

Abstract
Radiation modulates both tumor cells and immune cells in the tumor microenvironment to exert its anti-tumor activity; however, the molecular connection between tumor cells and immune cells that mediates radiation-exerted tumor suppression activity in the tumor microenvironment is largely unknown. We report here that radiation induces rapid activation of the p65/p50 and p50/p50 NF-κB complexes in human soft tissue sarcoma (STS) cells. Radiation-activated p65/p50 and p50/p50 bind to the TNFα promoter to activate its transcription in STS cells. Radiation-induced TNFα induces tumor cell death in an autocrine manner. A sublethal dose of Smac mimetic BV6 induces cIAP1 and cIAP2 degradation to increase tumor cell sensitivity to radiation-induced cell death in vitro and to enhance radiation-mediated suppression of STS xenografts in vivo. Inhibition of caspases, RIP1, or RIP3 blocks radiation/TNFα-induced cell death, whereas inhibition of RIP1 blocks TNFα-induced caspase activation, suggesting that caspases and RIP1 act sequentially to mediate the non-compensatory cell death pathways. Furthermore, we determined in a syngeneic sarcoma mouse model that radiation up-regulates IRF3, IFNβ, and the T cell chemokines CCL2 and CCL5 in the tumor microenvironment, which are associated with activation and increased infiltration of Th1/Tc1 T cells in the tumor microenvironment. Moreover, tumor-infiltrating T cells are in their active form since both the perforin and FasL pathways are activated in irradiated tumor tissues. Consequently, combined BV6 and radiation completely suppressed tumor growth in vivo. Therefore, radiation-induced NF-κB functions as a molecular link between tumor cells and immune cells in the tumor microenvironment for radiation-mediated tumor suppression.
Keywords: NF-κB, radiation, TNFα, cytotoxic T lymphocyte, Smac mimetic
INTRODUCTION
Radiotherapy is the mainstay local treatment for many types of solid cancers, and about two thirds of cancer patients require radiation at least once during the course of their disease treatment. However, local failure is frequent due to tumor recurrence, and survival rate after radiotherapy for many human cancers is not high [1-3]. Therefore, any improvement in radiotherapy efficacy will benefit a large number of human patients [3]. A direct consequence of radiation is DNA damage and DNA damage-mediated cell death. Recent studies, however, indicate that multiple biological effects play a key role in radiation-induced tumor suppression. In addition, radiation may increase various signaling transduction pathways in the tumor cells to increase the tumor cell sensitivity to chemotherapeutic agents [3, 4]. Interestingly, radiation may also alter the inflammatory tumor microenvironment through stimulation of host immune cells to suppress tumor growth and progression [4-9]. However, the molecular mechanisms underlying radiation-mediated biological and immunological effects are still elusive.
Low levels of constitutive NF-κB activity are present in many types of human cancers, and NF-κB activation often promotes tumor growth and progression [10-13]. This provides a strong rationale for anticancer strategies that inhibit NF-κB signaling [14, 15]. In fact, several hundred NF-κB inhibitors have been developed [16]. However, compelling experimental data have also shown that NF-κB may harbor dual functionality by acting to promote apoptosis and senescence in multiple types of cells, including cancer cells [17-34]. In addition to its essential function in tumor cells, the NF-κB pathway also exerts an essential function in the regulation of the immune response [35-37]. Particularly, NF-κB is known to be essential for T cell differentiation and function [35, 38]. T cell-based cancer immunotherapy has shown durable efficacy in human cancer patients [39] and has been approved for treatment of human cancers. Therefore, blocking NF-κB activation might impair T cell activation and function in cancer patients, and its side effects on T cells in the tumor microenvironment should be considered with great care. The molecular mechanisms underlying these contrasting functions of NF-κB in cell death and proliferation are currently unknown. In this study, we determine that radiation-activated NF-κB functions as a molecular link between tumor cells and immune cells to exert radiation-induced tumor suppression.
RESULTS
Smac mimetic BV6 is effective in sensitization of human tumor cells to radiation-induced cell death
Human soft tissue sarcoma cell lines HT1080, MPNST724, SK-LMS1, and SW684 were irradiated and cultured for 24 hours. Analysis of cell death indicates that all four cell lines exhibited limited responses to radiation in a one day assay (Figure 1A). Further analysis with two of the tumor cell lines indicate that these tumor cells are sensitive to a dose of 10 Gy or higher after two or more days post radiation in vitro (Figure 1B). To test whether inhibition of IAPs increase STS cell sensitivity to radiation [21, 40-44], cells were cultured in the presence of BV6. Analysis of cell death revealed that BV6 induces STS cell death in a dose-dependent manner (Figure 1C). To test the efficacy of BV6 as a sensitizer of radiation-induced cell death, human sarcoma cells were either untreated or irradiated, followed by culture in the absence or presence of a sublethal dose of BV6. A sublethal dose of BV6 significantly increased the sensitivity of all four human sarcoma cell lines to radiation-induced cell death (Figure 1D).
Figure 1. BV6 increases the sensitivity of human soft tissue sarcoma cells to radiation.
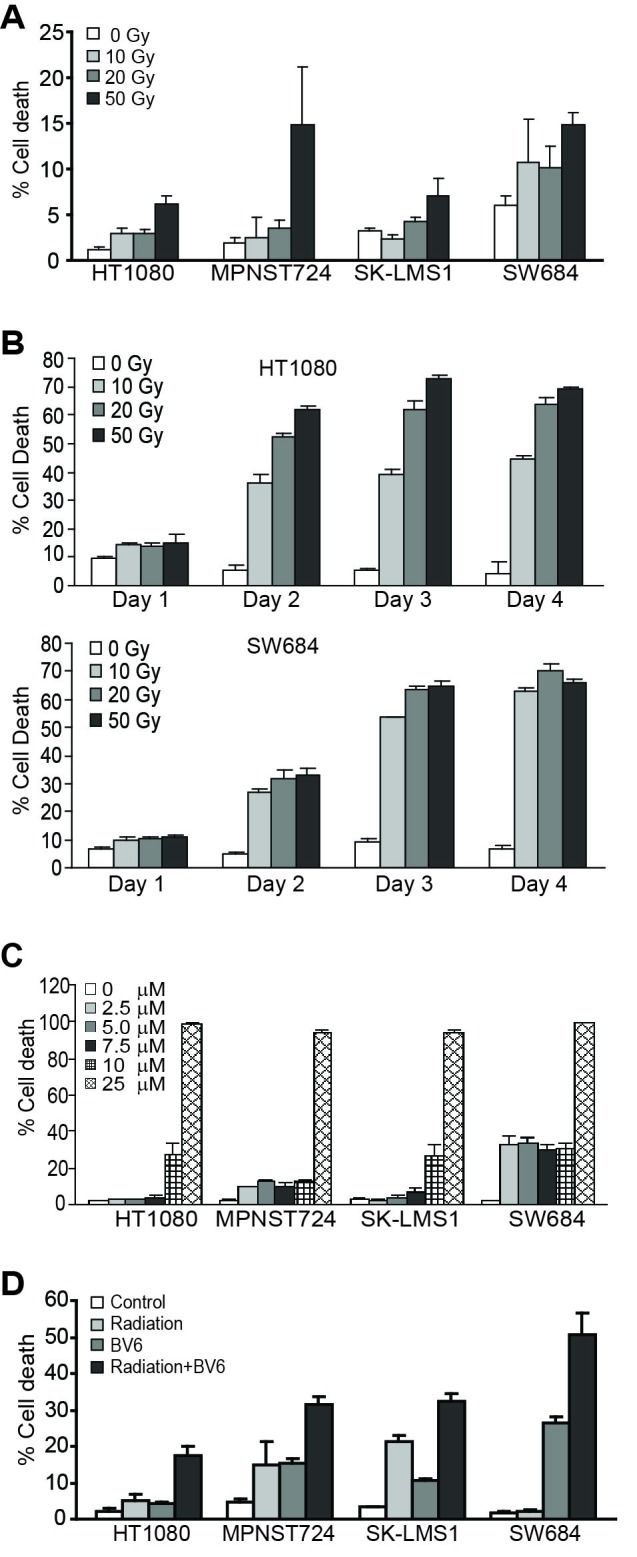
A. Tumor cells were irradiated at the indicated doses and cultured for 24 hours. Cells were stained with PI and analyzed by flow cytometry. Percent cell death is expressed as % PI+ cells. Column: mean; Bar: SD. B. Tumor cells were irradiated with γ-ray at the indicated doses and cultured for 1, 2, 3 and 4 days. Cells were then analyzed as in A. C. Tumor cells were cultured in the presence of BV6 at the indicated doses for 24 hours. Cell death was determined as in A. D. Tumor cells were either untreated or irradiated (50 Gy) and then cultured in the absence or presence of BV6 (5 μM) for 24h. Cell death was determined as in A.
To determine whether this observation can be extended to other types of human cancer cells, we also examined the effects of radiation and BV6 on human colon carcinoma cells. The human colon carcinoma cell lines SW620 and LS411N are relatively resistant to radiation in a one day assay (Figure 2A). BV6 also exhibits cytotoxicity to these two human colon carcinoma cell lines (Figure 2B). Consistent with observations in human sarcoma cell lines, a sublethal dose of BV6 increased the sensitivity of both SW620 and LS411N cell lines to radiation-induced cell death (Figure 2C).
Figure 2. BV6 increases the sensitivity of human colon carcinoma cells to radiation-induced cell death.
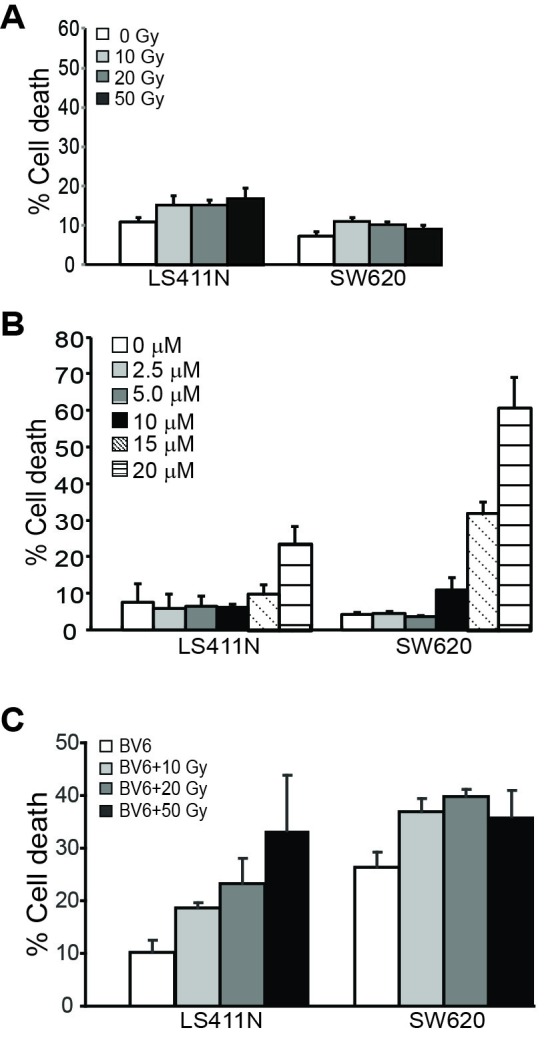
A.. Tumor cells were irradiated at the indicated doses and cultured for 24 hours. Cells were stained with PI and analyzed by flow cytometry. Percent cell death was expressed as % PI+ cells. Column: mean; Bar: SD. B. Tumor cells were cultured in the presence of BV6 at the indicated doses for 24 hours. Cell death was determined as in A. C. Tumor cells were irradiated at the indicated dose and then cultured in the presence of BV6 (5 μM) for 24 hours. Cell death was determined as in A.
cIAP1 protein level indicates poor prognosis of human CRC patients
BV6 is a Smac mimetic that induces IAPs degradation [21, 43, 44]. BV6 treatment resulted in rapid degradation of cIAP1 and cIAP2 in human STS and colon carcinoma cells (Figure 3). Next, we made use of a human colon cancer tissue microarray and stained for cIAP1 proteins. Kaplan-Meier analysis of the 235 human colorectal cancer specimens revealed that the cIAP1 protein level is inversely correlated with disease-specific survival and positively correlated with cancer recurrence (Figure 3B). Patients with high cIAP1 protein levels had a significantly lower survival time as compared to patients with medium to low or undetectable cIAP1 protein levels in the tumor cells. Furthermore, patients with high cIAP1 protein levels in the tumor cells also exhibited a significantly higher recurrence rate as compared to patients with medium to low and undetectable cIAP1 protein levels in the tumor cells (Figure 3B).
Figure 3. cIAP1 protein level is correlated with shorter survival time and earlier recurrence in human colorectal cancer patients.

A. Human sarcoma (top panel) and colon carcinoma (bottom panel) cells were treated with BV6 (5 μM) for the indicated time and analyzed by Western blotting analysis for cIAP1 and cIAP2 levels. β-actin was used as a normalization control. B. TMA slides containing human colorectal cancer specimens (N=235) were stained for cIAP1 protein level. The stained specimens were then statistically analyzed for correlations between cIAP1 protein levels and patient survival time (top panel) or cancer recurrence time (bottom panel). Each variable is indicated by colored lines in the plot.
BV6 activates the non-canonical but not the canonical NF-κB pathway
cIAP1 and cIAP2 are also E3 ligases that mediate NF-κB activation [19, 21, 45]. BV6 induced rapid IκBα phosphorylation in human sarcoma cells. Time-dependent p100 processing to p52 was also observed in both sarcoma cell lines (Figure 4A). Similar patterns were also observed in the human colon carcinoma LS411N and SW620 cell lines (Figure 4B). As expected, the positive control TNFα induced activation of the canonical NF-κB as both the p65 and p50 subunits are bound to the DNA probe (Figure 4C). However, BV6 treatment did not induce detectable p65 or p50 binding to the DNA (Figure 4C). Similar outcomes were observed in the human colon carcinoma cells (Figure 4D). A complimentary approach was used to validate the EMSA results. SW620 cells were untreated or treated with TNFα, BV6, or both TNFα and BV6 and analyzed for nuclear p65 subcellular localization. In untreated cells, p65 protein is primarily localized in the cytoplasm (Figure 5a1 & 5a2). As expected, TNFα treatment dramatically increased nuclear p65 translocation (Figure 5b1 & 5b2, 5d1 & 5d2), but BV6 treatment did not increase p65 nuclear translocation (Figure 5c1 & 5c2).
Figure 4. BV6 activates the alternate but not the canonical NF-κB.

A. The indicated human sarcoma cells were treated with BV6 (5 μM) for the indicated time. Cells were then analyzed by Western blotting analysis for p100/p52, pIκBα, and IκBα. B. The indicated human colon carcinoma cells were treated with BV6 (5 μM) for the indicated time and then analyzed for p100/p52, pIκBα, and IκBα as in A. C. The indicated human sarcoma cells were treated with BV6 (5μM, 1 and 2h), TNFα (100 U/ml, 1h), or both as indicated. Nuclear extracts were prepared from the tumor cells and analyzed for canonical NF-κB activity using EMSA with NF-κB consensus sequence-containing DNA probes (Santa Cruz Biotech). Anti-p65 and anti-p50 antibodies were used to identify the canonical NF-κB-DNA complexes. D. SW620 cells were treated with BV6 (5 μM, for 0.5, 1, and 2h) and TNFα (1 h) and analyzed for NF-κB activation as in C.
Figure 5. BV6 does not induce NF-κB nuclear translocation.
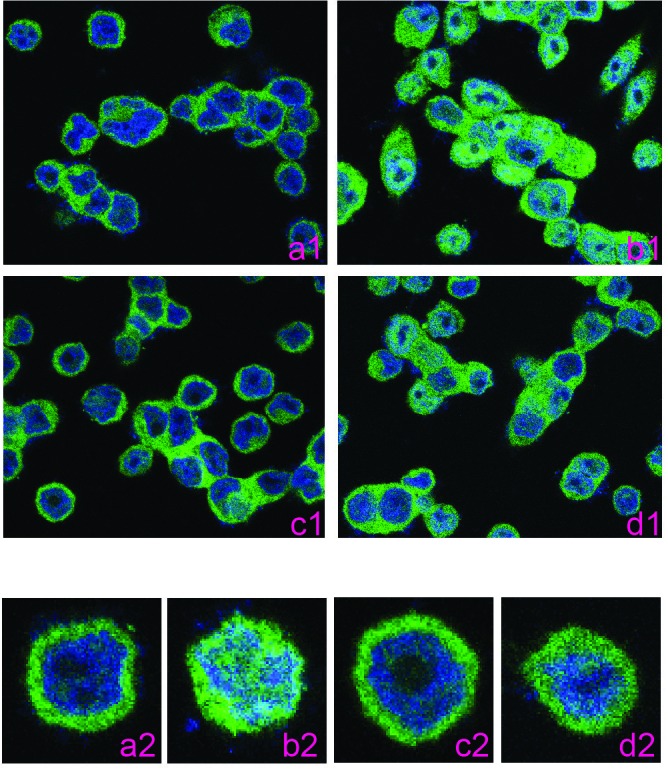
SW620 cells were either untreated (a), treated with TNFα (b, 100 U/ml), BV6 (c, 5 μM) or both TNFα and BV6 (d) for 60 min. Cells were fixed, permeabilized, and stained with p65-specific antibody, followed by fluorescent dye-conjugated 2nd antibody. The images were obtained with a confocal microscope. a1-d1 are low amplification images and a2-d2 are high amplification images.
Radiation activates the canonical p65/p50 and p50/p50 NF-κB
All four human STS cells exhibit weak constitutive NF-κB activity (Figure 6A). However, radiation induces rapid NF-κB activation in all four cell lines within 60 minutes post radiation (Figure 6A). NF-κB subunit-specific antibody-based supershift assays indicate that no p52, RelB, or cRel DNA-binding activity was detectable in irradiated human sarcoma cells (Figure 6B). Two types of NF-κB-DNA complexes were detected. The C1 complex represents the canonical NF-κB p65/p50 heterodimer, whereas the C2 complex represents the NF-κB p50/p50 homodimer (Figure 6B). Therefore, we have determined that radiation activates both the canonical NF-κB p65/p50 heterodimer and the NF-κB p50/p50 homodimer.
Figure 6. Radiation activates the canonical NF-κB.

A. Tumor cells were irradiated at a dose of 50 Gy and nuclear extracts were prepared at 1 and 2 h after culture. Nuclear extracts were then analyzed for canonical NF-κB activity using EMSA with NF-κB consensus sequence-containing DNA probe as in Figure 3. Anti-p65 and anti-p50 antibodies were used to identify the canonical NF-κB-DNA complexes. C1: p65/p50 heterodimer complex; C2: p50/p50 homodimer complex. B. The tumor cells were irradiated and cultured for 1 and 2h, respectively. Nuclear extracts were then analyzed for NF-κB activation using EMSA as in A. Antibodies that are specific for the five indicated NF-κB Rel subunits (p65, p50, p52, RelB, and cRel) were used to identify the NF-κB-DNA complexes.
Radiation induces activation of canonical NF-κB that directly regulates TNFα expression to induce tumor cell death
Analysis of the human TNFα promoter DNA sequence identified three potential NF-κB-binding DNA sequences (Figure 7A). Analysis of DNA-protein interactions revealed that radiation-induced canonical NF-κB binds to all three NF-κB binding sites in human STS cells (Figure 7B). In probes 1 and 2 regions, both p65/p50 and p50/p50 complexes were detected. In probe 3 region, only the p65/p50 complex was detected (Figure 7B). Consistent with NF-κB activation and binding to the TNFα promoter region, radiation induced an increase in TNFα mRNA level (Figure 7C).
Figure 7. Radiation-induced NF-κB directly regulates TNFα transcription that acts in concert with BV6 to induce tumor cell death.

A. The human TNFα promoter structure. The three NF-κB-binding consensus sequence elements are indicated at the bottom of the bar. B. Tumor cells were irradiated at a dose of 50 Gy and then cultured for 90 min. Nuclear extracts were prepared and analyzed for NF-κB activation using EMSA with the three DNA probes of the human TNFα promoter as indicated in A. C. Tumor cells were irradiated with the indicated dose and cultured for 24h. Cells were then analyzed for human TNFα mRNA level using quantitative PCR. D. Tumor cells were treated as indicated for 24h and analyzed for cell death using PI staining and flow cytometry analysis. E. Tumor cells were irradiated and then cultured in the presence of BV6 (5 μM). IgG or anti-TNFα neutralization antibody (200 μg/ml) was added to the culture, respectively, for 24h. Cell death was analyzed by PI staining and flow cytometry analysis. *p < 0.05.
Radiation up-regulates TNFα expression to induce tumor cell death
Tumor-producing TNFα has been shown to induce tumor cell death in an autocrine manner [19, 21]. However, we observed that all four human sarcoma cell lines are not sensitive to TNFα-induced cell death (Figure 7D). A sublethal dose of BV6 significantly increased the sensitivity of human sarcoma cells to TNFα-induced cell death (Figure 7D). Next, tumor cells were irradiated and cultured in the presence of BV6 and a TNFα neutralization antibody. TNFα neutralization antibody significantly decreased radiation and BV6-induced cell death (Figure 7E).
Radiation activates the canonical NF-κB to activate FAS transcription
NF-κB is a known FAS transcription activator [46-48]. Analysis of the FAS promoter DNA sequences identified one potential NF-κB-binding consensus sequence (Figure 8A). EMSA analysis revealed that radiation-induced canonical NF-κB binding to this DNA element. Use of p65- and p50-specific antibodies indicates that NF-κB p65/p50 and p50/p50 complexes bind to the FAS promoter in MPNST724 cells, whereas only p65/p50 heterodimer binds to the FAS promoter in HT1080 cells (Figure 8A). Quantitative PCR analysis indicates that radiation increases Fas expression level (Figure 8B) and flow cytometry analysis reveals that radiation increases Fas protein level on the surface of tumor cells (Figure 8C).
Figure 8. Radiation induces NF-κB to regulate FAS transcription.
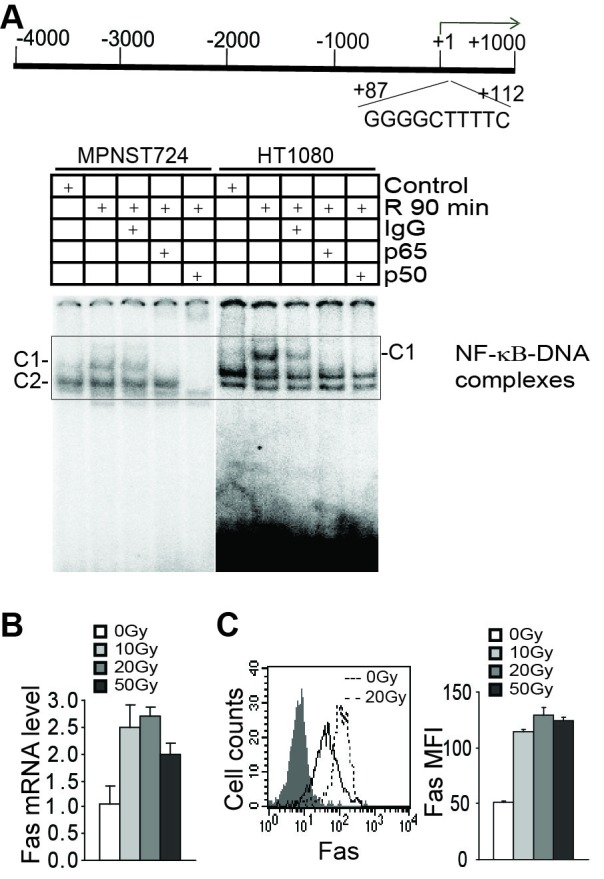
A. The human FAS promoter structure. The NF-κB-binding consensus sequence element is indicated at the bottom of the bar. Tumor cells were irradiated at a dose of 50 Gy and then cultured for 90 min. Nuclear extracts were prepared and analyzed for NF-κB activation using EMSA with the DNA probe of the human FAS promoter as indicated in the top panel. B. HT1080 tumor cells were irradiated with the indicated doses and cultured for 24 hours. Cells were then analyzed for human Fas mRNA levels using quantitative PCR. C. HT1080 tumor cells were irradiated with the indicated doses for 24 hours. The cell surface Fas was stained with Fas-specific antibody and analyzed by flow cytometry. Representative plots are presented at the left. Fas MFI was quantified and presented at the right.
Radiation induces both apoptosis and necroptosis of tumor cells
To test the relative contributions of apoptosis and necroptosis in radiation-induced cell death, we made use of the caspase inhibitor Z-VAD-fmk, an allosteric RIP1 kinase inhibitor Necrostain-1 (Nec-1), and the mixed linage kinase-like (MLKL) protein inhibitor Necrosulfonamide (NSA). Z-VAD effectively blocked radiation and BV6-induced cell death in three of the four human sarcoma cells. However, Nec-1 and NSA also inhibited radiation and BV6-induced cell death in the same three human sarcoma cell lines (Figure 9A). Similarly, Z-VAD blocked TNFα and BV6-induced cell death in three of the four STS cell lines. Nec-1 blocked cell death in HT1080 and SW684 cells, whereas NSA inhibited cell death in MPNST724 and SW684 cells (Figure 9B).
Figure 9. Radiation and TNFα induce apoptosis and necroptosis.

A. Tumor cells were irradiated (R) at a dose of 50 Gy and then cultured in the presence of BV6 (B, 5 μM) plus Z-VAD (Z, 10 μM), Nec-1 (10 μM), and NSA (2 μM), either alone or in combinations, for 24 hours. Cell death was determined by PI staining and flow cytometry analysis. Column: mean, bar: SD. B. Tumor cells were cultured in the presence of TNFα (T, 100 U/ml) and BV6 (B, 5 μM) plus Z-VAD (Z, 10 μM), Nec-1 (10 μM), and NSA (2 μM), either alone or in combinations, for 24h, and analyzed for cell death as in A. Column: mean; Bar: SD.
BV6 modulates both the apoptosis and necroptosis pathways to increase TNFα-induced cell death
Analysis of human tumor cells after BV6 treatment indicates that a sublethal dose of BV6 alone is insufficient to induce caspase activation (Figure 10A). TNFα alone also failed to induce caspase activation. However, a combined treatment with TNFα and BV6 activated caspases 8, 9, and 3, and induced PARP cleavage. To dissect the relative roles of apoptosis and necroptosis in tumor cell death, we treated tumor cells with BV6 and TNFα in the presence of apoptosis and necroptosis inhibitors. As expected, inhibition of apoptosis with Z-VAD blocked activation of caspases 8, 9, and 3, and blocked activation of apoptosis as indicated by lack of PARP cleavage (Figure 10B). Interestingly, Nec-1 also reduced caspase activation in both HT1080 and SW684 cells and decreased apoptosis as indicated by decreased PARP cleavage (Figure 10B). These observations suggest that BV6 and TNFα synergistically induce apoptosis and necroptosis that sequentially mediate tumor cell death.
Figure 10. TNFα and BV6 cooperate to activate sequential necroptosis and apoptosis.

A. Tumor cells were cultured in the presence of TNFα (100 U/ml), BV6 (5 μM), or both for the indicated time and analyzed by Western blotting analysis for the indicated caspases. PARP was used as an apoptosis indicator. B. Tumor cells were cultured in the presence of TNFα (100 U/ml), BV6 (5 μM), plus Z-VAD (10 μM), Nec-1 (10 μM), or NSA (2 μM), respectively for 4 h, and analyzed by Western blotting for the indicated caspases and PARP.
BV6 enhances radiation-induced xenograft tumor growth suppression in vivo
HT1080 cells were transplanted to nude mice. The tumor xenografts were then irradiated with a dose of 8 Gy, followed by BV6 treatment. The human sarcoma xenografts grew rapidly in vivo (Figure 11A). The post hoc pairwise comparisons showed that there were statistically significant differences between groups within days and between days within group. On day 4, the radiation group and the radiation+BV6 group had significantly lower mean tumor volume than the control group. On day 6, the radiation only, BV6 only, and radiation+BV6 had significantly lower mean tumor volumes than the control group, and the radiation+BV6 group had a significantly lower mean tumor volume than BV6 only (p < 0.01). Overall, combined radiation and BV6 eradicated three of the four tumors (Figure 11A).
Figure 11. BV6 enhances radiation-mediated tumor growth suppression in vivo.

A. HT1080 cells (2.5×106/mouse) were transplanted to athymic nude mice. Ten days after tumor injection, the established xenograft tumors were either untreated or irradiated using an external beam of radiation at a dose of 8 Gy (n=4 each treatment group). The mice were then either untreated or injected with BV6 i.v. (5 mg/kg body weight) every two days for four treatments. Tumors were dissected and presented at the left panel. The tumor xenograft growth kinetics are presented in the right panel. B. Xenograft tumor tissues, as shown in A, were dissected from mice and analyzed by immunohistochemical staining for in situ TUNEL. The brown color indicates apoptotic cell death. a: tumor from control mice, b: tumor from BV6-treated mice, c:tumor from radiated mice, and d: tumor from combined BV6 and radiation-treated mice. C. Xenograft tumor tissues, as shown in A, were dissected from mice and analyzed by immunohistochemical staining for Ki67 protein levels. The brown color indicates proliferating Ki67-positive tumor cells. a: tumor from control mice, b: tumor from BV6-treated mice, c:tumor from radiated mice, and d: tumor from combined BV6 and radiation-treated mice. D. Mouse sarcoma CMS4-met cells (2.5×105/mouse) were transplanted to BALB/c mice. Ten days after tumor injection, the established tumors were either untreated or irradiated using an external beam of radiation at a dose of 8 Gy (n=4 each treatment group). The mice were then either untreated or injected with BV6 (5 mg/kg body weight) every two days for four treatments. The tumor growth was measured over time. Shown are the tumor growth kinetics. E. CMS4-met cells were injected to BALB/c mice and treated as in A except the radiation dose was increased to 20Gy. Shown are the tumor growth kinetics.
Analysis of apoptotic cell death of the tumor tissues by an in situ TUNEL assay indicates that radiation increases apoptosis of tumor cells as compared to the control and BV6-treated tumors (Figure 11B). In the combined radiation and BV6-treated tumor tissues, almost all cells are apoptotic (Figure 11Bd). Analysis of tumor cell proliferation with Ki67 reveals that radiation also decreases the xenograft tumor cell proliferation, and combined radiation and BV6 treatment exhibited greater suppression of tumor cell proliferation than radiation and BV6 alone (Figure 11Cd).
BV6 enhances radiation-induced syngeneic tumor growth suppression in vivo
Recent studies indicate that the host immune system plays a key role in radiation-mediated tumor suppression [1, 2, 4, 49, 50]. We then extend our study to an immune competent mouse tumor model. Mouse sarcoma CMS4-met cells were injected into syngeneic mice. The established tumors were irradiated with an external beam of radiation (8 Gy) and treated with BV6 as in the xenograft tumor model. Surprisingly, although both radiation and BV6 suppressed the xenograft tumor growth (Figure 11A), neither radiation nor BV6 suppressed the synergetic tumor growth in this immune competent syngeneic mouse tumor model (Figure 11D). Although a radiation dose of 8 Gy was commonly used in mouse tumor models [5], recent studies have shown that a higher dose of external beam radiation (20-40 Gy) is more effective at inducing an anti-tumor immune response [49]. We therefore increased the radiation dose to 20 Gy. One mouse in the radiation group and one mouse in the combined treatment group did not respond to radiotherapy due to a missed external beam of radiation of the tumor and therefore were excluded from the study. Analysis of tumor growth kinetics indicates that tumor volume was steadily increased across the measurement days in the control and BV6-treated mice. A slight increase in tumor volume was seen from day 0 to 2 with little to no change from day 2 to 8 in the radiation group. No change in tumor volume was seen across the measurement days in the combined radiation and BV6 group, indicating complete tumor suppression (Figure 11E). Within day 2 there were no significant differences between the 4 groups. Within day 4, the combined radiation and BV6 group had significantly lower tumor volume than the BV6 only group (p < 0.01). Within day 6, the combined radiation and BV6 group had significantly lower tumor volume than the BV6 only and control groups, and the radiation only group had significantly lower tumor volume than the BV6 only group. Within day 8, the combined radiation and BV6 group had significantly lower tumor volume than the BV6 only and control groups (p < 0.01).
Radiation induces a Th1/Tc1 T cell response in the tumor microenvironment in the syngeneic tumor-bearing mice
We next used quantitative PCR to analyze tumor tissues for the effects of radiation on the expression of signature genes for T cells and T cell linage-specific transcription factors. Tumors were excised and extracted for total RNA. Real-time RT-PCR analysis revealed that radiation induces an increase of CD4 and CD8 T cells (Figure 12A). Furthermore, the expression levels of IFNβ and t-Bet, the signature genes for Tc1/Th1 T cells, are also dramatically induced by radiation (Figure 12B). We also observed that radiation up-regulates perforin and granzyme B, two essential effectors of the perforin pathway. FasL is also up-regulated in the tumor microenvironment (Figure 12A). We next sought to determine whether radiation activates the STING pathway [49, 51] to activate T cells in the sarcoma tumor microenvironment. Quantitative PCR analysis revealed that IRF3 and IFNβ, two key mediators of the STING pathway, is up-regulated (Figure 12C). It has been reported that NF-κB is a transcriptional activator of the T cell chemokines CCL2 and CCL5 [52]. Analysis of the tumor tissues revealed that radiation up-regulates both CCL2 and CCL5 (Figure 12D). In addition, consistent with what was observed in irradiated tumor cell lines in vitro, Fas and TNFα are up-regulated by radiation in the tumor microenvironment (Figure 12E). Taken together, our data suggest that radiation not only activates the STING pathway to induce Th1/Tc1 T cell activation but also activates the canonical NF-κB to increase CCL2 and CCL5 to induce Th1/Tc1 T cell tumor infiltration.
Figure 12. Radiation induces T cell immune responses in the tumor microenvironment.
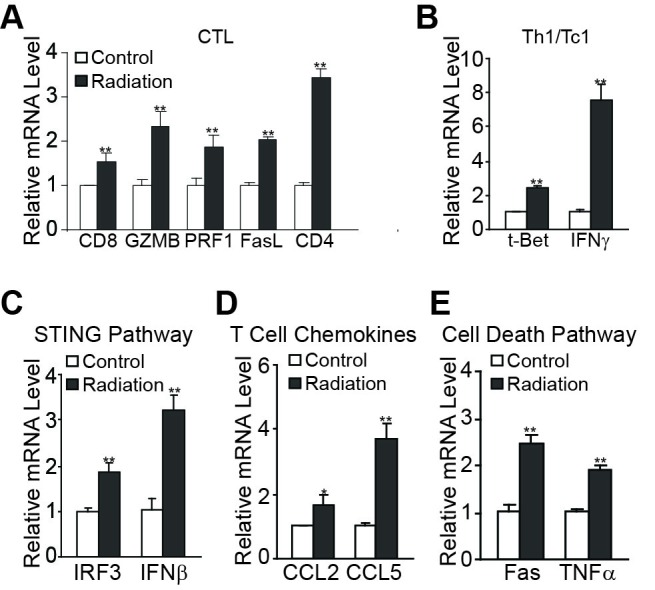
Tumors of CMS4-met tumor-bearing mice were irradiated at a dose of 20Gy as shown in Figure 11E. Tumors were excised 8 days after radiation and RNA was isolated from the tumor tissues of untreated control and irradiated mice and analyzed by quantitative PCR for signature genes of T cells (A.), Th1/Tc1 T cells (B.), the STING pathway (C.), T cell chemokines (D.), and TNF cell death pathways (E.). RNA from tumor tissues of three irradiated mice was pooled and RNA from tumor tissues of three untreated mice was pooled and used to prepare cDNA. The expression levels of genes in the control group were arbitrarily set to 1 for each subset of genes. The expression level of genes in the irradiated group is expressed as fold change over the control. *p<0.05, **p<0.01.
DISCUSSION
Radiation is known to activate NF-κB [34, 53-56], and NF-κB has been shown to confer tumor cell radioresistance [54]. However, NF-κB has also been shown to be required for radiation-induced cell death [34, 52]. In this study, we demonstrated that radiation rapidly induces activation of the canonical NF-κB. We further demonstrated that radiation activates both the p65/p50 heterodimer and p50/p50 homodimer of the canonical NF-κB that directly binds to the TNFα and FAS promoters, respectively, to activate their expression. NF-κB-activated TNFα then induces tumor cell death in an autocrine mechanism, whereas increased Fas in the tumor cells may increase tumor cell sensitivity to FasL-mediated cytotoxicity of tumor-reactive CTLs in the tumor microenvironment [57]. Therefore, we determine that radiation suppresses tumor cell growth at least in part through activation of the NF-κB-TNFα/Fas axis in the target tumor cells.
In agreement with early reports that BV6 induce cIAP1 and cIAP2 degradation to increase NIK stability to activate the alternate NF-κB [21, 58], we observed that BV6 induces the alternate NF-κB activation. However, the alternate NF-κB activation occurs at a late stage after BV6 treatment. The role of alternate NF-κB in BV6-mediated sarcoma and colon carcinoma cell death requires further study.
It is known that TNFα induces both apoptosis and necroptosis and these two cell death pathways often compensate for each other [59, 60]. Strikingly, we demonstrated that BV6 enhances TNFα function in the induction of tumor cell apoptosis and necroptosis. However, the BV6 and TNFα-induced apoptosis and necroptosis pathways do not compensate for each other since blocking either caspases alone or RIP1/RIP3 alone still inhibits cell death. It has also been shown that cIAP1 and cIAP2 use their E3 ligase activity to ubiquitylate RIP1 in order to mediate caspase 8 activation [61]. RIP1 is critical for caspase-8 activation-induced by Smac mimetic [61]. In this study, we observed that inhibition of RIP1 but not RIP3 diminishes caspase 8 activation, whereas inhibition of RIP1 or RIP3 both blocks radiation/TNFα-induced cell death. These results suggest that RIP1 is required for both apoptosis and necroptosis induced by radiation or TNFα and that RIP1 acts upstream of caspase 8.
The host immune system plays an essential role in radiation-mediated tumor suppression [1, 2, 4, 49, 50, 56]. In this study, we observed that radiation induced a dramatic increase in both CD4 and CD8 T cells in the tumor microenvironment. Furthermore, the tumor-infiltrating T cells are preferentially Tc1 and Th1 phenotypes as characterized by the increased expression of signature genes tBet and IFNβ. These tumor-infiltrating T cells are also activated by radiation since effector molecules such as perforin, granzyme B, and FasL are all high in the radiation-treated tumor than in untreated control tumors. It has recently been shown that antigen presenting cells take dsDNA fragments from irradiated tumor cells to activate the STING pathway in order to activate tumor-reactive CTLs [49, 51]. We observed here that radiation induces IRF3 and IFNβ, two key mediators of the STING pathway, in the tumor microenvironment. Therefore, radiation may activate the STING pathway to induce Tc1/Th1 T cell activation in the sarcoma tumor microenvironment.
In addition, it is possible that radiation-induced NF-κB activation in tumor cells might also be the link to radiation-induced T cell infiltration and activation in the tumor microenvironment [35]. It has recently been shown that NF-κB enhances the expression of T cell chemokines CCL2 and CCL5 to increase T cell tumor infiltration and tumor rejection in mouse tumor models and human lung cancer patients [52]. NF-κB activation in dying cells also determines cross-prming of CD8+ T cells [31]. We observed that radiation rapidly activates NF-κB in human sarcoma cells and increases both CD4+ and CD8+ T cells in the tumor microenvironment. Increased T cell tumor infiltration is correlated with increased CCL2 and CCL5 expression in the tumor tissues. It is thus likely that, in addition to the activation of the STING pathway to activate T cells, NF-κB may also up-regulate the T cell chemokines CCL2 and CCL5 in the tumor cells to attract T cell infiltration into the tumor microenvironment. Therefore, it is likely that radiation not only induces activation of the STING pathways to activate the Th1/Tc1 T cells but also up-regulates NF-κB to activate CCL2 and CCL5 to attract the activated Th1/Tc1 cells infiltration into the tumor microenvironment to suppress tumor development. CTL infiltration level in human sarcoma varies greatly from patients to patients. Analysis of tumor tissues from six human sarcoma patients indicates that half of the tumor tissues exhibit few to no CTL infiltration (Figure 13). Therefore, radiation-induced CTL infiltration may be a significant contributor for its tumor suppression efficacy.
Figure 13. CD8+ cytotoxic T lymphocyte infiltration levels in human sarcoma.

Six Human fibrosarcoma tissues were stained with CD8-specific mAb. Brown color indicates tumor-infiltrating CD8+ CTLs. Brown color indicates CD8-specific staining, and the tissues were counterstained with hematoxylin. Each panel represents representative image of specimen from one patient.
We propose that radiation initiates at least four bystander events (Figure 14): 1) radiation activates the canonical NF-κB p65/p50 and p50/p50 complexes to up-regulate TNFα expression. Increased TNFα in the tumor microenvironment induces tumor apoptosis and necroptosis in an autocrine manner; 2) radiation-activated canonical NF-κB p65/p50 and p50/p50 complexes activate Fas expression to increase tumor cells to apoptosis induction by FasL of tumor-infiltrating Tc1 cells [5, 57, 62]; 3) radiation-activated NF-κB also up-regulates CCL2 and CCL5 in tumor cells to attract activated T cell infiltration to the tumor microenvironment; and 4) radiation activates the STING pathway to induce T cell activation. On the other hand, RIP1 acts as molecular switch that determines whether TNFα induces cell death or survival. cIAP1/2 ubiquitylates RIP1 in the TNFR cytoplasmic complex (complex I) and the ubiquitylated RIP1 prevents the formation of the death complex IIb to promote cell survival [59, 63]. BV6 induces cIAP1 and cIAP2 degradation. Degradation of cIAP1 and cIAP2 leads to two events: first, caspase 9 activation; and second, deubiquitylation of RIP1. Deubiquitylated RIP1 dissociates from the membrane-bound TNFR complex I to form cytosolic complex IIb that mediates both caspase-dependent apoptosis and RIP3-dependent necroptosis. Thus, radiation-induced NF-κB functions as a key molecular link between tumor cells and immune cells in the tumor microenvironment for radiation-mediated tumor suppression.
Figure 14. Model of radiation modulation of NF-κB and CTL anti-tumor immune response in the tumor microenvironment.
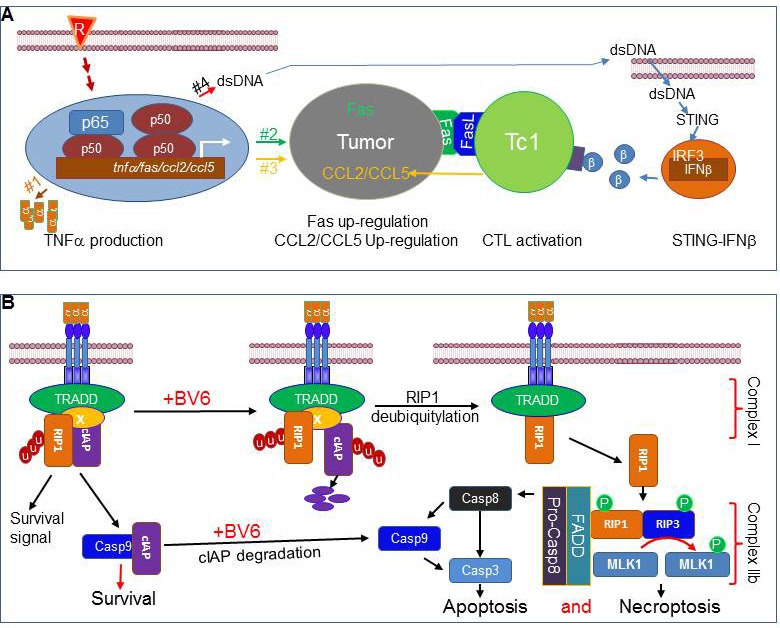
A. Radiation initiates at least four molecular effects: 1) radiation activates the canonical NF-κB p65/p50 and p50/p50 complexes that directly bind to the TNFα promoter to activate its transcription; 2) radiation-activated canonical NF-κB p65/p50 and p50/p50 complexes also directly bind to the FAS promoter to activate its transcription to increase tumor cell sensitivity to apoptosis induction by FasL of tumor-infiltrating Tc1 cells; 3) radiation-activated NF-κB also up-regulates the T cell chemokines CCL2 and CCL5 to attract T cell infiltration to the tumor microenvironment; and 4) radiation-generated dsDNA fragments activate the STING pathway, as characterized by increased IRF3 and IFNβ expression, to induce T cell activation. B. TNFα in the tumor microenvironment induces tumor apoptosis and necroptosis in an autocrine manner, and RIP1 is a key molecular switch that determines whether TNFα induces cell death or survival signaling pathways. cIAP1 and cIAP2 ubiquitylates RIP1 in the TNFR cytoplasmic complex (complex I) and the ubiquitylated RIP1 promotes cell survival by preventing the formation of the death complex IIb. BV6 induces cIAP1 and cIAP2 degradation. Degradation of cIAP1 and cIAP2 leads to activation of caspase 9. Degradation of cIAP1 and cIAP2 also leads to deubiquitylation of RIP1. Deubiquitylated RIP1 dissociates from the membrane-bound TNFR complex I to form cytosolic complex IIb that mediates both caspase-dependent apoptosis and RIP3-dependent necroptosis.
MATERIALS AND METHODS
Mice and tumor cells
Athymic nude mice (nu/nu) were obtained from Taconic Farm Inc. BALB/c mice were obtained for Charles River Laboratory. All mouse studies were carried out according to an approved protocol by Georgia Regents University Institutional Animal Care and Use Committee. All human tumor cell lines used in this study were obtained from American Type Culture Collection (Manassas, VA). ATCC has characterized these cells by morphology, immunology, DNA fingerprint, and cytogenetics.
Reagents
BV6 was provided by Genetech Inc. TNFα were obtained from R&D System. Z-VAD was obtained from ApexBio. Necrostatin-1 (Nec1) was obtained from Selleckchem. Necrosulfonamide (NSA) was obtained from Calbiochem.
Radiation
Tumor cells were suspended in culture medium and irradiated with the MarK 1 Irradiator (Shepherd & Associates, San Fernando, CA). Tumors of tumor bearing mice were irradiated using the SARRP Irradiator System (Xstrahl Life Sciences Suwanee, GA). Mice were anesthetized under constant isoflurane/O2 flow and tumors were localized with the SARRP imaging system. Tumors were then irradiated with external beams of x-ray at the designated doses.
Immunohistochemistry
Colon cancer tissue microarray (TMA) slides were provided by the National Cancer Institute (NCI)-sponsored cooperative human tissue network. The TMAs were designed by NCI statisticians for high statistical power for examination of associations of markers with tumor stage, clinical outcome, and other clinic-pathologic variables. TMA slides were stained with anti-cIAP antibody (R&D System), and counterstained with hematoxylin (Richard-Allan Scientific, Kalamazoo, MI) as previously described [64].
Tumor cell death assay
For cell death analysis, cells were stained with Propidium Iodide and analyzed by flow cytometry as previously described [64].
Western blotting analysis
Western blotting analysis was performed as previously described [65]. Anti-cleaved caspase 8 (Cat#: AF705, 0.5μg/ml) and anti-cIAP1 (Cat#: AF8181, 1:500) were obtained from R&D systems. Anti-caspase 3 (Cat#: 9661, 1:1000), anti-caspase 9 (Cat#: 9501, 1:500), anti-PARP (Cat#: 9541, 1:250), anti-xIAP (Cat#: 2042, 1:500), anti-p100/p52 (Cat#: 4882, 1:1000), anti-IκBα (Cat#: 4814, 1:2000), anti-pIκBα (Cat#: 2859, 1:1000) antibodies were obtained from Cell Signaling. Anti-cIAP2 (Cat#: NB110-57030, 1:250) was obtained from Novus. Anti-β-actin (Cat#: A5441, 1:5000) was obtained from Sigma (St Louis, MO).
Electrophoresis mobility shift assay (EMSA) of NF-κB activation
NF-κB activation was analyzed using NF-κB probe (Santa Cruz Biotech) (Table S1), and probes with NF-κB consensus sequences of the TNFα and Fas promoter regions (Table S1) as previously described [66]. Briefly, the end-labeled probes were incubated with nuclear extracts for 20 min at room temperature. Anti-p65, p52, p50, RelB and cRel antibodies (Santa Cruz Biotech) were included to identify specific NF-κB-DNA complexes. DNA-protein complexes were separated by electrophoresis in 6% polyacrylamide gels and identified using a phosphoimage screen (Molecular Dynamics) and the images were acquired using a Strom 860 imager (Molecular Dynamics).
Subcellular localization analysis
SW620 cells were cultured for 24 hours and were untreated, treated with BV6 (5μM), treated with TNFα (100 U/ml), or treated with BV6 and TNFα for one hour. Cells were fixed with 4% paraformaldehyde for 30 minutes, rinsed with PBS, blocked for one hour, and incubated with anti-p65 antibody (Santa Cruz) for one hour. The cells were then washed three times with PBS and incubated with a Cy5-conjugated anti-rabbit IgG for one hour. The cells were washed four times with PBS and followed by incubation with HOECHST for 5 minutes. The cells were analyzed by confocal microscopy.
Gene expression analysis
Fresh tumor tissues were homogenized in Trizol (Life Technologies) to isolate total RNA. cDNA was synthesized from total RNA and used for analysis of gene expression using gene-specific primers (Table S1) in the StepOne Plus Real-Time PCR System (Applied Biosystems).
Tumor tissue proliferation and apoptosis
Tumor tissues were embedded in paraffin and sections were stained with anti-Ki67 antibody (BD Biosciences), and the ApopTag® Plus Peroxidase in situ Apoptosis Kit (Millipore, Billerica, MA), respectively according to the manufacturer's instructions.
TNFα neutralization
Tumor cells were irradiated and then cultured in the presence of BV6 plus either IgG (20 μg/ml) or anti-TNFα mAb (Biolegend, 20 μg/ml) for 24h. Cell death was then measured by PI staining and flow cytometry.
Statistical analysis
Statistical analysis was performed using SAS 9.4 and statistical significance was assessed using an alpha level of 0.05. A repeated measures mixed model was used to examine differences in tumor volume between treatment groups across days. Kaplan-Meier analysis was used to examine differences in time to recurrence and survival time by cIAP1 protein level. Student's t test was also used to compare differences between control and treatment groups. A p<0.05 was taken as statistically significant.
SUPPLEMENTARY TABLE
Acknowledgments
We thank Dr. Hong Zhang for assistance in external beam radiation of tumors and Ms. Kimya Jones for assistance in immunohistochemistry.
Footnotes
CONFLICTS OF INTEREST
The authors have declared that no conflict of interest exists
GRANT SUPPORT
NIHCA182518 (to K.L.), NIHU54CA168512 (to R.E.P.).
Editorial note
This paper has been accepted based in part on peerreview conducted by another journal and the authors' response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
- 1.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, Herati RS, Mansfield KD, Patsch D, Amaravadi RK, Schuchter LM, Ishwaran H, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–377. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang C, Wang X, Soh H, Seyedin S, Cortez MA, Krishnan S, Massarelli E, Hong D, Naing A, Diab A, Gomez D, Ye H, Heymach J, Komaki R, Allison JP, Sharma P, et al. Combining radiation and immunotherapy: a new systemic therapy for solid tumors? Cancer Immunol Res. 2014;2:831–838. doi: 10.1158/2326-6066.CIR-14-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11:239–253. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- 4.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124:687–695. doi: 10.1172/JCI67313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chakraborty M, Abrams SI, Camphausen K, Liu K, Scott T, Coleman CN, Hodge JW. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J Immunol. 2003;170:6338–6347. doi: 10.4049/jimmunol.170.12.6338. [DOI] [PubMed] [Google Scholar]
- 6.Lee H, Rotolo JA, Mesicek J, Penate-Medina T, Rimner A, Liao WC, Yin X, Ragupathi G, Ehleiter D, Gulbins E, Zhai D, Reed JC, Haimovitz-Friedman A, Fuks Z, Kolesnick R. Mitochondrial ceramide-rich macrodomains functionalize Bax upon irradiation. PLoS One. 2011;6:e19783. doi: 10.1371/journal.pone.0019783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Barros M, Thin TH, Maj J, Cordon-Cardo C, Haimovitz-Friedman A, Fuks Z, Kolesnick R. Impact of stromal sensitivity on radiation response of tumors implanted in SCID hosts revisited. Cancer Res. 2010;70:8179–8186. doi: 10.1158/0008-5472.CAN-10-1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garnett-Benson C, Hodge JW, Gameiro SR. Combination regimens of radiation therapy and therapeutic cancer vaccines: mechanisms and opportunities. Semin Radiat Oncol. 2015;25:46–53. doi: 10.1016/j.semradonc.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gameiro SR, Ardiani A, Kwilas A, Hodge JW. Radiation-induced survival responses promote immunogenic modulation to enhance immunotherapy in combinatorial regimens. Oncoimmunology. 2014;3:e28643. doi: 10.4161/onci.28643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Simone V, Franze E, Ronchetti G, Colantoni A, Fantini MC, Di Fusco D, Sica GS, Sileri P, MacDonald TT, Pallone F, Monteleone G, Stolfi C. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene. 2015;34:3493–3503. doi: 10.1038/onc.2014.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Donatis GM, Pape EL, Pierron A, Cheli Y, Hofman V, Hofman P, Allegra M, Zahaf K, Bahadoran P, Rocchi S, Bertolotto C, Ballotti R, Passeron T. NF-kB2 induces senescence bypass in melanoma via a direct transcriptional activation of EZH2. Oncogene. 2015 doi: 10.1038/onc.2015.331. [DOI] [Google Scholar]
- 12.Xi G, Hayes E, Lewis R, Ichi S, Mania-Farnell B, Shim K, Takao T, Allender E, Mayanil CS, Tomita T. CD133 and DNA-PK regulate MDR1 via the PI3K- or Akt-NF-kappaB pathway in multidrug-resistant glioblastoma cells in vitro. Oncogene. 2016;35:241–50. doi: 10.1038/onc.2015.78. [DOI] [PubMed] [Google Scholar]
- 13.Bonavia R, Inda MM, Vandenberg S, Cheng SY, Nagane M, Hadwiger P, Tan P, Sah DW, Cavenee WK, Furnari FB. EGFRvIII promotes glioma angiogenesis and growth through the NF-kappaB, interleukin-8 pathway. Oncogene. 2012;31:4054–4066. doi: 10.1038/onc.2011.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 16.Gilmore TD, Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006;25:6887–6899. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- 17.Brady RR, Loveridge CJ, Dunlop MG, Stark LA. c-Src dependency of NSAID-induced effects on NF-kappaB-mediated apoptosis in colorectal cancer cells. Carcinogenesis. 2011;32:1069–1077. doi: 10.1093/carcin/bgr077. [DOI] [PubMed] [Google Scholar]
- 18.Stark LA, Reid K, Sansom OJ, Din FV, Guichard S, Mayer I, Jodrell DI, Clarke AR, Dunlop MG. Aspirin activates the NF-kappaB signalling pathway and induces apoptosis in intestinal neoplasia in two in vivo models of human colorectal cancer. Carcinogenesis. 2007;28:968–976. doi: 10.1093/carcin/bgl220. [DOI] [PubMed] [Google Scholar]
- 19.Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, Brink R, Leverkus M, Tergaonkar V, Schneider P, Callus BA, Koentgen F, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131:682–693. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 20.Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, Harran P, Wang X. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell. 2007;12:445–456. doi: 10.1016/j.ccr.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Cui H, Schroering A, Ding JL, Lane WS, McGill G, Fisher DE, Ding HF. NF-kappa B2 p100 is a pro-apoptotic protein with anti-oncogenic function. Nat Cell Biol. 2002;4:888–893. doi: 10.1038/ncb872. [DOI] [PubMed] [Google Scholar]
- 23.Ishikawa H, Carrasco D, Claudio E, Ryseck RP, Bravo R. Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-kappaB2. J Exp Med. 1997;186:999–1014. doi: 10.1084/jem.186.7.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW. Control of the senescence-associated secretory phenotype by NF-{kappa}B promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–2136. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jing H, Kase J, Dorr JR, Milanovic M, Lenze D, Grau M, Beuster G, Ji S, Reimann M, Lenz P, Hummel M, Dorken B, Lenz G, Scheidereit C, Schmitt CA, Lee S. Opposing roles of NF-{kappa}B in anti-cancer treatment outcome unveiled by cross-species investigations. Genes Dev. 2011;25:2137–2146. doi: 10.1101/gad.17620611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klein U, Ghosh S. The Two Faces of NF-kappaB Signaling in Cancer Development and Therapy. Cancer Cell. 2011;20:556–558. doi: 10.1016/j.ccr.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crescenzi E, Pacifico F, Lavorgna A, De Palma R, D'Aiuto E, Palumbo G, Formisano S, Leonardi A. NF-kappaB-dependent cytokine secretion controls Fas expression on chemotherapy-induced premature senescent tumor cells. Oncogene. 2011;30:2707–2717. doi: 10.1038/onc.2011.1. [DOI] [PubMed] [Google Scholar]
- 28.Karl S, Pritschow Y, Volcic M, Hacker S, Baumann B, Wiesmuller L, Debatin KM, Fulda S. Identification of a novel pro-apopotic function of NF-kappaB in the DNA damage response. J Cell Mol Med. 2009;13:4239–4256. doi: 10.1111/j.1582-4934.2009.00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jennewein C, Karl S, Baumann B, Micheau O, Debatin KM, Fulda S. Identification of a novel pro-apoptotic role of NF-kappaB in the regulation of TRAIL- and CD95-mediated apoptosis of glioblastoma cells. Oncogene. 2012;31:1468–1474. doi: 10.1038/onc.2011.333. [DOI] [PubMed] [Google Scholar]
- 30.Thoms HC, Loveridge CJ, Simpson J, Clipson A, Reinhardt K, Dunlop MG, Stark LA. Nucleolar targeting of RelA(p65) is regulated by COMMD1-dependent ubiquitination. Cancer Res. 2010;70:139–149. doi: 10.1158/0008-5472.CAN-09-1397. [DOI] [PubMed] [Google Scholar]
- 31.Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, Green DR, Oberst A, Albert ML. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science. 2015;350:328–334. doi: 10.1126/science.aad0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rovillain E, Mansfield L, Caetano C, Alvarez-Fernandez M, Caballero OL, Medema RH, Hummerich H, Jat PS. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene. 2011;30:2356–2366. doi: 10.1038/onc.2010.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu X, Zimmerman MA, Bardhan K, Yang D, Waller JL, Liles GB, Lee JR, Pollock R, Lev D, Ware CF, Garber E, Bailly V, Browning JL, Liu K. Lymphotoxin beta receptor mediates caspase-dependent tumor cell apoptosis in vitro and tumor suppression in vivo despite induction of NF-kappaB activation. Carcinogenesis. 2013;34:1105–1114. doi: 10.1093/carcin/bgt014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berger R, Jennewein C, Marschall V, Karl S, Cristofanon S, Wagner L, Vellanki SH, Hehlgans S, Rodel F, Debatin KM, Ludolph AC, Fulda S. NF-kappaB is required for Smac mimetic-mediated sensitization of glioblastoma cells for gamma-irradiation-induced apoptosis. Mol Cancer Ther. 2011;10:1867–1875. doi: 10.1158/1535-7163.MCT-11-0218. [DOI] [PubMed] [Google Scholar]
- 35.Ruefli-Brasse AA, French DM, Dixit VM. Regulation of NF-kappaB-dependent lymphocyte activation and development by paracaspase. Science. 2003;302:1581–1584. doi: 10.1126/science.1090769. [DOI] [PubMed] [Google Scholar]
- 36.Espinosa L, Margalef P, Bigas A. Non-conventional functions for NF-kappaB members: the dark side of NF-kappaB. Oncogene. 2015;34:2279–2287. doi: 10.1038/onc.2014.188. [DOI] [PubMed] [Google Scholar]
- 37.Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012:4. doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gerondakis S, Fulford TS, Messina NL, Grumont RJ. NF-kappaB control of T cell development. Nat Immunol. 2014;15:15–25. doi: 10.1038/ni.2785. [DOI] [PubMed] [Google Scholar]
- 39.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 41.Phillips AH, Schoeffler AJ, Matsui T, Weiss TM, Blankenship JW, Zobel K, Giannetti AM, Dueber EC, Fairbrother WJ. Internal motions prime cIAP1 for rapid activation. Nat Struct Mol Biol. 2014;21:1068–1074. doi: 10.1038/nsmb.2916. [DOI] [PubMed] [Google Scholar]
- 42.Paschall AV, Zimmerman MA, Torres CM, Yang D, Chen MR, Li X, Bieberich E, Bai A, Bielawski J, Bielawska A, Liu K. Ceramide targets xIAP and cIAP1 to sensitize metastatic colon and breast cancer cells to apoptosis induction to suppress tumor progression. BMC Cancer. 2014;14:24. doi: 10.1186/1471-2407-14-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kavanagh E, Rodhe J, Burguillos MA, Venero JL, Joseph B. Regulation of caspase-3 processing by cIAP2 controls the switch between pro-inflammatory activation and cell death in microglia. Cell Death Dis. 2014;5:e1565. doi: 10.1038/cddis.2014.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Varfolomeev E, Alicke B, Elliott JM, Zobel K, West K, Wong H, Scheer JM, Ashkenazi A, Gould SE, Fairbrother WJ, Vucic D. X chromosome-linked inhibitor of apoptosis regulates cell death induction by proapoptotic receptor agonists. J Biol Chem. 2009;284:34553–34560. doi: 10.1074/jbc.M109.040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Varfolomeev E, Goncharov T, Maecker H, Zobel K, Komuves LG, Deshayes K, Vucic D. Cellular Inhibitors of Apoptosis Are Global Regulators of NF-kappaB and MAPK Activation by Members of the TNF Family of Receptors. Sci Signal. 2012;5:ra22. doi: 10.1126/scisignal.2001878. [DOI] [PubMed] [Google Scholar]
- 46.Liu F, Bardhan K, Yang D, Thangaraju M, Ganapathy V, Waller JL, Liles GB, Lee JR, Liu K. NF-kappaB Directly Regulates Fas Transcription to Modulate Fas-mediated Apoptosis and Tumor Suppression. J Biol Chem. 2012;287:25530–25540. doi: 10.1074/jbc.M112.356279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Urresti J, Ruiz-Meana M, Coccia E, Arevalo JC, Castellano J, Fernandez-Sanz C, Galenkamp KM, Planells-Ferrer L, Moubarak RS, Llecha-Cano N, Reix S, Garcia-Dorado D, Barneda-Zahonero B, Comella JX. Lifeguard inhibits Fas ligand-mediated endoplasmic reticulum-calcium release mandatory for apoptosis in type II apoptotic cells. J Biol Chem. 2016;291:1221–34. doi: 10.1074/jbc.M115.677682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galenkamp KM, Carriba P, Urresti J, Planells-Ferrer L, Coccia E, Lopez-Soriano J, Barneda-Zahonero B, Moubarak RS, Segura MF, Comella JX. TNFalpha sensitizes neuroblastoma cells to FasL-, cisplatin- and etoposide-induced cell death by NF-kappaB-mediated expression of Fas. Mol Cancer. 2015;14:62. doi: 10.1186/s12943-015-0329-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, Huang X, Gajewski TF, Chen ZJ, Fu YX, Weichselbaum RR. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41:843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang H, Deng L, Chmura S, Burnette B, Liadis N, Darga T, Beckett MA, Lingen MW, Witt M, Weichselbaum RR, Fu YX. Radiation-induced equilibrium is a balance between tumor cell proliferation and T cell-mediated killing. J Immunol. 2013;190:5874–5881. doi: 10.4049/jimmunol.1202612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre ML, Gajewski TF. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hopewell EL, Zhao W, Fulp WJ, Bronk CC, Lopez AS, Massengill M, Antonia S, Celis E, Haura EB, Enkemann SA, Chen DT, Beg AA. Lung tumor NF-kappaB signaling promotes T cell-mediated immune surveillance. J Clin Invest. 2013;123:2509–2522. doi: 10.1172/JCI67250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brach MA, Hass R, Sherman ML, Gunji H, Weichselbaum R, Kufe D. Ionizing radiation induces expression and binding activity of the nuclear factor kappa B. J Clin Invest. 1991;88:691–695. doi: 10.1172/JCI115354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Veuger SJ, Hunter JE, Durkacz BW. Ionizing radiation-induced NF-kappaB activation requires PARP-1 function to confer radioresistance. Oncogene. 2009;28:832–842. doi: 10.1038/onc.2008.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu H, Mohan S, Natarajan M. Radiation-Triggered NF-kappaB Activation is Responsible for the Angiogenic Signaling Pathway and Neovascularization for Breast Cancer Cell Proliferation and Growth. Breast Cancer (Auckl) 2012;6:125–135. doi: 10.4137/BCBCR.S9592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hellweg CE. The Nuclear Factor kappaB pathway: A link to the immune system in the radiation response. Cancer Lett. 2015;368:275–289. doi: 10.1016/j.canlet.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 57.Caldwell SA, Ryan MH, McDuffie E, Abrams SI. The Fas/Fas ligand pathway is important for optimal tumor regression in a mouse model of CTL adoptive immunotherapy of experimental CMS4 lung metastases. J Immunol. 2003;171:2402–2412. doi: 10.4049/jimmunol.171.5.2402. [DOI] [PubMed] [Google Scholar]
- 58.Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh WC, Mak TW, Korneluk RG, Cheng G. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. 2008;9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15:362–374. doi: 10.1038/nri3834. [DOI] [PubMed] [Google Scholar]
- 60.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 61.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 62.Afshar-Sterle S, Zotos D, Bernard NJ, Scherger AK, Rodling L, Alsop AE, Walker J, Masson F, Belz GT, Corcoran LM, O'Reilly LA, Strasser A, Smyth MJ, Johnstone R, Tarlinton DM, Nutt SL, et al. Fas ligand-mediated immune surveillance by T cells is essential for the control of spontaneous B cell lymphomas. Nat Med. 2014;20:283–290. doi: 10.1038/nm.3442. [DOI] [PubMed] [Google Scholar]
- 63.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 64.Bardhan K, Paschall AV, Yang D, Chen MR, Simon PS, Bhutia YD, Martin PM, Thangaraju M, Browning DD, Ganapathy V, Heaton CM, Gu K, Lee JR, Liu K. IFNgamma induces DNA methylation-silenced GPR109A expression via pSTAT1/p300 and H3K18 acetylation in colon cancer. Cancer Immunol Res. 2015;3:795–805. doi: 10.1158/2326-6066.CIR-14-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paschall AV, Yang D, Lu C, Choi JH, Li X, Liu F, Figueroa M, Oberlies NH, Pearce C, Bollag WB, Nayak-Kapoor A, Liu K. H3K9 Trimethylation Silences Fas Expression To Confer Colon Carcinoma Immune Escape and 5-Fluorouracil Chemoresistance. J Immunol. 2015;195:1868–82. doi: 10.4049/jimmunol.1402243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Simon PS, Sharman SK, Lu C, Yang D, Paschall AV, Tulachan SS, Liu K. The NF-kappaB p65 and p50 homodimer cooperate with IRF8 to activate iNOS transcription. BMC Cancer. 2015;15:770. doi: 10.1186/s12885-015-1808-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.