighboring populations. A detectable signature of admixture between taxa with strong reproductive isolating barriers has also been shown in a number of other recent studies, such as between annual sunflowers species (Sambatti et al. 2012), and in Capsella (Brassicaceae), where introgression occurs between species with different ploidy levels (Slotte et al. 2008).
Instead of clustering by life history, genome‐wide SNPs show strong phylogeographic structure. The genetic composition of populations varies latitudinally, evident in the phylogeny, and as a significant association between latitude and STRUCTURE assignment values and latitude and PC1 scores. This contrasts with a previous microsatellite study by Oneal et al. (2014), where no detectable latitudinal gradient was observed across M. guttatus populations. Overall, our genome‐wide data imply that patterns of genetic variation are strongly shaped by the underlying geography of Western North America. We resolve two major geographic clades, with phylogeographic structure corresponding to the complex Pleistocene glacial history of the region (Soltis et al. 1997; Brunsfeld et al. 2001; Shafer et al. 2010). The topology of the phylogeny provides direct support for the Leading Edge hypothesis of postglacial expansion (Soltis et al. 1997), where organisms with widespread distributions across the Pacific states have migrated northwards from southern refugia as Pleistocene ice sheets retreated. We can refute the alternative North‐South recolonization hypothesis, where migration occurs from additional refugia in the surrounding unglaciated islands (Shafer et al. 2010), as populations from Vancouver and Graham Islands are in a derived position in the phylogenetic tree. Instead, there are likely multiple southern refugia, including south of the Klamath‐Siskiyou Mountains. This pattern of northwards colonization exclusively from southern refugia has been found in diverse organisms such as mule deer (Latch et al. 2009) and salamanders (Steele and Storfer 2007), and suggests shared responses to Pleistocene climatic cycles among distinct taxa.
THE ROLE OF A LARGE CHROMOSOMAL INVERSION IN MAINTAINING ECOTYPIC DIFFERENCES
This population genomic analyses reveal a stark contrast between the genome‐wide profile that reflects geography, and loci under divergent selection in the inversion that cluster by life history. The FST‐outlier analysis reveals that loci within the linkage group 8 inversion represent a genomic region with major allele frequency differences between life history ecotypes. The best explanation for the divergence of inversion loci between ecotypes of M. guttatus, in conjunction with the paucity of other genomic regions showing allele frequency differences, is that loci within the inversion are responsible for range‐wide adaptive divergence across populations of M. guttatus. An adaptive role for the inversion is supported by QTL for numerous traits that colocalize with the inversion, in replicate mapping populations (Hall et al. 2006; Hall et al. 2010; Lowry and Willis 2010; Friedman and Willis 2013; Friedman et al. 2015). Moreover, reciprocal transplant studies using near isogenic lines for the inversion show strong selection for inversion loci in the environment of each ecotype (Lowry and Willis 2010). An adaptive role for the inversion is also supported by Oneal et al. (2014), who found a strong association between inversion loci and environmental variation.
The finding of an adaptive role for an inversion in Mimulus supports the local adaptation hypothesis for inversion spread (Kirkpatrick and Barton 2006). In this model, inversions capture and protect sets of genes involved in local adaptation from recombination, and thus contribute to adaptive divergence. An alternative adaptive hypothesis for inversion spread is that the position of the breakpoints confers an advantage, rather than the alleles within the inversion (Guerrero et al. 2012). Under this model, the breakpoints directly change reading frames or expression patterns (Puig et al. 2004). Teasing apart the molecular mechanism by which the inversion confers an adaptive benefit will require additional work (described below). An adaptive role for inversions contrasts with much of the early literature on the evolutionary importance of chromosomal inversions, which emphasized their direct effect on hybrid sterility, caused by chromosomal heterozygotes producing unbalanced gametes (Dobzhansky 1970; White 1978). As such, inversions were predominantly considered an agent contributing to reproductive isolation and speciation. These effects are very different to what is observed with the LG8 inversion in M. guttatus. Not only are there no reports of an effect on hybrid sterility (Lowry and Willis 2010), it does not prevent homogenizing gene flow between geographically proximal ecotypes at collinear regions of the genome.
Overall, ecotypes of M. guttatus do not appear to be far along the speciation continuum, despite considerable morphological and ecological divergence. This is particularly interesting because the genomic analyses suggest that there has been considerable time for divergence to have occurred since the origin of the inversion. The strongest evidence for an old origin for the inversion is that STRUCTURE plots reveal the signature of geographic subdivision within the inversion loci for both ecotypes, and this mirrors the pattern observed across the rest of the genome. Further evidence comes from the presence of substantial nucleotide diversity in both inversion orientations (one of which must be derived), supporting the inversion being old enough to have accumulated a large number of mutations. As such, it seems very unlikely that the inversion is of recent origin, sweeping through existing populations. Instead it seems more plausible that the inversion is old, potentially arising early in the origin of the ecotypes, and has subsequently experienced much the same demographic history as the collinear regions of the genome. To explicitly test the age of the inversion will require a greater number of inversion SNPs, which could be used to construct a well‐resolved dated phylogeny, or used for other methods for estimating inversion age based on nucleotide diversity (e.g., Fang et al. 2012).
Our phylogeny is not able to resolve the overall direction of life history evolution in M. guttatus—we cannot tell whether the annual or perennial form is derived or ancestral. However, our genomic analyses do allow us to infer the likely origin of the inversion. The observed reduction in pairwise nucleotide diversity at inversion loci in perennials, relative to annuals and that observed between ecotypes, suggests that the perennial orientation is derived from the annual orientation, and only captured a subset of genetic diversity. An alternative to this scenario is that recent selective sweeps within the perennials have caused the reduction in genetic diversity at inversion loci (Cruickshank and Hahn 2014). However, this would require multiple selective sweeps that follow the geographic structure observed across the rest of the genome. Further evidence comes from recent comparative linkage mapping in two species from different sections of the Mimulus genus. Fishman et al. (2014) show that marker orders within M. lewisii are collinear with the annual inversion orientation in M. guttatus. Thus, the evidence suggests the inversion originated in a perennial population prior to range expansion.
GENOMIC ANALYSES OF DIVERGING ECOTYPES
Our study in Mimulus is one of an increasing number that show that strong selection causing adaptive divergence within species, may not produce reciprocally monophyletic groups in genome‐wide phylogenetic analyses. In three‐spined sticklebacks, a nuclear gene phylogeny shows populations with different lateral plate morphologies are nonmonophyletic, but with a clear geographic genetic break corresponding to divergence between the Pacific and the Atlantic (Colosimo et al. 2005). In this case, the genetic basis of the phenotype is due to the same alleles being repeatedly recruited by selection from standing genetic variation. In the Senecio lautus complex (Asteraceae) in Australia, phylogenetic analyses with 2770 SNP loci revealed strong geographic structure, with populations adapted to different habitats (dunes, headlands, alpine) spread throughout the phylogenetic tree (Roda et al. 2013). They implicated parallel evolution, involving some shared alleles and some unique genomic regions, across different areas of the species’ range (Roda et al. 2013). These two studies illustrate two different scenarios by which this phylogenetic pattern can occur.
In contrast to sticklebacks and Senecio, we implicate a different evolutionary history by which loci underlying adaptive divergence have become widespread in M. guttatus. The most plausible scenario is that the inversion arose on a single occasion capturing adaptive variation, with the ecotypes subsequently colonizing the appropriate habitats, and gene flow between adjacent populations overwriting any genome‐wide signature of life history. This is probable given the high gene flow observed between populations of M. guttatus (Fig. S2), and its ability to move through the landscape and rapidly colonize new habitats. An alternative scenario of independent origins of the inversion throughout the species range seems unlikely, as numerous origins would be necessary to explain the observed pattern.
Confirming the number of origins of the inversion and locating the breakpoints and the genes within the inversion that have been under selection during adaptive divergence represent the next major goals for inversion studies in M. guttatus. However, these goals are a particular challenge when fine‐mapping cannot be used, due to reduced recombination within the inversion. The first stage in achieving these goals is to create a better genome assembly, particularly in the inversion region, and map the breakpoints. This will enable confirmation that the position of the breakpoints is shared across all populations, as expected with a single origin of the inversion. We are hopeful that with extensive genomic and transcriptomic sequencing, the causal loci underlying population divergence between ecotypes can be characterized (Korbel et al. 2007; Schlötterer et al. 2014). In light of knowing the breakpoints, and having an improved reconstruction of the inversion haplotypes, it will be possible to directly infer the age of the inversion (e.g., Huynh et al. 2010; Fang et al. 2012), and study the effect of reduced recombination on patterns of genetic diversity and the decay of linkage disequilibrium around the breakpoints (e.g., Davis et al. 2011). Moreover, it will be possible to assess the extent of gene flow between ecotypes within the inversion, mediated by double crossovers or gene conversion.
Our results from M. guttatus provide exceptional support for the local adaptation mechanism of inversion spread, with strong habitat‐mediated selection for loci captured by the chromosomal inversion maintaining life history ecotypes. We have demonstrated the remarkable ability of an inversion to enable ecotypes to persist in near sympatry, with recurrent gene flow, over the species’ full 4000 km+ native range. We anticipate that genomic surveys of other widespread species will reveal inversion polymorphisms to be a common means to maintain adaptive variation across heterogeneous environments.
Supporting information
Figure S1. Phylogeographic structure across populations of M. guttatus revealed by genome‐wide SNPs.
Figure S2. Patterns of admixture among populations of M. guttatus.
Figure S3. Evidence for postglacial range expansion in the phylogeny of M. guttatus.
Figure S4. Latitudinal gradient in genome‐wide genetic variation across populations of M. guttatus.
Figure S5. Patterns of genetic structure at inversion loci.
Table S1. Populations of M. guttatus sampled for genetic analyses.
Table S2. Analyses of molecular variance for genome‐wide and inversion loci in populations of M. guttatus.
ACKNOWLEDGMENTS
This research was supported by Syracuse University, and National Science Foundation grant (DEB‐1354259) to JF. Research by ADT is supported by the Natural Environment Research Council (NE/L011336/1). We thank the many individuals who supplied additional samples for analysis (Table S1). Next‐generation sequencing library preparation was assisted by S. Mitchell and C. Acharya. We thank M. den Bakker for assistance growing plants, A. Edmonds and iPlant staff for help with computing, D. Thomas for help with phylogenetic analyses, and K. Lohse for useful discussions about inversion biology. For comments on the manuscript, we thank D. Althoff, S. Barrett, R. Ennos, D. Lowry, J. Puzey, and J. Willis. ADT and JF conceived the study. ADT made the NGS libraries, and performed the analyses. Both authors contributed to the writing of the manuscript and approved the final version. No competing financial interests to report.
DATA ARCHIVING
Raw sequence reads are available in the Sequence Read Archive (SRA) as project SRP055962. The matrix of aligned SNPs are available at Dryad: doi:10.5061/dryad.5h032. The Bayesian phylogenetic trees are available from TreeBase, with accession number 17291.
LITERATURE CITED
Associate Editor: D. Moeller
Handling Editor: T. Lenormand
- Ayres, D. L. , Darling A., Zwickl D. J., Beerli P., Holder M. T., Lewis P. O., Huelsenbeck J. P., Ronquist F., Swofford D. L., Cummings M. P., et al. 2012. BEAGLE: an application programming interface and high‐performance computing library for statistical phylogenetics. Syst. Biol. 61:170–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourret, V. , Kent M. P., Primmer C. R., Vasemägi A., Karlsson S., Hindar K., McGinnity P., Verspoor E., Bernatchez L., and Lien S.. 2013. SNP‐array reveals genome‐wide patterns of geographical and potential adaptive divergence across the natural range of Atlantic salmon (Salmo salar). Mol. Ecol. 22:532–551. [DOI] [PubMed] [Google Scholar]
- Bradbury, P. J. , Zhang Z., Kroon D. E., Casstevens T. M., Ramdoss Y., and Buckler E. S.. 2007. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635. [DOI] [PubMed] [Google Scholar]
- Brandvain, Y. , Kenney A. M., Flagel L., Coop G., and Sweigart A. L.. 2014. Speciation and introgression between Mimulus nasutus and Mimulus guttatus . PLoS Genet. 10:e1004410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunsfeld, S. J. , Sullivan J., Soltis D. E., and Soltis P. S.. 2001. Comparative phylogeography of northwestern North America: a synthesis Pp. 319–339 in Silvertown J., and Antonovics J., eds. Integrating ecological and evolutionary processes in a spatial context. Blackwell Science, Oxford. [Google Scholar]
- Colosimo, P. F. , Hosemann K. E., Balabhadra S., Villarreal G., Dickson M., Grimwood J., Schmutz J., Myers R. M., Schluter D., and Kingsley D. M.. 2005. Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science 307:1928–1933. [DOI] [PubMed] [Google Scholar]
- Coluzzi, M. , Sabatini A., della Torre A., Di Deco M. A., and Petrarca V.. 2002. A polytene chromosome analysis of the Anopheles gambiae species complex. Science 298:1415–1418. [DOI] [PubMed] [Google Scholar]
- Cruickshank, T. E. , and Hahn M. W.. 2014. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 23:3133–3157. [DOI] [PubMed] [Google Scholar]
- Davis, J. K. , Mittel L. B., Lowman J. J., Thomas P. J., Maney D. L., Maney D.L., Martin C.L., NISC Comparative Sequencing Program, and Thomas J.W.. 2011. Haplotype‐based genomic sequencing of a chromosomal polymorphism in the white‐throated sparrow (Zonotrichia albicollis). J. Hered. 102:380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky, T. 1970. Genetics of the evolutionary process. Columbia Univ. Press, New York. [Google Scholar]
- Drummond, A. , and Rambaut A.. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshire, R. J. , Glaubitz J. C., Sun Q., Poland J. A., Kawamoto K., Buckler E. S., and Mitchell S. E.. 2011. A robust, simple genotyping‐by‐sequencing (GBS) approach for high diversity species. PLoS One 6:e19379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno, G. , Regnaut S., and Goudet J.. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14:2611–2620. [DOI] [PubMed] [Google Scholar]
- Excoffier, L. , and Lischer L. H. E.. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10:564–567. [DOI] [PubMed] [Google Scholar]
- Falush, D. , Stephens M., and Pritchard J. K.. 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, Z. , Pyhajarvi T., Weber A. L., Dawe R. K., Glaubitz J. C., González J. J. S., Ross‐Ibarra C., Doebley J., Morrell P. L., and Ross‐Ibarra J.. 2012. Megabase‐scale inversion polymorphism in the wild ancestor of maize. Genetics 191:883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder, J. F. , Egan S. P., and Nosil P.. 2012. The genomics of speciation‐with‐gene‐flow. Trends Genet. 28:342–350. [DOI] [PubMed] [Google Scholar]
- Friedman, J. , Twyford A. D., Willis J. H., and Blackman B. K.. 2015. The extent and genetic basis of phenotypic divergence in life history traits in Mimulus guttatus . Mol. Ecol. 24:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, J. , and Willis J. H.. 2013. Major QTLs for critical photoperiod and vernalization underlie extensive variation in flowering in the Mimulus guttatus species complex. New Phytol. 199:571–583. [DOI] [PubMed] [Google Scholar]
- Fishman, L. , Willis J. H., Wu C. A., and Lee Y.‐W.. 2014. Comparative linkage maps suggest that fission, not polyploidy, underlies near‐doubling of chromosome number within monkeyflowers (Mimulus; Phrymaceae). Heredity 112:562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaubitz, J. C. , Casstevens T. M., Lu F., Harriman J., Elshire R. J., Sun Q., and Buckler E. S.. 2014. TASSEL‐GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One 9:e90346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompert, Z. , and Buerkle C. A.. 2011. A hierarchical bayesian model for next‐generation population genomics. Genetics 187:903–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero, R. F. , Rousset F., and Kirkpatrick M.. 2012. Coalescent patterns for chromosomal inversions in divergent populations. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367:430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, M. C. , Basten C. J., and Willis J. H.. 2006. Pleiotropic quantitative trait loci contribute to population divergence in traits associated with life history variation in Mimulus guttatus . Genetics 172:1829–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, M. C. , Lowry D. B., and Willis J. H.. 2010. Is local adaptation in Mimulus guttatus caused by trade‐offs at individual loci? Mol. Ecol. 19:2739–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten, U. , Wright K. M., Jenkins J., Shu S., Yuan Y., Wessler S. R., Schmutz J., Willis J. H., and Rokhsar D. S.. 2013. Fine‐scale variation in meiotic recombination in Mimulus inferred from population shotgun sequencing. Proc. Natl. Acad. Sci. USA 110:19478–19482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, A. A. , and Rieseberg L. H.. 2008. Revisiting the impact of inversions in evolution: from population genetic markers to drivers of adaptive shifts and speciation? Annu. Rev. Ecol. Evol. Syst. 39:21–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holeski, L. , Monnahan P., Koseva B., McCool N., Lindroth R. L., and Kelly J. K.. 2014. A high‐resolution genetic map of yellow monkeyflower identifies chemical defense QTLs and recombination rate variation. G3 4:813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson, D. H. , and Bryant D.. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23:254–267. [DOI] [PubMed] [Google Scholar]
- Huynh, L.Y. , Maney D. L., and Thomas J. W.. 2010. Chromosome‐wide linkage disequilibrium caused by an inversion polymorphism in the white‐throated sparrow (Zonotrichia albicollis). Heredity 106:537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson, M. , and Rosenberg N. A.. 2007. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. [DOI] [PubMed] [Google Scholar]
- Joron, M. , Frezal L., Jones R. T., Chamberlain N. L., Lee S. F., Haag C. R., Whibley A., Becuwe M., Baxter S. W., Ferguson L., et al. 2011. Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477:203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, M. , and Barton N.. 2006. Chromosome inversions, local adaptation and speciation. Genetics 173:419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel, J. O. , Urban A. E., Affourtit J. P., Godwin B., Grubert F., Simons J. F., Kim P. M., Palejev D., Carriero N. J., Du L., et al. 2007. Paired‐end mapping reveals extensive structural variation in the human genome. Science 318:420–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimbas, C. B. , and Powell J. R.. 1992. Drosophila inversion polymorphism. CRC Press, Cleveland. [Google Scholar]
- Latch, E. K. , Heffelfinger J. R., Fike J. A., and Rhodes O. E. Jr. 2009. Species‐wide phylogeography of North American mule deer (Odocoileus hemionus): cryptic glacial refugia and postglacial recolonization. Mol. Ecol. 18:1730–1745. [DOI] [PubMed] [Google Scholar]
- Lenormand, T. 2002. Gene flow and the limits to natural selection. Trends Ecol. Evol. 17:183–189. [Google Scholar]
- Lewis, P. O. 2001. A likelihood approach to estimating phylogeny from discrete morphological character data. Syst. Biol. 50:913–925. [DOI] [PubMed] [Google Scholar]
- Li, H. , and Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado, P. , and Rozas J.. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. [DOI] [PubMed] [Google Scholar]
- Lowry, D. B. , and Willis J. H.. 2010. A widespread chromosomal inversion polymorphism contributes to a major life history transition, local adaptation, and reproductive isolation. PLoS Biol. 8:e1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry, D. B. , Modliszewski J. L., Wright K. M., Wu C. A., and Willis J. H.. 2008a. The strength and genetic basis of reproductive isolating barriers in flowering plants. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 363:3009–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry, D. B. , Rockwood R. C., and Willis J. H.. 2008b. Ecological reproductive isolation of coast and inland races of Mimulus guttatus . Evolution 62:2196–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesom, G. 2012. Taxonomy of Erythranthe sect. Simiola (Phrymaceae) in the USA and Mexico. Phytoneuron 40:1–123. [Google Scholar]
- Oneal, E. , Lowry D. B., Wright K. M., Zhu Z., and Willis J. H.. 2014. Divergent population structure and climate associations of a chromosomal inversion polymorphism across the Mimulus guttatus species complex. Mol. Ecol. 23:2844–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens M., and Donnelly P.. 2000. Inference of population structure using multilocus genotype data. Genetics 155:945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig, M. , Caceres M., and Ruiz A.. 2004. Silencing of a gene adjacent to the breakpoint of a widespread Drosophila inversion by a transposon‐induced antisense. Proc. Natl. Acad. Sci. USA 101:9013–9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell, S. , Neale B., Todd‐Brown K., Thomas L., Ferreira M. A. R., Bender D., Maller J., Sklar P., de Bakker P. I. W., Daly M. J., et al. 2007. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am. J. Hum. Genet. 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzey, J. , and Vallejo‐Marín M.. 2014. Genomics of invasion: diversity and selection in introduced populations of monkeyflowers (Mimulus guttatus). Mol. Ecol. 23:4472–4485. [DOI] [PubMed] [Google Scholar]
- Raj, A. , Stephens M., and Pritchard J. K.. 2013. Variational inference of population structure in large SNP datasets. Genetics 197:573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roda, F. , Ambrose L., Walter G. M., Liu H. L., Schaul A., Lowe A., Pelser P. B., Prentis P., Rieseberg L. H., and Ortiz‐Barrientos D.. 2013. Genomic evidence for the parallel evolution of coastal forms in the Senecio lautus complex. Mol. Ecol. 22:2941–2952. [DOI] [PubMed] [Google Scholar]
- Roff, D. A. 2002. Life history evolution. Sinauer Associates, Sunderland, MA. [Google Scholar]
- Ronquist, F. , Teslenko M., van der Mark P., Ayres D. L., Darling A., Höhna S., Larget B., Liu L., Suchard M. A., and Huelsenbeck J. P.. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambatti, J. B. M. , Strasburg J. L., Ortiz‐Barrientos D., Baack E. J., and Rieseberg L. H.. 2012. Reconciling extremely strong barriers with high levels of gene exchange in annual sunflowers. Evolution 66:1459–1473. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C. , Tobler R., Kofler R., and Nolte V.. 2014. Sequencing pools of individuals—mining genome‐wide polymorphism data without big funding. Nat. Rev. Genet. 15:749–763. [DOI] [PubMed] [Google Scholar]
- Shafer, A. B. A. , Cullingham C. I., Côté S. D., and Coltman D. W.. 2010. Of glaciers and refugia: a decade of study sheds new light on the phylogeography of northwestern North America. Mol. Ecol. 19:4589–4621. [DOI] [PubMed] [Google Scholar]
- Slatkin, M. 1987. Gene flow and the geographic structure of natural populations. Science 236:787–792. [DOI] [PubMed] [Google Scholar]
- Slotte, T. , Huang H., Lascoux M., and Ceplitis A.. 2008. Polyploid speciation did not confer instant reproductive isolation in Capsella (Brassicaceae). Mol. Biol. Evol. 25:1472–1481. [DOI] [PubMed] [Google Scholar]
- Soltis, D. E. , Gitzendanner M. A., Strenge D. D., and Soltis P. S.. 1997. Chloroplast DNA intraspecific phylogeography of plants from the Pacific Northwest of North America. Plant Syst. Evol. 206:353–373. [Google Scholar]
- Steele, C. A. , and Storfer A.. 2007. Phylogeographic incongruence of codistributed amphibian species based on small differences in geographic distribution. Mol. Phylogenet. Evol. 43:468–479. [DOI] [PubMed] [Google Scholar]
- Sweigart, A. L. , and Willis J. H.. 2003. Patterns of nucleotide diversity in two species of Mimulus are affected by mating system and asymmetric introgression. Evolution 57:2490–2506. [DOI] [PubMed] [Google Scholar]
- Thomas, D. C. , Surveswaran S., Xue B., Sankowsky G., Mols J. B., Keßler P. J. A., and Saunders R. M. K.. 2012. Molecular phylogenetics and historical biogeography of the Meiogyne‐Fitzalania clade (Annonaceae): generic paraphyly and late Miocene‐Pliocene diversification in Australasia and the Pacific. Taxon 61:559–575. [Google Scholar]
- Umina, P. A. , Weeks A. R., Kearney M. R., McKechnie S. W., and Hoffmann A. A.. 2005. A rapid shift in a classic clinal pattern in Drosophila reflecting climate change. Science 308:691–693. [DOI] [PubMed] [Google Scholar]
- White, M. 1978. Modes of speciation. W. H. Freeman, San Francisco. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Phylogeographic structure across populations of M. guttatus revealed by genome‐wide SNPs.
Figure S2. Patterns of admixture among populations of M. guttatus.
Figure S3. Evidence for postglacial range expansion in the phylogeny of M. guttatus.
Figure S4. Latitudinal gradient in genome‐wide genetic variation across populations of M. guttatus.
Figure S5. Patterns of genetic structure at inversion loci.
Table S1. Populations of M. guttatus sampled for genetic analyses.
Table S2. Analyses of molecular variance for genome‐wide and inversion loci in populations of M. guttatus.