

Abstract
Tenascin‐W is a matricellular protein with a dynamically changing expression pattern in development and disease. In adults, tenascin‐W is mostly restricted to stem cell niches, and is also expressed in the stroma of solid cancers. Here, we analyzed its expression in the bone microenvironment of breast cancer metastasis. Osteoblasts were isolated from tumor‐free or tumor‐bearing bones of mice injected with MDA‐MB231‐1833 breast cancer cells. We found a fourfold upregulation of tenascin‐W in the osteoblast population of tumor‐bearing mice compared to healthy mice, indicating that tenascin‐W is supplied by the bone metastatic niche. Transwell and co‐culture studies showed that human bone marrow stromal cells (BMSCs) express tenascin‐W protein after exposure to factors secreted by MDA‐MB231‐1833 breast cancer cells. To study tenascin‐W gene regulation, we identified and analyzed the tenascin‐W promoter as well as three evolutionary conserved regions in the first intron. 5′RACE analysis of mRNA from human breast cancer, glioblastoma and bone tissue showed a single tenascin‐W transcript with a transcription start site at a noncoding first exon followed by exon 2 containing the ATG translation start. Site‐directed mutagenesis of a SMAD4‐binding element in proximity of the TATA box strongly impaired promoter activity. TGFβ1 induced tenascin‐W expression in human BMSCs through activation of the TGFβ1 receptor ALK5, while glucocorticoids were inhibitory. Our experiments show that tenascin‐W acts as a niche component for breast cancer metastasis to bone by supporting cell migration and cell proliferation of the cancer cells.
Keywords: tenascin, metastatic niche, breast cancer, bone, gene regulation
Short abstract
What's new?
Once breast cancer metastasizes, it is generally incurable. Proteins in the extracellular matrix play a crucial role in launching the tumor cells to a new site. These authors investigated one such protein, tenascin‐W, which can be found surrounding not only tumor cells but also in bone tissue. Among other things, they studied how breast cancer cells affected tenascin‐W expression. The tumor cells induced bone marrow stromal cells to make more tenascin‐W, suggesting that the protein may pave the way for the cancer to spread to the bone.
Abbreviations
- α‐MEM
minimal essential medium alpha
- BMSCs
bone marrow‐derived stromal cells
- BrdU
5‐bromo‐2‐deoxyuridine
- DCC
dextran‐coated charcoal
- ECM
extracellular matrix
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- L2G
Luc‐2eGFP
- nGRE
negative glucocorticoid response element
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- SBE
Smad‐binding element
- SEAP
secreted embryonic alkaline phosphatase
- TBP
TATA‐binding protein
- TNC
tenascin‐C
- TNW
tenascin‐W
- TSS
transcription start site
Despite great progress in the treatment of breast cancer, metastatic disease is not curable and the treatment options remain palliative. Bone is the most frequent site of metastatic lesions and occurs in 80% of women with advanced breast cancer.1, 2 Therefore, it is important to investigate the mechanism for this osteotropism as well as the interactions of the cancer cells with the bone microenvironment with the ultimate aim being to define new treatment options. The microenvironment, including the extracellular matrix (ECM) surrounding primary tumors as well as metastases, has been found to be an important factor determining tumor cell behavior.3, 4, 5, 6 Tenascin‐C (TNC) and tenascin‐W (TNW) are two ECM proteins that are highly expressed in the stroma of most solid tumors7, 8 and a crucial role for TNC in breast cancer metastasis to the lung has been demonstrated.9, 10 Currently, there is no information on TNW expression or a potential role in breast cancer metastasis, the goal of the work described here.
Prominent expression of TNW has been reported in developing bone where it was shown to be particularly abundant in the stem cell niche of the cambium, the location of osteoblast progenitors.11 Therefore, we decided to investigate TNW expression in the bone environment in the MDA‐MB231 xenograft model of breast cancer metastasis.12 We used MDA‐MB231‐1833 cells which have bone tropism following intracardial injection.12 We found that MDA‐MB231‐1833 tumors induced TNW in situ in the bone stroma. Moreover, in a coculture model of MDA‐MB231‐1833 cells with human bone marrow‐derived stromal cells (BMSCs), we also observed increased levels of TNW. To provide mechanistic insight to this observation, we investigated the signaling pathways inducing TNW in BMSCs and characterized the gene structure of the human TNW gene. We identified a crucial effect of TGF‐beta signaling in the regulation of TNW expression in human BMSCs, which in turn will provide a congenial microenvironment for tumor cell growth.
Material and Methods
Bone metastasis model
The breast cancer cell line MDA‐MB231‐SCP1833 was kindly provided by Prof. J. Massagué (Memorial Sloan Kettering Cancer Center, New York, NY). These cells were transduced with a lentiviral vector encoding Luc‐2eGFP genes (L2G) as described in Ref. 13. MDA‐MB231‐SCP1833 L2G cells were harvested from subconfluent cell culture plates, washed in phosphate‐buffered saline (PBS) and injected into the left ventricle (0.5 × 106 in 100 μl PBS) of 8‐week‐old female NOD SCID mice. Successful injections were verified by the pumping of arterial blood into the syringe and imaging with a bioluminescence imager (NightOWL, Berthold Technologies, Bad Wildbad, Germany). Bone marrow metastases were monitored by in vivo imaging over 20 days after which long bones were excised for cell sorting or immunostaining.
Bone marrow cell suspensions from tumor‐free or tumor‐bearing mice (n = 6–10 samples) were obtained by grinding the bone with mortar and pestle and digestion of the bone powder for 1 hr at 37 °C with 1 mg/ml collagenase (Roche diagnostics, Rotkreuz, Switzerland), 1 mg/ml dispase (Roche diagnostics) and 50 KU/ml DNAse (Sigma‐Aldrich, Buchs, Switzerland) into a single‐cell suspension. Stromal and hematopoietic cell fractions were enriched via a discontinuous percoll density gradient separation using 1.065 and 1.115 g/l (GE Healthcare Bio‐Sciences, Uppsala Sweden). Remaining red blood cells were lysed (140 mM NH4Cl and 17 mM Tris‐base, pH 7.4) and cells were stained and sorted directly into RNA extraction buffer (Qiagen, Hilden, Germany) using a MoFlo cell sorter (Beckman Coulter, Brea, CA). The osteoblast population was defined as GFP−TR119−CD45−SCA1−CD51+ cells. RNA was extracted with Pico Pure RNA Isolation Kit (at. KIT0204, Arcturus, Foster City, CA) and cDNA prepared with the Ovation Pico Kit (cat. 3302, NuGen, Bemmel, The Netherlands) following standard procedures and used for quantitative real‐time polymerase chain reaction (qRT‐PCR, see below).
Cell culture
Fibrosarcoma HT1080 cells (CCL‐121, ATCC), MDA‐MB231 (HTB‐26, ATCC) and MDA‐MB231‐SCP1833 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) and 10% fetal bovine serum (FBS). Human BMSCs immortalized with the hTERT/GFP system have been described previously.14 BMSCs were cultured in Eagle's minimal essential medium alpha (α‐MEM) with 2 mM l‐glutamine and 10% FBS. To strip glucocorticoids from serum, 2.5 g of dextran‐coated charcoal (DCC; Sigma‐Aldrich) was added to 125 ml of serum and mixed gently overnight at 4 °C. DCC was removed by centrifugation followed by sterile filtration.
For co‐culture assays, 6 × 103 BMSCs and 3 × 103 MDA‐MB231‐SCP1833 cells were seeded per 1 cm2 into poly‐l‐lysine‐coated eight‐well chamber slides (BD Falcon, Franklin Lakes, NY). In parallel each cell line was cultured individually at a density of 3 × 103 cells/cm2. For transwell co‐culture assays, cells were cultured in wells containing inserts separated by a polycarbonate membrane with 0.4‐µm pores (Costar, Corning Amsterdam, Netherlands). MDA‐MB231‐SCP1833 or BMSCs were plated in the upper chamber (5 × 103 cells in 0.5 ml medium) and BMSCs or MDA‐MB231‐SCP1833 (5 × 104 cells in 1.5 ml) were cultured on 10‐mm round glass coverslips coated with fibronectin (5 μg/ml, for 1 hr) placed in the bottom chamber. Cells were cultured in α‐MEM/10% FBS and maintained for 7 days with medium changes every 2 days.
4T1 (CRL‐2539, ATCC) and 4T1.2 cells were cultured in α‐MEM/10% FBS. To produce conditioned medium of 4T1, 4T1.2, MDA‐MB231 and MDA‐MB231‐SCP1833 cells, cultures were grown to 80% confluence in α‐MEM/10% FBS. Then the medium was switched to serum‐free α‐MEM containing 0.2% BSA for 48 hr. To test for the effect of conditioned medium on BMSCs, they were seeded at a density of 1 × 105 in six‐well plates overnight at 37 °C to reach 70% confluence. After washing with PBS they were either maintained in α‐MEM containing 0.2% BSA as control or exposed to conditioned medium mixed in a ratio of 1:1 with fresh serum‐free medium plus 0.2% BSA.
Immunostaining
Long bones were fixed for 48 hr in 10% formalin, decalcified in 0.5 M EDTA (pH 7.5) for 3 days at 4 °C, dehydrated in 30% sucrose in PBS overnight and embedded in OCT. Antigen retrieval of 8‐µm sections was performed in citrate buffer 10 mM pH 6, 0.5% Tween20 (2 hr) followed by blocking with 3% BSA/0.2% Triton in PBS. Cultured cells were fixed with 4% formaldehyde in PBS for 15 min at RT, permeabilized in cold 100% methanol for 2 min at −20 °C, washed twice with PBS and blocked with 0.01% Tween/1% BSA in PBS. Slides with bone sections or cells were stained with rabbit‐anti‐mTNW11 and the mouse monoclonal anti‐hTNW56O15 followed by secondary antibodies Alexa Fluor 568 (Invitrogen, Life Technlogies Europe, Zug, Switzerland). Slides were mounted with ProLong Gold containing DAPI (Invitrogen) and images acquired using an Axio Imager Z2 LSM700 confocal microscope (Zeiss, Jena, Germany).
RNA isolation and quantitative real‐time PCR
Total RNA was isolated from BMSCs treated with 5 ng/ml TGFβ1 (R&D Systems) in α‐MEM/0% FBS for 24 hr, with or without 10 µM SB‐431542 inhibitor (Sigma) added to the cell cultures 1 hr before the addition of TGFβ1. Total RNA was extracted from BMSCs using RNeasy Mini Kits and QIAshredder (Qiagen). RNA was transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Life Technologies Europe, Zug, Switzerland) and qRT‐PCR analysis was performed with SYBR green PCR SuperMix‐UDG/ROX (Invitrogen). The level of expressed genes was measured by the relative quantification (ΔΔCt method) or by the relative standard curve quantification (ΔCt method) using either human TBP (TATA binding protein) or mouse GAPDH (glyceraldehyde‐3‐phosphate dehydrogenase) genes as internal reference. For semiquantitative RT‐PCRs bands were visualized by agarose gel electrophoresis. Primers used are listed in Supporting Information Table 1.
Cell proliferation and migration assays
TNW protein was purified as described.11 For proliferation assays the 5‐bromo‐2‐deoxyuridine (BrdU) chemiluminescent enzyme incorporation kit was used (Roche diagnostics). Tumor cells were seeded in a 96‐well black plate at a density of 1 × 104 per well and incubated overnight. After the addition of different concentrations of TNW protein (0–10 µg/ml) in serum‐free DMEM cell proliferation was quantified 48 hr after plating and the addition of 10 µM BrdU for 2 hr at 37 °C. Cells were fixed and stained and the incorporated BrdU detected in a chemiluminescence microplate reader (Perkin Elmer, Waltham, MA).
Cell migration assays were performed using 24‐transwell chambers (8‐µm pore size; Costar). The underside of the polycarbonate membrane was precoated with different concentrations of TNW recombinant protein (0–10 µg/ml) for 2 hr at 37 °C and subsequently blocked for 30 min at 37 °C with 1% BSA/PBS. MDA‐MB231‐SCP1833 cells were cultured in the upper chamber in serum‐free DMEM (1 × 105 cells in 1 ml). Serum‐free medium was added to the bottom chamber (0.6 ml) and cells were allowed to migrate for 24 hr. Cells were scraped off the top of the membrane using cotton swabs. Cells that had migrated across the membrane were fixed in 4% formaldehyde and stained with 1% crystal violet and total cell‐covered areas were quantified using the ImageJ software as described before.15
Transcription start site identification and cloning of the reporter constructs
5′RACE was performed using total RNA from normal bone (OriGene, Rockville, MD), breast cancer tissue (Clontech) and glioblastoma (OriGene) using the 5′/3′RACE Kit (2nd Generation, Roche diagnostics). Briefly, 1 µg of total RNA was reverse‐transcribed into first‐strand cDNA using the human tenascin‐W‐specific primer hSP1. To identify the 5′ end of the TNW mRNA two gene‐specific primers hSP2 and hSP3 were used (primers are listed in Supporting Information Table 1). DNA products were cloned and sequenced.
Different promoter constructs, all containing exon 1 (77 bp) preceded by different lengths of human tenascin‐W promoter sequences, were amplified from human genomic DNA isolated from HEK293 cells resulting in the following constructs: pSEAP‐TNW (−1800 bp), pSEAP‐TNW (−957 bp), pSEAP‐TNW (−512 bp), pSEAP‐TNW (−320 bp), pSEAP‐TNW (−252 bp), pSEAP‐TNW (−148 bp), pSEAP‐TNW (−79 bp), pSEAP‐TNW (−59 bp), pSEAP‐TNW (−35 bp), and pSEAP‐TNW (+77 bp). See Supporting Information Table 1 for the list of primers used. The primers included NheI and XhoI restriction sites for directional cloning into the MCS of pSEAP2‐basic (Clontech, Mountain View, CA). The three conserved intronic DNA fragments were amplified from HEK293 genomic DNA as template and inserted into NheI/HindIII sites of the pSEAP‐basic vector. Further deletions of the second conserved region were created. All primers used and their intronic locations are listed in Supporting Information Table 1. The three intronic regions were also cloned into the NheI and MluI sites 5′ of pSEAP‐TNW (−79 bp) creating the plasmids pSEAPI/‐79, pSEAPII/‐79 and pSEAPIII/‐79.
Site‐directed mutagenesis of the SMAD4‐binding site (gcctAGACagg) was performed using a scrambled sequence (AGAGTGATCA), which does not display any known cis‐acting sequence. Overlapping primers used for PCR including the scrambled sequences underlined are listed in Supporting Information Table 1. Following transformation of the DpnI‐digested PCR, plasmid DNA was isolated and the mutations confirmed by sequencing.
Cell transfection and reporter gene assays
HT1080 cells were cultured in DMEM/10% FCS. For reporter assays cells were plated at 5 × 104 cells/well in 12‐well plates overnight to reach 60–70% confluence. Cells were transfected by jetPEI (Polyplus, Strasbourg, France) with 0.6 µg/well of total DNA (pSEAP reporter construct and pMetLuc for normalization mixed at 1:20 molar ratio) in DMEM/0.3% FCS. Reporter activity was measured 24 hr after transfection. Alternatively, cells were transiently transfected in 3% FBS or 3% DCC‐treated FBS for 24 hr. Secreted alkaline phosphatase (SEAP) activity in the culture medium was determined using the SEAP Reporter Gene Assay Kit (Roche diagnostics) and for normalization the Ready‐To‐Glow secreted luciferase reporter system (Clontech) and measured in a luminometer (Mithras LB940; Berthold technologies, Bad Wildbad, Germany). SEAP values were normalized to Luciferase values to control for transfection efficiency. The normalized luminescence values were then standardized by the background activity (empty vector). The pSEAP‐TNW‐79 bp construct was included in all experiments and was used to calibrate different experiments. Error bars represent the standard error of the mean (SEM) between all replicates of each experiment.
Statistical analyses
Data are represented as means and SD or SEM as stated in the figure legends. Statistical analysis using a two‐tailed t‐test was carried out with SigmaPlot for Windows version 12.0. All the experiments shown are the means of three replicates. The difference between two groups was statistically significant when p < 0.05.
In silico analyses
The genomic location of the human tenascin‐W gene and its evolutionary conservation was examined using the UCSC genome browser (http://genome.ucsc.edu/). MatInspector (http://www.genomatix.de) was used to identify predicted transcription factor‐binding sites in the genomic sequences.
Results
Tenascin‐W is induced in bone metastases of MDA‐MB231‐1833 breast cancer cells
We investigated the expression of TNW in the bone stroma of MDA‐MB‐231‐SCP1883 tumor‐bearing mice. Osteoblasts were isolated from tumor‐free or tumor‐bearing bones as described in Material and Methods and RNA was extracted from this host cell population. Using RT‐PCR mouse TNW mRNA levels were upregulated fourfold to fivefold in osteoblasts isolated from bones harboring metastases compared with osteoblasts from nontumor‐bearing control bone (Fig. 1 a). Immunostaining of the long bones shows that TNW is expressed in the region surrounding the GFP‐expressing tumor cells (Fig. 1 b). Using species‐specific monoclonal antibodies, we verified that TNW is specifically unregulated in the host tissue as no human TNW was detectable. Thus, the MDA‐MB231‐1833 cells are not a source of TNW, but are surrounded by a host‐derived TNW‐rich ECM in the bone metastatic niche.
Figure 1.
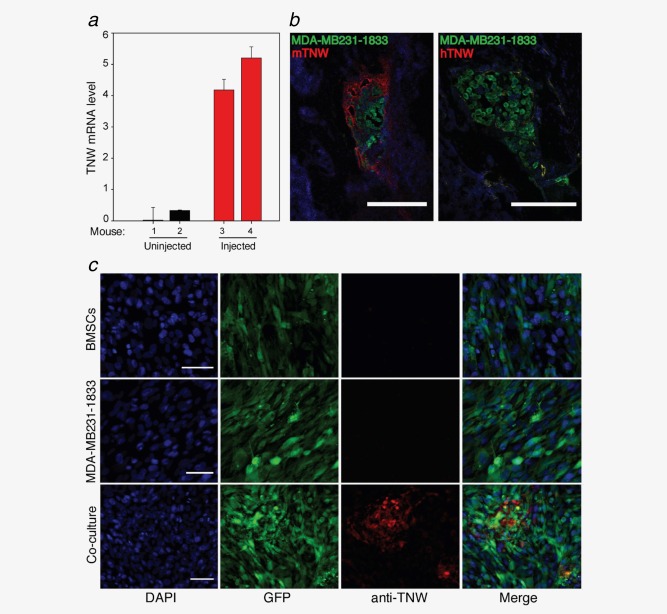
TNW is upregulated in bone metastasis of breast cancer. (a) TNW transcript levels in RNA isolated from osteoblasts sorted from two individual tumor‐free (1, 2, black) or two tumor‐bearing mice (3, 4, red). TNW levels relative to GAPDH were calculated using the relative standard curve method (ΔCt). Averages ± SD of two independent experiments in triplicate are shown. (b) Tissue sections of tibia show GFP‐MDA‐MB231‐1833 metastases (green). Staining of TNW was detectable with anti‐mouse TNW only (red, mTNW, left panel) but not with anti‐human TNW (hTNW, right panel). Scale bar, 100 µm. (c) Immunofluorescence staining for TNW of human bone marrow stromal cells (GFP‐BMSCs) and human breast cancer cells (GFP‐MDA‐MB231‐ 1833) cultured alone, or in co‐culture reveals TNW protein (red) expression after 7 days of co‐culture. Nuclei were labeled with DAPI (blue). Scale bars, 50 µm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Breast cancer cells release factors that induce tenascin‐W expression in human BMSCs
Prompted by the striking correlation of TNW expression with breast cancer metastases, co‐culture assays were established to investigate whether paracrine interaction between MDA‐MB231‐1833 and immortalized human BMSCs can induce TNW expression. Indeed, BMSCs are targeted by homing signals and they represent a significant cellular source not only of osteoblasts but also of tumor‐associated myofibroblasts that support tumor cell growth.16 Monocultures and co‐cultures of the two cell lines were subjected to immunofluorescence staining to detect TNW protein expression. Interestingly, TNW was exclusively detectable in co‐culture conditions and neither the tumor cells nor the BMSCs alone yielded any TNW staining (Fig. 1 c).
To examine whether the expression of TNW in co‐cultures depends on direct cell–cell contacts, transwell co‐culture assays were performed (Fig. 2). Each cell type was cultured in the bottom well of transwell culture dishes (Fig. 2 a), either alone or with the other cell type cultured in an upper well (Fig. 2 b). Upper and lower compartments were separated by a polycarbonate membrane with 0.4‐µm pores to allow the diffusion of soluble factors, but not the transmigration of cells (Fig. 2 b). For each condition, the cells in the bottom well were analyzed by immunostaining for TNW expression after 7 days in culture. This confirmed that neither cell type alone produced TNW, but TNW was induced in BMSCs in the presence of tumor cells in the upper chamber and not vice versa. Thus, in this in vitro system, tumor cells can induce the expression of TNW in the bone‐derived BMSCs and this depends on the release of a soluble factor (or factors) secreted by the tumor cells.
Figure 2.
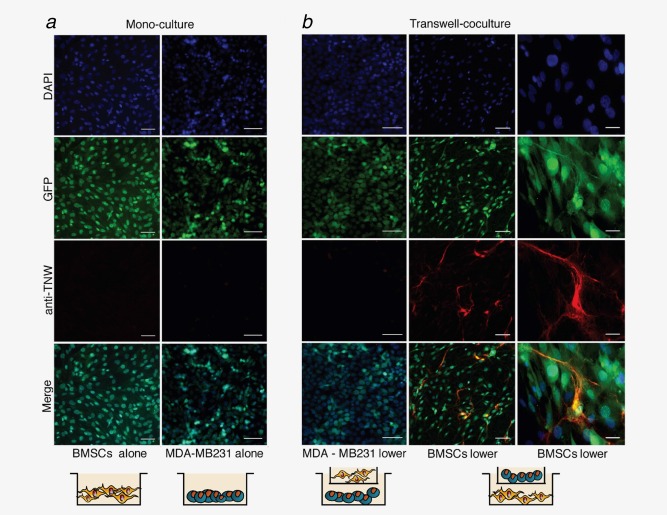
Soluble factors secreted by MDA‐MB231‐1833 breast cancer cells stimulate TNW expression in BMSCs. (a) Monocultures of GFP‐BMSCs (left panels) and GFP‐MDA‐MB231‐1833 cells (right panels) maintained for 7 days in culture. Nuclei were stained with DAPI (blue). Staining with human anti‐TNW monoclonal antibody does not detect any TNW expression. (b) MDA‐MB231‐1833 cells or BMSCs were seeded in the bottom well of transwell chambers (MDA‐MB231‐1833 lower; BMSCs lower) and exposed to the other cell type in the upper chamber in an indirect co‐culture system. Under these conditions, TNW protein expression (red) was detected exclusively in BMSCs and not in MDA‐MB231‐1833 cells. Scale bars: 100 µm for all panels except for the magnification shown in the right panels representing 20 µm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Transcriptional regulation of tenascin‐W gene expression
To get a hint on the possible factor and signaling pathway inducing TNW expression in BMSCs, we decided to isolate and study the TNW gene promoter and to identify regulatory sequences present in the TNW gene. In the case of human TNW, the gene name in the databases is TNN. The 5′part of the TNN gene, as it is annotated in the UCSC browser, is shown in Figure 3 a. It shows a first noncoding exon separated by a large intron from exon 2, which contains the ATG translation start. Underneath the sequence tracks, the evolutionary conservation reveals high conservation of the exon sequences and the putative promoter region as well as of three conserved regions in the first intron (Fig. 3 a). To experimentally confirm the predicted transcription start site (TSS) we performed 5′RACE experiments (Fig. 3 c). To assess potential transcript variants in different tissues that are known to express TNW, we used total RNA isolated from glioblastoma and breast cancer tissues which have high TNW expression levels17, 18 as well as RNA from healthy bone tissue, as TNW is known to be associated with the osteogenesis process.19 PCR products were cloned and sequenced. From all RNA sources tested, we found a single TNW transcript with a TSS at the noncoding first exon. Thus, normal and cancer tissues tested here are using a common single TSS to initiate TNW transcription.
Figure 3.
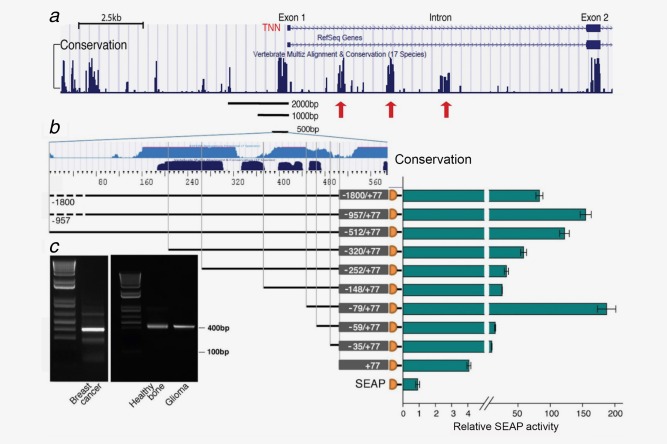
Experimental and computational analysis of the transcription start site and promoter activity of the TNW gene. (a) UCSC browser display of the 5′ region of the tenascin‐W (TNN) gene with the RefSeq genes depicting exon 1 and exon 2 separated by a large intron. In the Vertebrate Multiz Alignment & Conservation track below, note the areas of high conservation peaking in the upstream region next to exon 1, exon 1 and 2 and three regions within the first intron indicated by red arrows. (b) To characterize the TNW promoter, exon 1 (+77 bp) and different lengths of the 5′ flanking region as indicated were cloned upstream a promoterless SEAP vector. The UCSC Genome browser tracks ESPERR Regulatory Potential (seven species; light blue) and Vertebrate Multiz Alignment & Conservation (17 species; dark blue) of about 500 bp upstream of the TSS are depicted above the promoter constructs. Plasmid DNA constructs were transiently transfected in HT1080 cells for 24 hr. SEAP activity is normalized to a co‐transfected secreted luciferase plasmid and plotted relative to the promoterless SEAP vector control. Values are the average and SEM of three independent experiments. (c) Agarose gel electrophoresis shows the products resulting from 5′RACE of total RNA from breast cancer, healthy bone and glioma tissues as indicated. Cloning of the DNA bands (400 bp) and the subsequent sequencing revealed a single transcription start site adding a 79 bp first exon to exon 2 containing the ATG start codon. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
To identify regulatory regions present in the TNW promoter, promoterless SEAP (secreted embryonic alkaline phosphatase) reporter constructs containing exon 1 (+77) and different parts of the 5′ flanking region up to a length of 2 kb were cloned (Fig. 3 b). First we searched for a cell line that has the machinery to activate the TNW promoter reporter constructs. We tested the osteosarcoma cell lines U2OS, Saos‐2 and KRIB as well as the fibrosarcoma cell line HT1080 (not shown). As we obtained the highest reporter activity using HT1080 cells we continued our experiments with HT1080 cells. Transfection of the promoter constructs revealed that the main control region was contained within −512 bp of the TSS as longer constructs gave similar results. Thus, the promoter region coincides with the UCSC Genome browser tracks ESPERR Regulatory Potential (Fig. 3 b light blue) and the Vertebrate Multiple sequence Alignment & Conservation tracks (Fig. 3 b dark blue). However, further shortening of the promoter constructs to −320, −252 and −148 bp revealed a gradual loss of reporter activity. This could be related to the loss of binding sites for factors that enhance the promoter activity in a cooperative manner. One such factor could be SMAD1/5, as by MatInspector analysis a GC‐rich Smad‐binding element (SBE) was detected at position −230. An increase of reporter activity was observed by the deletion of the sequence from −148 to −79, pointing to the presence of a negative regulatory site in this region. Using MatInspector, a GATA‐binding element was detected in this region and may be responsible for the repression in a similar way as it was described for TNC.20 Interestingly, the short −79 bp minimal promoter exhibited the highest activity after transient transfection which was lost by the deletion of −79 to −59 bp, indicating important transcription factor‐binding sites within these 20 bp. Indeed, MatInspector analysis revealed a SMAD4 site at this location. The sequence of the TNW promoter and the first exon as well as potential transcription factor‐binding sites are given in Supporting Information Figure 1.
Analysis of the evolutionary conserved regions within the first intron
Further inspection of the TNN genomic locus using the UCSC genome browser revealed striking patterns of histone modifications within the first intron of the TNN gene, namely an H3K4 me1 enhancer chromatin signature, as well as H3K27 acetylation that defines nucleosome exclusion regions (Fig. 4 a). These epigenetic marks overlap with three evolutionarily conserved regions which we tested for a potential involvement in the regulation of TNW transcription (Fig. 4 b). We cloned the three intronic conserved modules I, II, and III upstream of the SEAP reporter gene and tested their effects on reporter gene activity. While the first conserved region (+1178/+1909) appeared to act as a silencer, the second conserved region (+2920/+3447) strongly enhanced transcription when compared to the pSEAP basic vector. The third region did not seem to influence the reporter gene activity when tested on its own, but in conjunction with the active region II, both region I as well as III seemed to silence the activity of region II (Fig. 4 b). Further dissection of the second conserved region showed that the sequence between +3299 and +3447 was mainly responsible for the strong activation (Fig. 4 b). Although we did not find any alternative TSS in the tissues analyzed, the possibility remains that this region might be an alternative promoter of the TNN gene.
Figure 4.
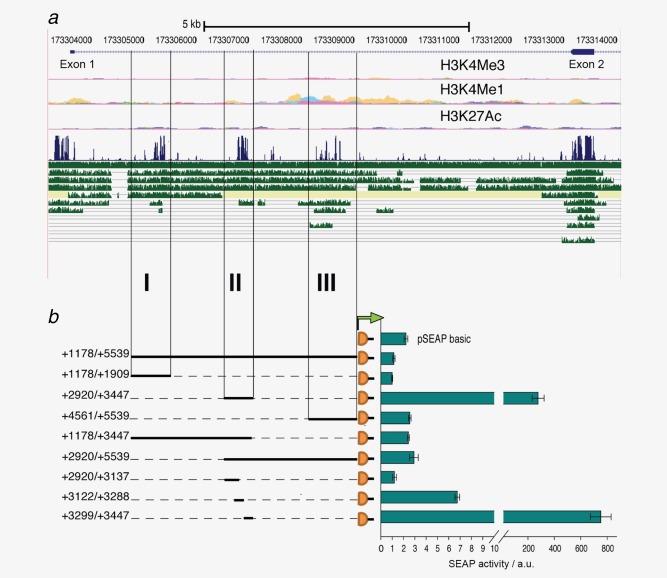
Analysis of the evolutionary conserved regions within the first intron. (a) UCSC browser display of the 5′ region of the tenascin‐W gene with absolute coordinates of chromosome 1 indicated above the RefSeq genes showing Exon 1 and 2 separated by a large Intron. Chromatin signatures shown below (H3K4Me3, H3K4Me1 and H3K27Ac) overlap with the intronic modules. Below the Vertebrate Multiz Alignment & Conservation track in blue, tracks in green show from top to bottom comparisons with rhesus, mouse, dog, horse, armadillo, opossum, platypus, lizard, chicken, X_tropicalis and stickleback orthologs. (b) To assess potential transcriptional activities, each conserved region I, II and III, combinations thereof and truncated versions as indicated were cloned upstream of the SEAP reporter gene. Transient transfections of the constructs in HT1080 cells were analyzed for SEAP activity shown in arbitrary units (a.u.) normalized to a co‐transfected secreted luciferase plasmid. The experiment was performed in triplicates and repeated three times (error bars = SEM). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Identification of negative and positive trans‐acting elements in the TNW gene
After the experimental determination of the most important regions of the TNW gene affecting promoter activity, we experimentally tested the identified putative transcription factor‐binding sites within the promoter and first intron regions. Computational predictions showed the presence of a SMAD4 nuclear transcription factor‐binding site at −61 bp from the TSS, next to a TATA box sequence located at −54 bp upstream of exon 1. This SMAD4‐binding site (gcctAGACagg) is located within the 20 bp region found to be crucial for the high activity of the proximal TNW promoter. To test whether this binding site is responsible for the promoter activity, site‐directed mutagenesis of the SMAD4 element was performed (Fig. 5 a). Indeed, the presence of the scrambled sequence (AGAGTGATCA) strongly impaired the SEAP reporter gene expression driven by the minimal basal promoter (−79 bp). SMAD4 is an intracellular mediator of the TGF‐β signaling pathway. Therefore, we assessed whether TGF‐β1 acts on endogenous TNW gene expression in BMSCs (Fig. 5 b). To examine the specificity of action of TGF‐β1 on TNW gene transcription we evaluated the efficiency of SB‐431542, a selective inhibitor of the TGF‐β1 receptor/ALK5, to inhibit TNW induction (Fig. 5 b). Preincubation of BMSCs with the ALK5 inhibitor abolished induction of TNW transcripts as assessed by qRT‐PCR. Thus, we identified TGF‐β1 as a factor inducing human TNW gene expression in BMSCs through activation of ALK5.
Figure 5.
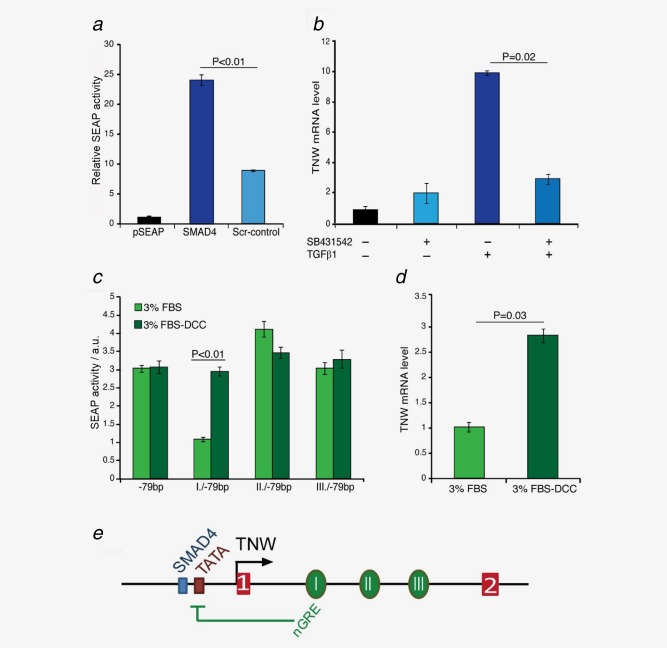
Regulation of TNW transcription by TGFβ1 and glucocorticoids. (a) The Smad4‐binding site (gcctAGACagg) was mutated by site‐directed mutagenesis and replaced with the scrambled sequence (Scr‐AGAGTGAT). Construct −79/+77 containing the normal (SMAD4) or modified Smad4 sequence (Scr‐control) was transfected in HT1080 cells and analyzed for SEAP activity normalized to the empty pSEAP as described in Fig. 3 b. (b) BMSCs were treated with SB‐431542‐DMSO (10 µM) or DMSO only for 1 h before the addition of TGFβ1 (5 ng/ml) for 24 h. TNW transcript levels were then analyzed by qRT‐PCR in comparison to the endogenous TBP. Relative mRNA levels were normalized to the nontreated control cells using the ΔΔCt method. (c) Constructs with the three intronic regions (I/II/III) cloned upstream the minimal TNW promoter (−79 bp/+77) were transfected in HT1080 cells in medium containing 3% FBS (light green bars) or glucocorticoid‐depleted 3% DCC‐FBS (dark green bars) for 24 hr. SEAP activity is shown in arbitrary units (a.u.) normalized to a co‐transfected secreted luciferase plasmid revealing that the intronic conserved region I inhibited the reporter activity in a glucocorticoid‐dependent manner. (d) BMSCs were cultured in untreated 3% FCS (light green bar) or in the presence of 3% charcoal/dextran‐treated FBS for 24 hr before measuring transcript levels of TNW by qRT‐PCR in comparison to the endogenous TBP. Relative mRNA levels were normalized to the cells grown in 3% FCS using the ΔΔCt method. All experiments were repeated at least three times (error bars = SEM). (e) Schematic model of the TNW gene regulation by a SMAD4 element preceding a TATA box in the proximal promoter region upstream of exon 1 (red box 1) and a negative glucocorticoid‐response element (nGRE) in the first conserved region within the first intron (green oval I). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Further sequence analysis of the intron showed a negative glucocorticoid response element (nGRE: tttttccaGGAGaga) located in the first conserved region. This prompted us to investigate whether glucocorticoids have an inhibitory effect on TNW transcription. For this purpose, we stripped endogenous steroids from the serum by using dextran‐coated charcoal (DCC‐FBS). Conserved intronic regions were cloned upstream of the −79 bp proximal promoter construct and transfected into HT1080 cells in the presence of 3% untreated FBS or DCC‐FBS for 24 hr (Fig. 5 c). Indeed the first intronic module (I/−79 bp) containing the nGRE exerted transcriptional repression of the TNW promoter measured by a twofold lower activity in the cells grown in untreated versus steroid‐stripped FBS. This was confirmed in BMSCs. In the presence of glucocorticoid‐depleted FBS, BMSCs showed higher TNW transcript levels than in normal FBS (Fig. 5 d). A model representing the main regulatory elements of the promoter and first intron of the TNW gene analyzed here is shown in Figure 5 e.
Cancer cells with bone tropism induce TNW in BMSCs and are enriched in TGF‐β1 transcripts
To investigate whether induction of TNW by the conditioned medium of MDA‐MB231‐1833 cells is caused by a paracrine action of TGF‐β1 secreted by the cancer cells we added the conditioned medium to BMSCs that were pretreated with SB‐431542 (Fig. 6 a). Indeed, the induction of TNW by the conditioned medium was significantly reduced by the inhibition of the TGF‐β1 receptor ALK5 confirming that TGF‐β1 is one of the MDA‐MB231‐1833 secreted factors inducing TNW. However, the inhibition was not complete and other factors may contribute to TNW induction, such as TNF‐α or IL8. Transcripts of both of these cytokines as well as TGF‐β1 are readily detectable by RT‐PCR in RNA isolated from MDA‐MB231‐1833 cells as shown on the agarose gel in Figure 6 a. To test whether the TNW‐inducing activity was specific for breast cancer cells with bone tropism, we compared the induction of TNW by conditioned medium of MDA‐MB231‐1833 cells with their parental cells MDA‐MB231 (Fig. 6 b). We also tested another cell line known to metastasize to bone,21 the mouse mammary cell line 4T1.2 in comparison to its parental 4T1 cells (Fig. 6 c). In both cases, the induction of TNW transcripts in BMSCs was higher for the bone metastatic subline than for the parental cells. Interestingly, this was accompanied by higher TGF‐β1 transcript levels in the bone metastatic lines as can be seen by the RT‐PCR products shown on the agarose gels in Figures 6 b and 6 c. These observations are consistent with a role for TGF‐β1 in the induction of TNW by cancer cells with bone tropism.
Figure 6.
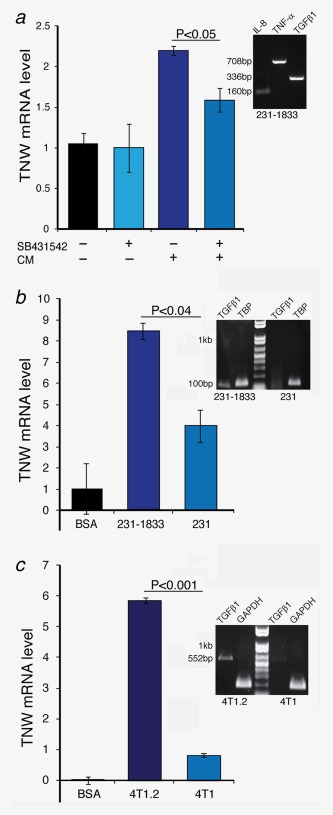
Induction of TNW in BMSCs by MDA‐MB231‐1833‐ and 4T1.2‐conditioned media. (a) BMSCs at 70% confluence were washed with PBS and maintained in serum‐free α‐MEM with or without the addition of 10 µM SB‐431542 inhibitor for 1 hr. Following SB‐431542 treatment, BMSCs were exposed to conditioned medium from MDA‐MB231‐1833 cells. Total RNA was extracted and TNW transcript levels were analyzed by qRT‐PCR in comparison to the endogenous hTBP. Relative mRNA levels were normalized to the nontreated control cells using the ΔΔCt method. The agarose gel to the right shows RT‐PCRs from RNA isolated from MDA‐MB231‐1833 cells to reveal the presence of hTGFβ1 (336bp), hIL‐8 (160bp) and hTNF‐α (708bp) transcripts in MDA‐MB231‐1833 cells. (b) BMSCs at 70% confluence were washed with PBS and either maintained in serum‐free α‐MEM/0.2% BSA as control (BSA) or conditioned media from MDA‐MB231 or MDA‐MB231‐1833 cells. Total RNA was extracted and TNW transcript levels were analyzed by qRT‐PCR in comparison to the endogenous TBP. Relative mRNA levels were normalized to the nontreated control cells using the ΔΔCt method. The agarose gel to the right shows RT‐PCRs from RNA isolated from MDA‐MB231‐1833 versus MDA‐MB231 cells to reveal the presence of hTGFβ1 (101bp) relative to hTBP (132). (c) BMSCs at 70% confluence were washed with PBS and either maintained in serum‐free α‐MEM/0.2% BSA as control (BSA) or conditioned media from 4T1.2 or 4T1 cells. Total RNA was extracted and TNW transcript levels were analyzed by qRT‐PCR in comparison to the endogenous mGAPDH. Relative mRNA levels were normalized to the nontreated control cells using the ΔΔCt method. The agarose gel to the right shows RT‐PCRs from RNA isolated from 4T1.2 versus 4T1 cells to reveal the presence of mTGFβ1 (552bp) relative to mGAPDH (189). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Tenascin‐W promotes breast cancer cell migration and proliferation in vitro
As the metastatic tumor cells in vivo are surrounded by TNW we investigated a potential functional role of TNW in tumor progression. MDA‐MB231‐1833 cells were treated with different concentrations of human TNW recombinant protein (5–10 µg/ml) and analyzed for proliferation and migration. Equal numbers of cells were plated on day 1. The next day we added serum‐free medium with or without TNW for another 24 hr followed by the measurement of BrdU incorporation over a 2‐hr period. There was a 3.5‐fold increase in BrdU incorporation in MDA‐MB231‐1833 cells cultured in the presence of TNW indicating that more cells are replicating in the presence of TNW than without addition of TNW (Fig. 7 a). Cell migration was assessed using transwell chamber assays. The underside of the filters was coated with TNW to investigate its role as chemoattractant. There was a 3.5‐fold increase in MDA‐MB231‐1833 cell migration toward TNW compared to control filters indicating a promigratory function of TNW for the MDA‐MB231‐1833 cells (Fig. 7 b).
Figure 7.
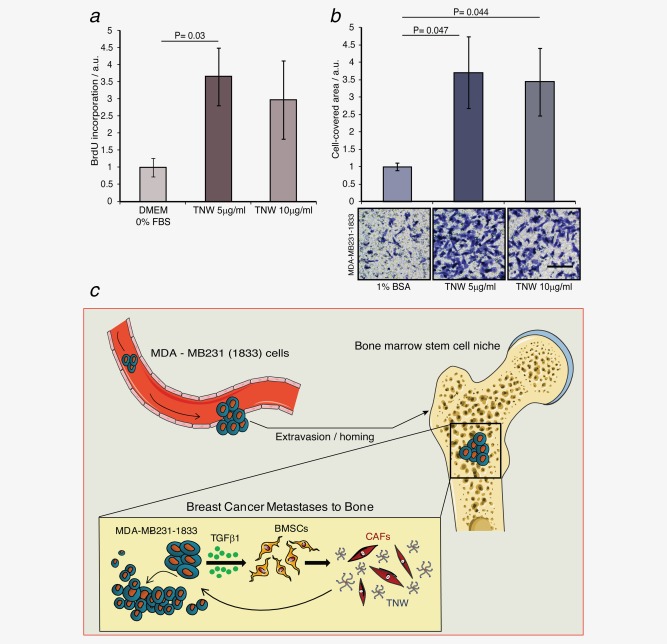
Effect of TNW on breast cancer cell proliferation and migration. (a) MDA‐MB231‐1833 cells were treated with different concentrations of recombinant human TNW protein (5–10 µg/ml) in serum‐free medium. DNA replication was measured by BrdU incorporation for 2 hr in arbitrary units (a.u.) 24 hr after exposure to TNW. (b) MDA‐MB231‐1833 cells were seeded in the upper chambers of transwell migration dishes and allowed to migrate for 24 hr toward TNW coated on the underside of the filters. Cells on the bottom side of the filters were stained and photographed (scale bar = 50 µm) and the cell‐covered area was quantified (error bars = SD). Migration is depicted in arbitrary units (a.u.) as determined by measuring the area of migrated cells using the Fiji distribution of ImageJ. Statistical analysis was assessed by paired Student's t‐test. p Values <0.05 are considered statistically significant. (c) Model for the role of TNW in breast cancer metastasis to bone: After extravasion and homing to the bone MDA‐MBA231 cells are releasing TGFβ1 which triggers BMSCs to differentiate into TNW‐secreting cancer‐associated fibroblasts (CAFs). TNW in turn is able to influence the growth and further invasion of the metastatic cancer cells. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Discussion
It is well known that paracrine signals released by breast cancer cells orchestrate the fate of stromal cells residing in the tumor microenvironment and vice versa.4, 22, 23 Stromal cells are targeted by tumor‐released factors and in response undergo myofibroblast‐like differentiation to a so‐called cancer‐associated fibroblast.24 We propose that breast cancer cells, after homing to the bone marrow via blood circulation, secrete TGFβ1 which induces changes in the bone marrow niche, including the deposition of TNW in the ECM. Exposure of MDA‐MB231‐1833 cells to TNW supports their migration and proliferation establishing a “vicious cycle” for cancer progression by promoting the growth and further invasive behavior of the breast cancer metastases (Fig. 7 c). We showed that a crucial signaling pathway for the induction of TNW is the TGFβ1 secreted by the tumor cells which induces Smad4‐dependent transcription of the TNW gene in the BMSCs. Indeed, TGF β1 has been found to be a major osteolytic factor secreted by MDA‐MB231 cells.25 Furthermore, downregulation of Smad4 in MDA‐MB‐231 cells strongly inhibited bone metastasis formation.26 In contrast to the induction of TNW expression by TGFβ1, we found that glucocorticoids have a negative impact on TNW expression. A similar antagonistic effect has also been described for the regulation of the TNW family member TNC and TNX.27, 28 Interestingly, anti‐inflammatory agents, including corticosteroids, are in use as cancer therapy.29 It is likely that such treatments also affect the ECM composition of the cancer microenvironment.
The actual situation in vivo is of course more complicated and it is clear that also osteoclasts are involved in the metastatic process.30, 31 It is known that upon stimulation of osteoblasts by cytokines released by the tumor cells, osteoblasts release RANKL which in turn promotes differentiation and activation of osteoclasts. Activated osteoclasts resorb the bone matrix with the consequent release of cytokines including TGFβ.32 These cytokines will act back on the osteoblasts as well as the tumor cells and in a vicious cycle keep the osteolytic lesions growing. Because of these findings, inhibition of osteoclasts by bisphosphonates as well as RANKL and TGFβ‐targeting agents are presently tested as adjuvant therapy for the treatment of breast cancer patients with bone metastases.32, 33, 34, 35, 36 The benefit of such treatments may include reduced TNW expression in the microenvironment of the metastatic cells and alleviate the tumor‐promoting effects exerted by TNW. Tumor‐promoting effects of TNW may include promotion of growth and migration as well as proangiogenic effects, as TNW was found to be expressed around tumor blood vessels in several types of cancers and to stimulate endothelial sprouting in culture.8, 17 Furthermore, with its tumor and metastasis‐specific expression, TNW may itself be a target for antibody‐mediated drug targeting of the bone metastatic niche.
Supporting information
Supporting Information
Acknowledgements
The authors thank Martin Degen for help with the cloning of the promoter constructs, Sandrine Bichet for help with immunostainings and Hubertus Kohler for FACS sorting of cells. They thank Matthias Chiquet and Richard P. Tucker for critical reading of the manuscript. R.C.‐E. received funds from the Swiss National Science Foundation grants number NF31003A_120235 and NF31003A_135584 as well as from the Swiss Cancer League grant number KFS‐2980‐08‐2012. N.E.H. was supported by grants SNF 310030‐138417 and 310030‐149751 and KFS 02743‐02‐201 and the Novartis Research Foundation.
References
- 1. Coleman RE, Rubens RD. The clinical course of bone metastases from breast cancer. Br J Cancer 1987;55:61–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ‐specific colonization. Nat Rev Cancer 2009;9:274–84. [DOI] [PubMed] [Google Scholar]
- 3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 4. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19:1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oskarsson T, Massague J. Extracellular matrix players in metastatic niches. EMBO J 2012;31:254–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Borovski T, De Sousa EMF, Vermeulen L, et al. Cancer stem cell niche: the place to be. Cancer Res 2011;71:634–9. [DOI] [PubMed] [Google Scholar]
- 7. Brellier F, Chiquet‐Ehrismann R. How do tenascins influence the birth and life of a malignant cell? Cell Mol Med 2012;16:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brellier F, Martina E, Degen M, et al. Tenascin‐W is a better cancer biomarker than tenascin‐C for most human solid tumors. BMC Clin Pathol 2012;12:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oskarsson T, Acharyya S, Zhang XH, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med 2011;17:867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'Connell JT, Sugimoto H, Cooke VG, et al. VEGF‐A and tenascin‐C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci USA 2011;108:16002–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scherberich A, Tucker RP, Samandari E, et al. Murine tenascin‐W: a novel mammalian tenascin expressed in kidney and at sites of bone and smooth muscle development. J Cell Sci 2004;117:571–81 [DOI] [PubMed] [Google Scholar]
- 12. Kang Y, Siegel PM, Shu W, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003;3:537–49. [DOI] [PubMed] [Google Scholar]
- 13. Liu H, Patel MR, Prescher JA, et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc Natl Acad Sci USA 2010;107:18115–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bourgine P, Le Magnen C, Pigeot S, et al. Combination of immortalization and inducible death strategies to generate a human mesenchymal stromal cell line with controlled survival. Stem Cell Res 2014;12:584–98. [DOI] [PubMed] [Google Scholar]
- 15. Brellier F, Ruggiero S, Zwolanek D, et al. SMOC1 is a tenascin‐C interacting protein over‐expressed in brain tumors. Matrix Biol 2011;30:225–33. [DOI] [PubMed] [Google Scholar]
- 16. Quante M, Tu SP, Tomita H, et al. Bone marrow‐derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011;19:257–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martina E, Degen M, Ruegg C, et al. Tenascin‐W is a specific marker of glioma‐associated blood vessels and stimulates angiogenesis in vitro. FASEB J 2010;24:778–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Degen M, Brellier F, Kain R, et al. Tenascin‐W is a novel marker for activated tumor stroma in low‐grade human breast cancer and influences cell behavior. Cancer Res 2007;67:9169–79. [DOI] [PubMed] [Google Scholar]
- 19. Martina E, Chiquet‐Ehrismann R, Brellier F. Tenascin‐W: an extracellular matrix protein associated with osteogenesis and cancer. Int J Biochem Cell Biol 2010;42:1412‐15. [DOI] [PubMed] [Google Scholar]
- 20. Ghatnekar A, Trojanowska M. GATA‐6 is a novel transcriptional repressor of the human tenascin‐C gene expression in fibroblasts. Biochim Biophys Acta 2008;1779:145–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eckhardt BL, Parker BS, van Laar RK, et al. Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol Cancer Res 2005;3:1–13. [PubMed] [Google Scholar]
- 22. Wan L, Pantel K, Kang Y. Tumor metastasis: moving new biological insights into the clinic. Nat Med 2013;19:1450–64. [DOI] [PubMed] [Google Scholar]
- 23. McMillin DW, Negri JM, Mitsiades CS. The role of tumour‐stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov 2013;12:217–28. [DOI] [PubMed] [Google Scholar]
- 24. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006;6:392–401. [DOI] [PubMed] [Google Scholar]
- 25. Pederson L, Winding B, Foged NT, et al. Identification of breast cancer cell line‐derived paracrine factors that stimulate osteoclast activity. Cancer Res 1999;59:5849–55. [PubMed] [Google Scholar]
- 26. Deckers M, van Dinther M, Buijs J, et al. The tumor suppressor Smad4 is required for transforming growth factor beta‐induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res 2006;66:2202–9. [DOI] [PubMed] [Google Scholar]
- 27. Talts JF, Weller A, Timpl R, et al. Regulation of mesenchymal extracellular matrix protein synthesis by transforming growth factor‐beta and glucocorticoids in tumor stroma. J Cell Sci 1995;108:2153–62. [DOI] [PubMed] [Google Scholar]
- 28. Sakai T, Furukawa Y, Chiquet‐Ehrismann R, et al. Tenascin‐X expression in tumor cells and fibroblasts: glucocorticoids as negative regulators in fibroblasts. J Cell Sci 1996;109:2069–77. [DOI] [PubMed] [Google Scholar]
- 29. Rayburn ER, Ezell SJ, Zhang R. Anti‐inflammatory agents for cancer therapy. Mol Cell Pharmacol 2009;1:29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen YC, Sosnoski DM, Mastro AM. Breast cancer metastasis to the bone: mechanisms of bone loss. Breast Cancer Res 2010;12:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roodman GD. Mechanisms of bone metastasis. N Engl J Med 2004;350:1655–64. [DOI] [PubMed] [Google Scholar]
- 32. Chiechi A, Waning DL, Stayrook KR, et al. Role of TGF‐ in breast cancer bone metastases. Adv Biosci Biotech 2013;4:15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Holten‐Verzantvoort AT, Bijvoet OL, Cleton FJ, et al. Reduced morbidity from skeletal metastases in breast cancer patients during long‐term bisphosphonate (APD) treatment. Lancet 1987;2:983–5. [DOI] [PubMed] [Google Scholar]
- 34. Dougall WC, Holen I, Gonzalez Suarez E. Targeting RANKL in metastasis. Bonekey Rep 2014;3:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clezardin P. Therapeutic targets for bone metastases in breast cancer. Breast Cancer Res 2011;13:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Onishi T, Hayashi N, Theriault RL, et al. Future directions of bone‐targeted therapy for metastatic breast cancer. Nat Rev Clin Oncol 2010;7:641–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information