

Summary
Background
Dysbiosis is associated with many diseases, including irritable bowel syndrome (IBS), inflammatory bowel diseases (IBD), obesity and diabetes. Potential clinical impact of imbalance in the intestinal microbiota suggests need for new standardised diagnostic methods to facilitate microbiome profiling.
Aim
To develop and validate a novel diagnostic test using faecal samples to profile the intestinal microbiota and identify and characterise dysbiosis.
Methods
Fifty‐four DNA probes targeting ≥300 bacteria on different taxonomic levels were selected based on ability to distinguish between healthy controls and IBS patients in faecal samples. Overall, 165 healthy controls (normobiotic reference collection) were used to develop a dysbiosis model with a bacterial profile and Dysbiosis Index score output. The model algorithmically assesses faecal bacterial abundance and profile, and potential clinically relevant deviation in the microbiome from normobiosis. This model was tested in different samples from healthy volunteers and IBS and IBD patients (n = 330) to determine the ability to detect dysbiosis.
Results
Validation confirms dysbiosis was detected in 73% of IBS patients, 70% of treatment‐naïve IBD patients and 80% of IBD patients in remission, vs. 16% of healthy individuals. Comparison of deep sequencing and the GA‐map Dysbiosis Test, (Genetic Analysis AS, Oslo, Norway) illustrated good agreement in bacterial capture; the latter showing higher resolution by targeting pre‐determined highly relevant bacteria.
Conclusions
The GA‐map Dysbiosis Test identifies and characterises dysbiosis in IBS and IBD patients, and provides insight into a patient's intestinal microbiota. Evaluating microbiota as a diagnostic strategy may allow monitoring of prescribed treatment regimens and improvement in new therapeutic approaches.
Introduction
Intestinal microbiota is generally comparable for individuals comprising the general adult population, with recent evidence supporting the gut microbiota as representing a healthy state defined as normobiosis.1, 2, 3 Notably, deviations from normobiosis can result in a transient or permanent microbiotic imbalance known as dysbiosis, which has been linked to several disorders, including Crohn's disease (CD), ulcerative colitis (UC), irritable bowel syndrome (IBS), obesity, nonalcoholic steatohepatitis, and type I and type II diabetes.4, 5, 6, 7, 8
Traditionally, evaluation of intestinal microbiota composition has been based on breath‐testing methods, small‐bowel culture techniques and culture‐independent techniques such as high‐throughput next‐generation sequencing.9, 10, 11 The use of these methods has significantly increased our understanding of the role of gut microbiota in health and disease10; for example, small intestinal bacterial overgrowth12 and altered intestinal microbiota13 are implicated in subgroups of patients with functional bowel disorders. Firm evidence for a causal role of microbiota composition on disease pathogenesis has, however, remained elusive due to inherent limitations in the diagnostic methods used. For instance, breath‐testing and culture techniques have not been validated, the majority of species cannot be cultured with standard methods, and the effect of potentially confounding polypharmacy has not been thoroughly evaluated.11, 14 Nevertheless, increasing awareness of the potential clinical impact of imbalance in the intestinal microbiota has led to a call for new standardised diagnostic methods, such as high‐throughput DNA sequencing, that facilitate profiling of the microbiome and possible differentiation between normobiosis and dysbiosis.15
Analysis of faecal samples from individuals with dysbiosis is anticipated to enable characterisation of the bacterial profile associated with different pathological conditions, thus aiding clinical diagnosis of pathological conditions and improving therapeutic regimens. Furthermore, detailed sequential profiling of intestinal microbiota over the course of a therapeutic regimen may allow for monitoring of inflammatory bowel disease (IBD) progression16 and the prediction of relapse, for example in CD.17 The ability to characterise the bacterial profiles both of normobiotic and dysbiotic patients may also help in evaluating the efficacy and further development of therapeutic approaches such as faecal microbiota transplantation (FMT), special diets and use of probiotics.17, 18, 19, 20, 21
In the present publication, a novel diagnostic test (GA‐map Dysbiosis Test, Genetic Analysis AS, Oslo, Norway) is evaluated that allows mapping of the intestinal microbiota profile for a selected set of bacteria, and used to identify and characterise dysbiosis in a clinical setting. The GA‐map Dysbiosis Test (GA‐test) is based on advances in DNA profiling using probes targeting variable regions (V3 to V7) of the bacterial 16S rRNA gene to characterise and identify bacteria present (Figure 1). The probes comprise a highly selective and specific bacterial probe set that is used with a unique algorithm to facilitate determination of dysbiosis level. The method provides a rapid, high‐throughput analysis of a large number of individual faecal samples. The breadth of knowledge gained from microbiome projects was used to develop a test aimed at characterising dysbiosis by deviation from a normobiotic state for use in a clinical diagnostic setting. For this purpose, the test was technically documented in accordance with EU requirements for an in vitro diagnostic test comprising the following intended use claim: ‘The GA‐test is intended to be used as a gut microbiota DNA analysis tool to identify and characterise dysbiosis’.
Figure 1.
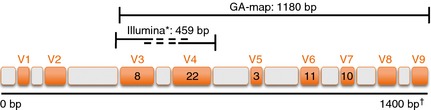
Target regions for the GA primer (1180 bp) and the Illumina primer (459 bp) showing variable (orange V1‐V9) and conserved (grey) regions in the bacterial 16S rRNA gene (1400 bp) utilised by the two methods. The numbers in V3 to V7 denote the number of GA probes targeting each variable region, in total 54 probes. *Illumina application note; http://res.illumina.com/documents/products/appnotes/appnote_16s_sequencing.pdf. †Position in E. coli (number of base pairs)40
Materials and methods
Human samples
Faecal samples were collected from 668 adults (aged 17–76; 69% women), including controls from healthy volunteers (n = 297) and patients with IBS (n = 236) and IBD (n = 135) (Table 1). Faecal samples were collected from hospitals in Norway, Sweden, Denmark and Spain (72%), as well as from workplaces in Oslo, Norway (28%), in an effort to achieve heterogeneity. The healthy donors had no clinical signs, symptoms or history of IBD, IBS or other organic gastrointestinal‐related disorders (e.g. colon cancer). Additional demographics are shown in Table 1, and sample inclusion and exclusion criteria are summarised in Data S1. The IBS samples were collected as part of prospective studies that used Rome II and III diagnostic criteria (depending on collection site) to identify IBS. The distribution of IBS subtypes was 44% IBS‐diarrhoea, 22% IBS‐alternating, 17% IBS‐constipation, 11% IBS‐unsubtyped and 4% IBS‐mixed. The diagnosis of IBD was based on clinical presentation confirmed by colonoscopy. Of the 135 IBD samples, 80 (59%) were treatment‐naïve patients and 55 (41%) were IBD patients in remission. The distribution of IBD types was 62% UC and 38% CD for the treatment‐naïve group, and 67% UC and 33% CD for the IBD in remission group. Informed consent was obtained for all samples along with approval from local scientific ethics committees. Samples were collected at home, office or hospital, and frozen within 3–5 days (for faecal sample collection, storage and processing, see Data S2).
Table 1.
Demographic information
| Categories | Total | Females (%) | Age (years)a | |
|---|---|---|---|---|
| Mean | Range | |||
| Healthy controls | 297 | 63 | 41 | 21–70 |
| Nordic | 254 | 64 | 42 | 21–70 |
| Danish | 19 | 63 | 42 | 23–61 |
| Spanish | 24 | 50 | 35 | 22–56 |
| IBSb | 236 | 78 | 40 | 17–76 |
| IBS‐D | 102 | 79 | 40 | 18–70 |
| IBS‐C | 41 | 85 | 42 | 22–73 |
| IBS‐M | 10 | 80 | 37 | 19–55 |
| IBS‐U | 25 | 88 | 41 | 19–68 |
| IBS‐A | 51 | 67 | 39 | 20–62 |
| IBD treatment‐naïve | 80 | 56 | 34 | 18–61 |
| CD | 30 | 50 | 33 | 19–53 |
| UC | 50 | 63 | 35 | 18–61 |
| IBD remissionc | 55 | 76 | 42 | 20–69 |
| CD | 18 | 72 | 38 | 20–59 |
| UC | 36 | 78 | 44 | 24–69 |
A, alternating; C, constipation; CD, Cohn's disease; D, diarrhoea; IBS, irritable bowel syndrome; IBD, inflammatory bowel disease; M, mixed; U, unsubtyped; UC, ulcerative colitis.
Precise ages were known for 99% of the total samples used.
IBStype known for 97% of the total IBS samples used.
CD/UC diagnosis known for 99% of the total IBD samples used.
Probe identification, selection, in silico and in vitro testing
To establish and optimise the most applicable bacterial probeset, data from previous IBD and IBS intestinal microbiota research was compiled based on pre‐defined search criteria (Data S3) to provide >500 bacterial observations associated with the occurrence of IBD and IBS. From a combined dataset of 496 16S rRNA gene sequences (consensus sequence[s] for each species, chosen from all available long 16S rRNA sequences and purified to avoid sequences errors) from 269 bacterial species, probes were designed to cover major bacterial observations made from the literature. All probes were designed according to Vebø et al.22 with a minimum melting temperature (T m) of 60 °C by the nearest‐neighbour method23 for the target group where the nucleotide 3′ end of the probe is a cytosine; nontarget group probe requirements were a T m of 30 °C or absence of a cytosine as the nucleotide adjacent to the 3′ end of the probe. Each probe was designed to target a bacterial species or group, i.e. Faecalibacterium prausnitzii (species), Lactobacillus (genus), Clostridia (class) and Proteobacteria (phylum), based on their 16S rRNA sequence (V3–V9). Probes that satisfied target detection and nontarget exclusion in silico were evaluated for cross‐labelling, self‐labelling and cross hybridisation before final validation was performed against bacterial strains in vitro.
After in vitro testing, a panel of 124 optimal probes was further selected using variable selection methods: variable importance in projection, selectivity ratio and interval partial least squares using data from a selection of healthy and IBS samples (data not shown). The variables (probes) were selected based on their ability to distinguish between samples isolated from healthy individuals and IBS patients. A final panel of 54 probes was selected covering the sites across V3 to V7 on the 16S rRNA sequence (Figure 1). Bacterial target specificity, tested with the 54‐probe set against 368 available single bacterial strains (Data S4), was performed to define the target bacteria for each probe. The probes detect bacteria within the six phyla; Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria, Tenericutes and Verrucomicrobia, covering 10 taxonomic bacterial classes and 36 genera (for more details on the bacterial targets for the 54 probes see Data S5).
Sample preparation and detection
The GA‐test is based on regular molecular biology techniques, comprising human faecal sample homogenisation and mechanical bacterial cell disruption; automated total bacterial gDNA extraction using magnetic beads; 16S rRNA PCR DNA amplification covering V3–V9; probe labelling by single nucleotide extension; hybridisation to complementary probes coupled to magnetic beads; and signal detection using BioCode 1000A 128‐Plex Analyzer (Applied BioCode, Santa Fe Springs, CA, USA). The method is described in detail in Data S2, and an overview to the whole process from sample preparation to result is shown in Figure 2.
Figure 2.
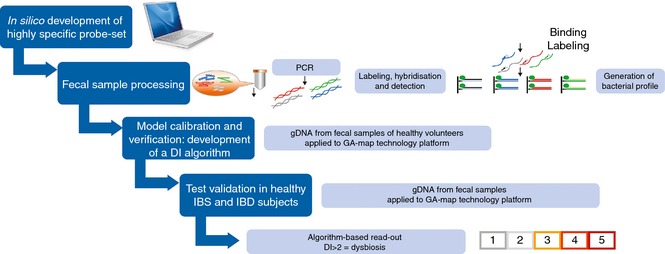
Flow chart illustrating the GA‐map Dysbiosis Test development, starting with in silico development of bacterial probe set, standardisation of laboratory analysis process, model calibration and verification in healthy individuals (normobiotic reference collection), and validation in healthy, IBS and IBD individuals. Derivation of a DI based on bacterial 16S rRNA DNA analysis in faecal samples demonstrates that a DI score >2 confirms microbiota profile deviations from the normobiotic reference collection.
Data pre‐processing
To ensure high quality assurance, several quality control criteria were applied to the detection data for each sample: (i) a bead count >2 for each probe; (ii) the hybridisation control (HYC) median signal >13 000; (iii) a median background signal <500 and (iv) a universal control median signal >4500. Normalisation was applied by first dividing the signal intensity of each probe in each sample by the signal intensity for HYC for that sample, and multiplying by 1000. This was done to adjust for sample differences due to pipetting or hybridisation. Subsequently, normalisation to adjust for run differences was applied by dividing the HYC‐normalised signal of each probe in each sample by the median HYC‐normalised signal of each probe for replicates of a synthetic DNA control (Data S2; Table S1), and multiplying by 1000. Prior to normobiotic microbiota profile calibration, normalised signal intensities below 15 were set to 0 to remove for low background noise and data was mean centred. Test and validation samples were normalised, and normalised signals below 15 were set to 0 before data were mean centred using mean probe signals from the normobiotic reference cohort.
Dysbiosis test development and validation
Principal component analysis (PCA)24 was used to build a normobiotic microbiota profile (model). The boundary between nondysbiotic and dysbiotic was determined by calculating confidence regions for the values of Hotelling's T‐square and Q statistics given by PCA scores in the model. Geometrically this corresponds to a rectangle with one corner located at the origin which classifies samples located within the rectangle as nondysbiotic and samples located outside as dysbiotic. Analysis of T‐square and Q statistics scaled by the confidence limit showed that the Euclidian distance from the origin had a log‐normal like distribution (data not shown). Euclidian distance from the origin was used to merge the two dimensions, and weighting was performed to capture the effect of T‐squared and Q statistics as appropriate. A single numeric representation of the degree of dysbiosis, defined as the Dysbiosis Index (DI), was derived from a log‐normal distribution by assigning estimated portions of the distribution to different values on a scale set from 0 to 5. A DI value of 2 was defined as class separation represented by the identified confidence limits; a DI of 2 or lower being the nondysbiotic region and a DI of 3 or higher being the dysbiotic region. The higher the DI above 2, the more the sample is considered to deviate from normobiosis, e.g. sample A with DI = 4 is farther away from the normobiotic reference cohort in the Euclidian space than sample B with DI = 3, thus A is more dysbiotic than B. The scale was optimised with emphasis on reducing technical variation between replicates, meaning that the integer part of the numeric output is decided by pre‐determined levels of the Euclidian distance.
To create the GA‐test, 211 healthy individuals were selected and randomly split into a training set (n = 165) designed to build models and a test set (n = 46) designed to tune parameters. Duplicate samples were run, and mean normalised signal was used for training and testing. Sample demographics for the two groups were similar (Table 2). In addition, a set of IBS patients were included in the test set (n = 127). A number of models were developed and evaluated, and the frequency of dysbiosis in the test set was used as measure of model performance. For the final PCA model, 15 principal components were used, and a 98% confidence limit was determined for T‐squared and Q statistics to define class separation. When the model is used to score other samples, values outside these limits are defined as dysbiotic.
Table 2.
Sample sets used for GA‐map Dysbiosis Test development and validation
| Cohort | Samples, n | Age, mean | Female (%) | Sample type, n | ||
|---|---|---|---|---|---|---|
| Healthy | IBS | IBD | ||||
| Training | 165 | 42 | 64 | 165 | – | – |
| Test | 173 | 40 | 73 | 46 | 127 | – |
| Validation | 287 | 39 | 71 | 43 | 109 | 135 |
| Full cohort | 625 | 40 | 70 | 254 | 236 | 135 |
External validation using an independent test set comprising healthy, IBS and IBD subjects (n = 287) was used to assess the clinical diagnostic performance of the model (Table 3). The validation set subjects were all from unique donors who had not been included in the healthy reference collection used for normobiotic profile calibration or in parameter tuning. Each sample was processed using the finalised algorithm which converts data for each sample into a single integer, i.e. the DI, which represents the degree of dysbiosis based on bacterial abundance and profile within a sample relative to the established normobiotic profile. A DI > 2 represents a potentially clinically relevant deviation in microbiotic profile from that of the normobiotic reference collection. Finally, the dysbiosis frequency was calculated. In addition, PCA was performed on the validation set to investigate differences in microbiota profile between the three subject groups.
Table 3.
Percentage dysbiosis and mean DI score in validation cohort
| Cohort | Total | Dysbiotic, % (95% CI) | DI, mean |
|---|---|---|---|
| Healthy controls | 43 | 16 (±11) | 1.72 |
| IBS | 109 | 73 (±8) | 2.98 |
| IBS‐D | 34 | 76 (±14) | 3.03 |
| IBS‐C | 26 | 73 (±17) | 3.00 |
| IBS‐M | 3 | 67 | 3.33 |
| IBS‐U | 25 | 72 (±18) | 3.04 |
| IBS‐A | 20 | 70 (±20) | 2.85 |
| IBD treatment‐naïve | 80 | 70 (±10) | 3.31 |
| CD | 30 | 80 (±14) | 3.60 |
| UC | 50 | 64 (±13) | 3.14 |
| IBD remission | 55 | 80 (±11) | 3.15 |
| CD | 18 | 89 (±14) | 3.65 |
| UC | 36 | 75 (±14) | 2.92 |
A, alternating; C, constipation; CD, Crohn's disease; D, diarrhoea; DI, Dysbiosis Index; IBD, inflammatory bowel disease; IBS, irritable bowel syndrome; M, mixed; U, un‐subtyped; UC, ulcerative colitis.
Technical performance
The EU directive for in vitro diagnostic tests was followed to ensure compliance with a CE‐marked test.25 The main technical parameters evaluated were precision and quantitative range of the test; both at probe signal level and at final output level (i.e. DI). At probe level, precision of signals [coefficient of variation [CV], %) varied with raw signal intensity. Signals below 500 IU were regarded as background noise; therefore, measurement of variance was not applicable. For signals above 500 IU precision was estimated to be 8.4%, using repeated runs for six donors over six faecal extractions per donor over 2 days (n = 328). A CV below 10% was set as a criterion in development of the DI algorithm. Based on repetitive measurements of 139 dysbiotic samples, 94% of the samples showed CVs below 10%. In addition, several in‐process test steps were evaluated (data not shown).
Faecal microbiota variation over time
Variation in microbiota over time was investigated both for normalised data across the selected probe set, and for the test result (DI). Faecal samples were collected from five donors (aged 24–38; 80% women) at a 1‐week interval for up to 14 weeks. PCA of normalised data was performed, and statistical assessment of variation in the signals for donor and sampling time (weekly) was conducted using R package ffmanova, an implementation of 50–50 multivariate analysis of variance.26
Comparison to Illumina deep sequencing
To compare the performance of deep sequencing and the GA‐test data for the gut microbiota, a total of 188 samples from 162 subjects (89 healthy subjects and 73 IBS subjects; from the training and test cohorts described in Table 2) were randomly selected. Sequencing was performed using the paired‐end 250 bp sequencing on the Illumina MiSeq platform27 at the Norwegian High Throughput Sequencing Center (UiO, Oslo, Norway). Demultiplexed Illumina readings were clustered into Operational Taxonomic Units (OTUs) using Qiime pipeline (v.1.7), StarCluster (http://star.mit.edu/cluster) and Amazon Web Services (https://aws.amazon.com, virtual machine identifier ami‐9bc9a7f2) at 97% sequence similarity. Standard tools and parameters for Qiime downstream analysis were used, such as uclust for OTU picking and Ribosomal Database Classifier for taxonomy assignment. A pre‐defined taxonomy map of reference sequence OTU to taxonomy was used rather than open‐reference picking and assignment, as the reference database of 16S rRNA sequences found in human gut is comprehensive. Thereafter, one representative read for each OTU group was extracted and aligned to create a phylogenetic tree and an OTU Biological Observation Matrix table was constructed (data not shown). The OTU table was rarefied to 5000 sequences to remove sample heterogeneity. Four samples which had less read count than the set threshold were excluded from further analysis.28
To compare the MiSeq sequence reads to the GA‐map Technology (GA‐Technology, Genetic Analysis AS, Oslo, Norway) probe signals, we identified probes that were specific for a maximum of two species or genera, and compared the normalised signals from the probes for each sample to the number of sequences of the corresponding sample and closest matching taxonomic bins found by MiSeq sequencing. If a genus found by sequencing corresponded to several probes, the sum of the probes was used in the comparison, and if a probe represented two genera the sum of sequences from both genera was used. Finally, correlation between deep sequencing data and GA‐technology data was calculated using Pearson correlation.
Furthermore, we applied the approach for defining the healthy reference collection, as described in the Dysbiosis test development section, to the Illumina sequence data set. In order to compare the results from this model with GA‐technology data, we constructed a new model using GA‐technology data limited to the same 188 samples. The samples were classified either as dysbiotic or nondysbiotic using both models, and the results compared by counting the number of samples that were classified equally across the models.
Statistical analysis
All data were analysed at GA (Genetic Analysis AS). Categorical data were expressed as the number of subjects (and percentage) with a specified condition or clinical variable, and the mean as appropriate. A test for association between the two technologies were performed using an independent t‐test based on Pearson's product moment correlation coefficient. The Mann–Whitney U‐test was used for testing DI values. All tests were two‐sided, and the chosen level of significance was P < 0.05. Analysis was done using the statistical computing language R version 3.0.229 and MATLAB 2011b (The MathWorks, Inc., Natick, MA, USA).
Results
Frequency of dysbiosis in healthy, IBS and IBD subjects
Validation of the developed GA‐test was performed by comparing frequency of dysbiosis in a set of 287 samples, including healthy individuals previously not included in the normobiotic profile calibration (n = 43) and patients with IBS (n = 109) and IBD (n = 135) (Table 2). The results in the validation cohort are given in Table 3. Of the 43 samples from healthy volunteers included in the validation cohort, seven (16%) were determined as being dysbiotic, with the distribution of DI scores for validation cohort shown in Figure 3. Among the IBS patients, 80 of 109 (73%) were determined as being dysbiotic. In the IBD cohort, 100 of 135 (74%) were determined as being dysbiotic, including 56 of 80 (70%) treatment‐naïve IBD patients, and 44 of 55 (80%) IBD patients in clinical remission. The distribution of DI between IBS and IBD patients was significantly different (P < 0.01) and more IBD patients than IBS patients had a DI >4 (Figure 3). Furthermore, both in treatment‐naïve IBD patients and in IBD patients in remission, the frequency of dysbiosis was higher in CD (80% and 89% respectively) than UC (64% and 75%), with significantly higher DI values in CD than UC (P = 0.03).
Figure 3.
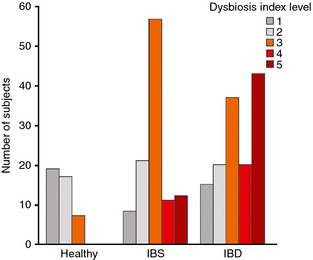
Distribution of DI scores 1–5 for the validation cohort as determined by GA‐map Dysbiosis Test, showing the increase in DI from healthy individuals through IBS patients and finally in IBD patients.
The test was also applied to a set of 43 available samples from healthy volunteers from Denmark (n = 19; aged 23–61; 63% women) and Spain (n = 24; aged 22–56; 50% women). Seven of the 19 Danish samples were determined as being dysbiotic with mean DI of 2.16, resulting in 37% dysbiotic (95% CI, ±22%) healthy volunteers in this cohort. Among the Spanish samples, 10 of 24 were determined as being dysbiotic with mean DI of 2.58, resulting in 42% dysbiotic (95% CI, ±20%). While the result for the Danish healthy cohort was not significantly different from the healthy validation samples (P > 0.05), we observed that 50% (5/10) of the dysbiotic samples in the Spanish samples showed a DI above 3.
Bacterial profile in dysbiosis
Applying PCA to the validation cohort using normalised data for all 54 probes demonstrated a relative clustering of samples by disease cohorts. The scores for the first two principal components (PC), accounting for 48% of the variance in the data, showed a tighter cluster for healthy subjects in the bottom right corner compared with a more diverse spread for subjects with IBD and IBS (Figure 4a). The sample distribution in the scores plot was found to be linked to the degree of dysbiosis, with a central cluster of nondysbiotic samples surrounded by samples with weak dysbiosis (DI = 3), and the samples with the most severe dysbiosis (DI = 5) scattered outside this cluster (Figure 4b). Both the first and second PC each separate the samples from healthy volunteers from IBS and IBD samples to a certain degree. The scatter of DI values implies that different bacteria dominate dysbiosis for different samples. To further investigate which bacterial groups were the main contributors to dysbiosis in IBD and IBS, differences in overall mean normalised signal between dysbiotic and nondysbiotic status for each of the 54 probes were calculated. The pre‐dominant bacteria contributing to dysbiosis within the IBS cohort were Firmicutes (Bacilli), Proteobacteria (Shigella/Escherichia), Actinobacteria and Ruminococcus gnavus (Figure 5a). Similarly, the pre‐dominant bacteria within the IBD cohort were Proteobacteria (Shigella/Escherichia), Firmicutes, specifically F. prausnitzii, and Bacteroidetes (Bacteroides and Prevotella) (Figure 5b). Interestingly, Proteobacteria (Shigella/Escherichia) was among the top five dysbiosis‐contributing bacterial groups for both IBS and IBD, implying similarities in dysbiosis between IBS and IBD. However, all bacterial groups that contributed most to dysbiosis in the IBS cohort showed increased probe signal intensity compared to nondysbiotic patients, while for the IBD cohort, both reduced (F. prausnitzii) and increased probe signal intensities were the main contributors to dysbiosis.
Figure 4.
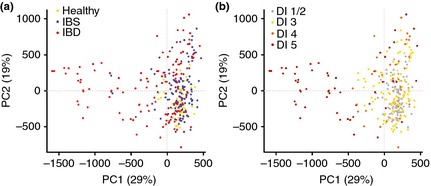
PCA scores for the first two principal components for validation cohort (n = 287) based on 54 probes. The two PCs account for 48% of the variation, and points are coloured according to (a) cohort: yellow – healthy, blue – IBS, and red – IBD; and (B) DI: grey = 1–2, orange = 3, red = 4, dark red = 5.
Figure 5.
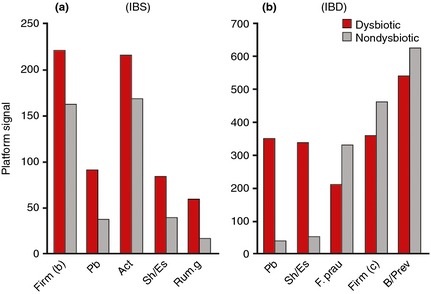
Mean normalised signal for top five probes sorted by absolute relative difference between dysbiotic (red) and nondysbiotic (grey) as determined by the GA‐map Dysbiosis Test for (a) IBS patients (n = 109), and (b) IBD patients (n = 135). Act, Actinobacteria; B/Prev, Bacteroides/Prevotella; Firm(b), Firmicutes (Bacilli); Firm (c), Firmicutes (Clostridia); F. prau, Faecalibacterium prausnitzii; Pb, Proteobacteria; Rum.g, Ruminococcus gnavus; Sh/Es, Shigella/Escherichia.
We found a single probe with a differential signal between samples from the Spanish and Scandinavian cohorts (P < 0.01; Benjamini–Hochberg correction). The probe targets Firmicutes (Streptococcus), and this signal was found to be elevated in the Spanish samples compared to the Scandinavian cohort. Figure 6 shows the pre‐dominant bacteria contributing to dysbiosis within the Spanish samples. As expected, Proteobacteria (Shigella/Escherichia) is again found to be a contributing bacteria in dysbiosis. In addition, Bacteroides stercoris and Bifidobacterium contribute to dysbiosis, which potentially could be linked to differences, in e.g. diet between Scandinavian countries and the Mediterranean region.
Figure 6.
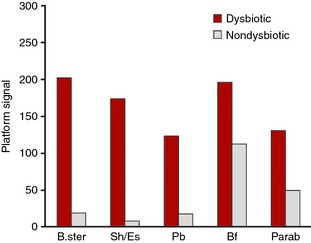
Mean normalised signal for probes sorted by absolute relative difference between dysbiotic (red) and nondysbiotic (grey) as determined by the GA‐map Dysbiosis Test for Spanish cohort (n = 24). Bf; Bifidobacterium, B. ster; Bacteroides stercoris, Parab; Parabacteroides, Pb; Proteobacteria, Sh/Es; Shigella/Escherichia.
Faecal microbiota variation over time
Faecal samples were collected from five individuals at 1‐week intervals for up to 14 weeks. PCA of the normalised data (n = 64) revealed that most variability in the longitudinal faecal microbiota analysis was related to inter‐individual variability; donors could clearly be distinguished by the three‐first and most important PCs in the score plot (Figure 7). The samples were clustered according to faecal donor independently of sample collection time. The three first PCs described 65% of the total variability in the faecal microbiota data.
Figure 7.
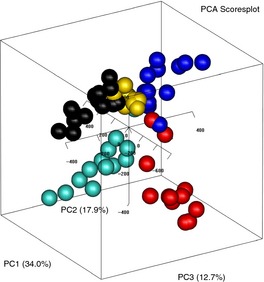
Scores for the first three principal components from PCA of normalised data from five healthy subjects collected weekly for up to 14 weeks (n = 64). One point is one sample for donor x taken at time point y. The first three PCs account for 65% of the variation, and points are coloured according to donor.
The significance of the PCs was analysed by ffmanova and performed using normalised data with only the main effects of donor and sampling time (weekly) included in the model. The results show that the average amount of variation between donors was greater than that within a donor (P < 0.001) with explained variances based on sums of squares of 0.48. The variation between sampling time points was not significant (P = 0.26), with explained variances based on sums of squares of 0.11. The low level of variation within one individual over time is crucial in utilising the test for monitoring changes during treatment for altering the microbiota profile.
Comparison to deep sequencing
The randomly selected set of 188 samples was sequenced using MiSeq Illumina to investigate similarities with GA‐technology profiles. Any reads that did not match a reference sequence at greater than or equal to 97% sequence identity were discarded according to a closed‐reference OTU‐picking protocol. A total of 7 564 142 reads were binned into 254 OTUs at higher taxonomic levels and 165 of these were identified at genus level. Of the 165 genera, 77 were found in more than 10% of samples.
After identifying the genera in the samples with MiSeq sequencing, a comparison was performed with the closest matching taxonomic bins detected by GA‐technology. In general, we found strong correlations between the GA‐technology signals and Qiime taxonomically assigned MiSeq reads (Table 4), where Alistipes, Bifidobacterium, Dialister, Lactobacillus/Pediococcus, R. gnavus and Shigella/Escherichia all had a Pearson correlation of r > 0.85. For some species the correlation was moderate, e.g. B. fragilis (r = 0.38), Ruminococcus albus/bromii (r = 0.31) and Streptococcus sanguinis/thermophilus (r = 0.49). However, since MiSeq sequencing did not allow for detection at species level, a direct comparison to the specific probe signals can be complicated if the specific species is not the dominating species in a genera. Interestingly, no correlation was found between the two methods for the species Mycoplasma hominis (r = −0.05). Using MiSeq sequencing, Mycoplasma genus was only detected in one sample, implying that MiSeq sequencing does not allow for the selective detection of this genus at all. In contrast, M. hominis was detected in a majority of the 188 samples with the GA‐technology. The highly specific GA‐technology probe detecting M. hominis binds to V6 on the 16S rRNA gene, a variable region not covered by MiSeq Illumina sequencing (Figure 1).
Table 4.
Correlation between normalised GA‐map signal data and MiSeq Illumina sequence data (97% sequence identity) for 188 healthy and IBS samples
| Taxonomic group | Correlation coefficient (r)a | P‐value |
|---|---|---|
| Mycoplasma hominis b | −0.05 | 0.50 |
| Ruminococcus albus/bromii c | 0.31 | <0.001 |
| Bacteroides fragilis c | 0.38 | <0.001 |
| Streptococcus sanguinis/thermophilus c | 0.49 | <0.001 |
| Phascolarctobacterium | 0.72 | <0.001 |
| Faecalibacterium prausnitzii | 0.75 | <0.001 |
| Streptococcus thermophilus | 0.78 | <0.001 |
| Akkermansia | 0.79 | <0.001 |
| Eubacterium | 0.79 | <0.001 |
| Megashera/dialister | 0.83 | <0.001 |
| Ruminococcus gnavus | 0.86 | <0.001 |
| Dialister | 0.88 | <0.001 |
| Alistipes | 0.90 | <0.001 |
| Bifidobacterium | 0.90 | <0.001 |
| Shigella/Escherichia | 0.93 | <0.001 |
| Lactobacillus/Pediococcus | 0.94 | <0.001 |
Correlation coefficients were determined for the closest matching taxonomic bins.
Mycoplasma only identified in one sample by MiSeq Illumina sequencing.
Illumina sequencing did not enable selective detection of species.
In addition, two new models were built using Illumina sequencing data and GA‐technology probe intensity data with eight and nine PCs, respectively. The number of PCs was selected by optimising the frequency of dysbiosis in test samples (at the most 20% of healthy individuals and 60% of IBS patients were determined as dysbiotic). The training data set consisted of 100 samples from healthy volunteers, and the test set included 15 healthy and 73 IBS samples. The results were compared across the two models and yielded 80% concordance.
Discussion
In this article, we demonstrate the performance of a novel gut microbiota test, aiming to identify and characterise dysbiosis by determining deviation from normobiosis. Such a diagnostic approach contrasts to direct diagnosis of a particular disease. Characteristic sets of bacteria are required in a healthy normobiotic gut microbiota, and deviation will represent a dysbiotic state. Quantitative measurement of deviation in bacterial microbiota makes it possible to characterise dysbiosis in samples from IBS and IBD patients based on a single diagnostic algorithm targeting normobiosis.
Ideal enabling technologies will be those that can profile the microbiome as a whole and, at the same time, reliably target deviations (and their degree) from normobiosis. Notably, gut microbiota also harbour a range of transient colonisers with no diagnostic value that have the potential to generate obscure diagnostic results. Furthermore, recent evidence suggests that species‐level information is important in gut microbiota diagnostics.30 Techniques with a low error rate which target a wide range of variable positions in the 16S rRNA gene would therefore be preferable for discriminating between normobiosis and dysbiosis.
The present test is a broad‐spectrum, reproducible, precise, high throughput, easy to use method of quantifying the extent of dysbiosis that is especially suitable for clinical use. This test gives an algorithmically derived DI based on bacterial abundance and profile within a sample. This DI is an indicator of the degree to which an individual's microbiome deviates from that of a healthy reference collection and could potentially be highly relevant in clinical diagnosis and monitoring of the progression of conditions such as IBD and IBS. The stability of the human gut microbiota is another important feature if microbial characterisation is to play a role in diagnosis, treatment and prevention of disease. Faith et al.31 showed that, in an individual's microbiota, 60% of the bacterial strains persisted over the course of 5 years. Our data also suggest that there is little variation in an individual's gut microbiota over time, since we found only a low within individual variation in weekly sampling over 14 weeks.
High‐throughput sequencing is an excellent tool for exploratory analyses of the gut microbiota, and is widely used. A limitation to this technology is the relatively short read‐lengths used for sequencing, only allowing for a limited region of the 16S rRNA gene to be exploited (usually V3 and/or V4) (Figure 1); thus, less than 50% of obtained sequences can be annotated at genus level.32 The lack of detection of Mycoplasma with MiSeq Illumina sequencing, detected in a majority of the samples using a probe targeting V6 with the GA‐map test, further illustrates the limitations of using only limited variable regions of the 16S rRNA gene. Moreover, since MiSeq sequencing does not allow for detection at species level, a direct comparison to the specific probe signals can be challenging if the specific species is not the dominating species in a genera. Even so, it is possible to gain important insights to an individuals' gut microbiota using high‐throughput sequencing. Compared to the GA‐technology, this technology is superior towards exploring novel bacterial biomarkers, and gaining in‐depth information regarding all bacteria present in a sample. However, in terms of the human gut microbiota, the main patterns have been explored,32 and GA‐technology has consolidated on this information in designing 54 highly specific DNA probes exploiting a broad range of gene variability (V3–V7). These 54 probes have further been converted into a diagnostic test but without the laborious data‐analysis required following high‐throughput sequencing. Therefore, the GA‐technology provides a unique opportunity to study changes in gut microbiota profiles potentially associated with gastrointestinal‐related disorders. Our results show agreement between the two technologies regarding determining dysbiosis, as well as strong correlations in detecting several bacteria. However, results also show weak correlations for some specific species, possibly due to lack of selective species detection by MiSeq Illumina sequencing.
The GA‐test identifies a high frequency of dysbiosis in IBS and IBD patients and low frequency in healthy individuals. Both IBD patients in remission and treatment‐naïve IBD patients reported DI scores well above the threshold of two with a dysbiosis frequency of 80% and 70%, respectively. IBS patients, defined according to Rome II and III‐criteria (depending on collection site), showed a dysbiosis frequency of 73%, confirming previous observations,30, 33, 34 while the frequency of dysbiosis in healthy individuals was 16%. The normobiotic reference collection comprised healthy Scandinavian individuals, which may be a potential limitation of the test. We found slightly increased DI in healthy controls from Denmark (DI ≥ 3 in 33%, n = 19) and Spain (DI ≥ 3 in 42%, n = 24); however, the sample size is too small to allow any definitive conclusions to be drawn regarding differences in frequency of dysbiosis or microbiota between the populations. Further investigation is needed with increased sample numbers from across Europe to firmly establish the broad clinical utility of the test.
The intestinal microbiome is a dynamic environment in which the relative balance of the composition of pro‐ and anti‐inflammatory bacterial species is known to be highly relevant.35 For example, the microbial signature of Firmicutes species present in the intestinal tract in patients with UC differs significantly from that in CD patients.36 Compared with CD patients in long‐term remission, patients with relapsing CD have lower levels of all Firmicutes species, and a bacterial profile significantly predictive of relapse for up to 1 year before infliximab withdrawal.17
Dysbiosis is associated with many diseases, including IBS, IBD, obesity and diabetes,4, 5, 6, 7, 8 and has also been implicated in depression and autism.37, 38 In recent years, new treatment options have emerged with respect to restoring the balance of the microbiota in dysbiotic patients. FMT is now regarded as the most effective treatment in relapsing Clostridium difficile colitis,18, 39 and is currently being studied in phase I to IV clinical trials in many of the aforementioned conditions (CD, phase II/III NCT01793831; UC, phase I NCT01947101, phase II NCT01896635, phase II/III NCT01790061; IBD including CD and UC, phase IV NCT02033408). A key barrier in the interpretation of FMT data has been the variability in bacterial composition of donor microbiota, not only related to pathogenic organisms but also to the composition of the normally occurring microbiota, further highlighting the importance of identifying a method to sufficiently characterise both pathogenic and nonpathogenic microbes. The ability to characterise an individual's microbiome and monitor alterations may allow for the prediction of therapeutic outcome or even relapse in such conditions.17 It may also help to explain why a patient is refractory to particular therapeutic regimens and aid adaptation of the regimen accordingly. Furthermore, rapid and reproducible detailed bacterial profiles from normobiotic and dysbiotic individuals may aid the continuation of innovative therapeutic approaches such as FMT.18 Thus, use of the test could prove clinically useful in determining dysbiosis, not only in IBS and IBD patients, but also in other conditions where knowledge about the microbiota profile might prove clinically useful, in the subsequent monitoring of prescribed treatment regimens, and in the evolution of new therapeutic approaches.
In conclusion, this is the first clinical test, aiming to identify and characterise dysbiosis based on faecal specimens. The diagnostic applicability of the test will have to await further clinical experience, also from international studies, as one might expect geographical deviations related to microbial patterns. Nevertheless, the present standardised and reproducible method represents, in particular, a step forward as a combined practical, ready to use, clinical and research tool. The method will allow us to gain more knowledge on the microbial component of intestinal disorders, and in general, provide the possibility of increasing our understanding of the part played by the microbiome in the disease process.
Authorship
Guarantor of the article: Professor Knut Rudi, Department of Chemistry, Biotechnology and Food Science, Norwegian University of Life Science, Aas, Norway.
Author contributions: C Casén, HC Vebø, FT Hegge and K Rudi conceived and designed the technical and clinical studies. M Sekelja and MK Karlsson analysed the data. C Casén and S Dzankovic administered sample collection and clinical data recording. E Ciemniejewska, S Dzankovic, C Frøyland and R Nestestog performed the laboratory work. L Engstrand, P Munkholm, OH Nielsen, G Rogler, M Simrén, L Öhman, MH Vatn and K Rudi contributed to strategic development decisions and clinical supervision. All authors contributed to writing the manuscript and approved the final submitted version.
Supporting information
Data S1. Sample inclusion and exclusion criteria.
Data S2. Faecal sample collection, storage and processing and data processing.
Data S3. NCBI MeSH search terms used to compile initial IBD and IBS intestinal microbiota bacterial observations.
Data S4. Probe set (54 probes): 368 strains used to test bacterial target specificity.
Data S5. Probes and bacterial target validation.
Acknowledgements
We thank N Pedersen, F‐A Halvorsen, J Sauar, A Røseth and L Böhn for clinical samples for IBS validation, P Ricanek and S Brackmann for clinical samples for IBD validation, and L Agreus, L Kjellstrom and A Andreasson for clinical samples for normobiotic validation, and Jordi Guardiola Capón for samples from healthy volunteers from Spain. We thank the GA Scientific Advisory Board (M H Vatn [Chair], L Engstrand, W Kruis, R Löfberg, O H Nielsen, G Rogler, and L Öhman) for contribution to discussion surrounding development of the dysbiosis test.
Declaration of personal interests: GR has been a consultant for Abbot, Abbvie, Augurix, Boehringer, Calypso, FALK, Genentech, Genetic Analysis, Essex, MSD, Novartis, Pfizer, Roche, UCB, Takeda, Tillots and Vifor; has received speaker honoraria from AstraZeneca, Abbott, Abbvie, FALK, MSD, Phadia, Tillots, UCB, and Vifor; and has received educational and research grants from Abbot, Abbvie, Ardeypharm, Essex/MSD, FALK, Flamentera, Novartis, Roche, Tillots, UCB and Zeller. PM has been a consultant for AstraZeneca, MSD, Tillots and Calpro. MHV has been an advisor for Genetic Analysis and organizer of the International Advisory Board of Genetic Analysis, a member of the Advisory Board of Tillots, and has received speaker honoraria from AstraZeneca, Abbott, MSD and Falk. OHN has been a member of the International Advisory Board of Genetic Analysis. KR is a board member of Genetic Analysis. LE has been an adviser for Genetic Analysis. LÖ has received speaker honoraria from Abbvie, UCB and Takeda, and is a member of the Advisory Board of Genetic Analysis, and has received an unrestricted research grant from AstraZeneca. MS has received unrestricted research grants from Danone, and served as a Consultant/Advisory Board member for Danone, Nestlé, Chr Hansen, Almirall, Albireo, AstraZeneca, Sucampo, and Shire. C Casén, HC Vebø, M Sekelja, FT Hegge, MK Karlsson, E Ciemniejewska, S Dzankovic, C Frøyland and R Nestestog are employees of Genetic Analysis AS. C Casén, HC Vebø, M Sekelja, FT Hegge, S Dzankovic, R Nestestog and K Rudi owns stocks and shares in Genetic Analysis AS. Genetic Analysis AS owns patent invented by HC Vebø and K Rudi: Methods of amplifying a target sequence of a 16S rRNA or 16S rDNA in a prokaryotic species, US patent No. 8889358, the 16S oligonucleotide primers used in this work. Genetic Analysis AS owns patents invented by M Sekelja, K Rudi and HC Vebø: Oligonucleotide probe set and methods of microbiota profiling, US application No 13/919056 describing a set of oligonucleotide probes used for profiling the microbiotia in the GI tract used in this work. Genetic Analysis AS owns patent invented by K Rudi: Nucleic acid detection method with US patent No. 6617138, describing the method of selective binding, labeling and hybridization of a oligonucelotide probe used in this work.
Declaration of funding interests: This study was funded by an unrestricted grant from Genetic Analysis AS and the EU FP7‐Health‐2012 project; IBD‐Charter Grant agreement no: 305676. Medical writing support was provided by Carl Felton of Prime Medica Ltd (Knutsford, Cheshire, UK) and funded by Genetic Analysis AS.
This article was accepted for publication after full peer‐review.
References
- 1. Arumugam M, Raes J, Pelletier E, et al Enterotypes of the human gut microbiome. Nature 2011; 473: 174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sekelja M, Berget I, Naes T, Rudi K. Unveiling an abundant core microbiota in the human adult colon by a phylogroup‐independent searching approach. ISME J 2011; 5: 519–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ding T, Schloss PD. Dynamics and associations of microbial community types across the human body. Nature 2014; 509: 357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006; 444: 1022–3. [DOI] [PubMed] [Google Scholar]
- 5. Manichanh C, Rigottier‐Gois L, Bonnaud E, et al Reduced diversity of faecal microbiota in Cohn's disease revealed by a metagenomic approach. Gut 2006; 55: 205–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Larsen N, Vogensen FK, van den Berg FW, et al Gut microbiota in human adults with type 2 diabetes differs from non‐diabetic adults. PLoS ONE 2010; 5: e9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 2014; 146: 1489–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hong SN, Rhee PL. Unraveling the ties between irritable bowel syndrome and intestinal microbiota. World J Gastroenterol 2014; 20: 2470–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gasbarrini A, Corazza GR, Gasbarrini G, et al Methodology and indications of H2‐breath testing in gastrointestinal diseases: the Rome Consensus Conference. Aliment Pharmacol Ther 2009; 29(Suppl 1): 1–49. [DOI] [PubMed] [Google Scholar]
- 10. Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev 2010; 90: 859–904. [DOI] [PubMed] [Google Scholar]
- 11. Simren M, Barbara G, Flint HJ, et al Intestinal microbiota in functional bowel disorders: a Rome foundation report. Gut 2013; 62: 159–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ford AC, Spiegel BM, Talley NJ, Moayyedi P. Small intestinal bacterial overgrowth in irritable bowel syndrome: systematic review and meta‐analysis. Clin Gastroenterol Hepatol 2009; 7: 1279–86. [DOI] [PubMed] [Google Scholar]
- 13. Salonen A, de Vos WM, Palva A. Gastrointestinal microbiota in irritable bowel syndrome: present state and perspectives. Microbiology 2010; 156: 3205–15. [DOI] [PubMed] [Google Scholar]
- 14. Vanner S. The small intestinal bacterial overgrowth. Irritable bowel syndrome hypothesis: implications for treatment. Gut 2008; 57: 1315–21. [DOI] [PubMed] [Google Scholar]
- 15. Budding AE, Grasman ME, Lin F, et al IS‐pro: high‐throughput molecular fingerprinting of the intestinal microbiota. FASEB J 2010; 24: 4556–64. [DOI] [PubMed] [Google Scholar]
- 16. Thorkildsen LT, Nwosu FC, Avershina E, et al Dominant fecal microbiota in newly diagnosed untreated inflammatory bowel disease patients. Gastroenterol Res Pract 2013; 2013: 636785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rajca S, Grondin V, Louis E, et al Alterations in the intestinal microbiome (dysbiosis) as a predictor of relapse after infliximab withdrawal in Cohn's disease. Inflamm Bowel Dis 2014; 20: 978–86. [DOI] [PubMed] [Google Scholar]
- 18. Kelly CP. Fecal microbiota transplantation – an old therapy comes of age. N Engl J Med 2013; 368: 474–5. [DOI] [PubMed] [Google Scholar]
- 19. Pedersen N, Vegh Z, Burisch J, et al Ehealth monitoring in irritable bowel syndrome patients treated with low fermentable oligo‐, di‐, mono‐saccharides and polyols diet. World J Gastroenterol 2014; 20: 6680–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pedersen N, Vinding K, Vegh Z, et al Gut microbiota in IBD patients with IBS before and after 6 weeks of low FODMAP diet. Gastroenterology 2014; 146: S‐241. [Google Scholar]
- 21. Macfarlane S, Cleary S, Bahrami B, Reynolds N, Macfarlane GT. Synbiotic consumption changes the metabolism and composition of the gut microbiota in older people and modifies inflammatory processes: a randomised, double‐blind, placebo‐controlled crossover study. Aliment Pharmacol Ther 2013; 38: 804–16. [DOI] [PubMed] [Google Scholar]
- 22. Vebø HC, Sekelja M, Nestestog R, et al Temporal development of the infant gut microbiota in immunoglobulin E‐sensitized and nonsensitized children determined by the GA‐map infant array. Clin Vaccine Immunol 2011; 18: 1326–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sugimoto N, Katoh M, Nakano S, Ohmichi T, Sasaki M. RNA/DNA hybrid duplexes with identical nearest‐neighbor base‐pairs have identical stability. FEBS Lett 1994; 354: 74–8. [DOI] [PubMed] [Google Scholar]
- 24. Mardia KV, Kent JT, Bibby JM. Multivariate Analysis. London: Academic Press, 1979. [Google Scholar]
- 25. Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices. Available at: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CONSLEG:1998L0079:20090807:en:PDF.
- 26. Langsrud Ø. 50‐50 multivariate analysis of variance for collinear responses. J R Stat Soc Ser D 2002; 51: 305–17. [Google Scholar]
- 27. Naseribafrouei A, Hestad K, Avershina E, et al Correlation between the human fecal microbiota and depression. Neurogastroenterol Motil 2014; 26: 1155–62. [DOI] [PubMed] [Google Scholar]
- 28. Lozupone CA, Knight R. Species divergence and the measurement of microbial diversity. FEMS Microbiol Rev 2008; 32: 557–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. R Development Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: the R Foundation for Statistical Computing. Available at: http://www.R-project.org/.
- 30. Jeffery IB, O'Toole PW, Ohman L, et al An irritable bowel syndrome subtype defined by species‐specific alterations in faecal microbiota. Gut 2012; 61: 997–1006. [DOI] [PubMed] [Google Scholar]
- 31. Faith JJ, Guruge JL, Charbonneau M, et al The long‐term stability of the human gut microbiota. Science 2013; 341: 1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aravindraja C, Viszwapriya D, Karutha PS. Ultradeep 16S rRNA sequencing analysis of geographically similar but diverse unexplored marine samples reveal varied bacterial community composition. PLoS ONE 2013; 8: e76724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Collins SM. A role for the gut microbiota in IBS. Nat Rev Gastroenterol Hepatol 2014; 11: 497–505. [DOI] [PubMed] [Google Scholar]
- 34. Karantanos T, Markoutsaki T, Gazouli M, Anagnou NP, Karamanolis DG. Current insights in to the pathophysiology of irritable bowel syndrome. Gut Pathog 2010; 2: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dupont HL. Review article: evidence for the role of gut microbiota in irritable bowel syndrome and its potential influence on therapeutic targets. Aliment Pharmacol Ther 2014; 39: 1033–42. [DOI] [PubMed] [Google Scholar]
- 36. Machiels K, Joossens M, Sabino J, et al A decrease of the butyrate‐producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014; 63: 1275–83. [DOI] [PubMed] [Google Scholar]
- 37. Dinan TG, Cryan JF. Melancholic microbes: a link between gut microbiota and depression? Neurogastroenterol Motil 2013; 25: 713–9. [DOI] [PubMed] [Google Scholar]
- 38. Finegold SM, Dowd SE, Gontcharova V, et al Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 2010; 16: 444–53. [DOI] [PubMed] [Google Scholar]
- 39. Sha S, Liang J, Chen M, et al Systematic review: faecal microbiota transplantation therapy for digestive and nondigestive disorders in adults and children. Aliment Pharmacol Ther 2014; 39: 1003–32. [DOI] [PubMed] [Google Scholar]
- 40. Mizrahi‐Man O, Davenport ER, Gilad Y. Taxonomic classification of bacterial 16S rRNA genes using short sequencing reads: evaluation of effective study designs. PLoS ONE 2013; 8: e53608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Sample inclusion and exclusion criteria.
Data S2. Faecal sample collection, storage and processing and data processing.
Data S3. NCBI MeSH search terms used to compile initial IBD and IBS intestinal microbiota bacterial observations.
Data S4. Probe set (54 probes): 368 strains used to test bacterial target specificity.
Data S5. Probes and bacterial target validation.