

Abstract
The human gut microbiota performs essential functions for host and well‐being, but has also been linked to a variety of disease states, e.g., obesity and type 2 diabetes. The mammalian body fluid and tissue metabolomes are greatly influenced by the microbiota, with many health‐relevant metabolites being considered ‘mammalian–microbial co‐metabolites’. To systematically investigate this complex host–microbial co‐metabolism, a systems biology approach integrating high‐throughput data and computational network models is required. Here, we review established top‐down and bottom‐up systems biology approaches that have successfully elucidated relationships between gut microbiota‐derived metabolites and host health and disease. We focus particularly on the constraint‐based modeling and analysis approach, which enables the prediction of mechanisms behind metabolic host–microbe interactions on the molecular level. We illustrate that constraint‐based models are a useful tool for the contextualization of metabolomic measurements and can further our insight into host–microbe interactions, yielding, e.g., in potential novel drugs and biomarkers. WIREs Syst Biol Med 2015, 7:195–219. doi: 10.1002/wsbm.1301
For further resources related to this article, please visit the WIREs website.
Conflict of interest: The authors have declared no conflicts of interest for this article.
INTRODUCTION
The Human Gut Microbiome
Humans harbor a complex ecosystem, the gut microbiota, in their intestines. The collective genome of the microbiota, the ‘microbiome’, contains 100–150 times as many genes as the human genome.1 The gut microbiota performs many important functions for the host, such as digestion of nutrients,2 maturation of the host immune system,3 maintenance of epithelial cell layer integrity,4 and protection against pathogens.5 Worldwide research efforts, including MetaHIT1 and the Human Microbiome Project,6 have elucidated the genetic repertoire and the phylogenetic composition of the human gut microbiome. These efforts have revealed that the human gut microbiome is dominated by two bacterial phyla, Bacteroidetes and Firmicutes, which account for more than 90% of the detected phylotypes.7, 8 Other microbial phyla found in the human gut microbiome include Actinobacteria, Proteobacteria, Fusobacteria, and Verrucomicrobia.9 Moreover, the concept of three enterotypes allowing stratification of human individuals according to their gut microbiome composition has been proposed.9
The gut microbiome is implicated in the etiology of many diseases. For instance, obesity has been directly linked to the gut microbiota.10 The obesity epidemic is partly caused by the high‐sugar, high‐fat diet consumed in developed countries; this diet is known to affect the composition of the gut microbiota.10 It has been proposed that there is a relationship between an increased ratio of Firmicutes to Bacteroidetes and an obese phenotype in humans and mice7, 11; however, this finding has not been consistently reproduced, and its importance remains unclear.10 Obesity‐related diseases, such as metabolic syndrome, type 2 diabetes, and cardiovascular disease as well as inflammatory bowel diseases, have also been associated with changes in the gut microbiome composition.10 Furthermore, there is increasing evidence that an altered gut microbiota is associated with autism12 and neurodegenerative diseases, such as Parkinson's disease.13 An underweight status and malnutrition in children have also been associated with altered gut microbiota development.14 It has been proposed that our ‘Western’ diet has resulted in a ‘dysbiotic’, less diverse microbiota, leading to the observed dramatic increase in lifestyle‐associated diseases.15 For instance, lower microbial gene richness has been linked to adiposity, higher insulin resistance, and inflammation.16 The gut microbiota contains ‘keystone’ species (e.g., Ruminococcus bromii) that are low in abundance but specialize in important functions, such as resistant starch degradation,17 and these species may be partially eliminated in human societies consuming the Western diet.15 However, the mechanisms underlying the relationship between the gut microbiota and disease states are still not completely understood. While certain genera are considered to be either protective against inflammation (e.g., Faecalibacterium)18 or proinflammatory (e.g., Escherichia),19 the role of most genera present in the gut in human health and well‐being remains unclear.
Mammalian–Microbial Co‐Metabolism and Impact on Human Health
Recent research efforts have moved beyond studying the composition (what is there) to investigating the functionality (what are they doing) of the human gut microbiota.20 While the same phyla are consistently found in the gut microbiomes of humans, interpersonal variation in type and abundance at the genus and species level is high.21 In fact, the gut microbiota varies greatly even between closely related individuals.22 In contrast, a human gut microbiome gene catalog assembled from 124 Europeans revealed a core set of approximately 300,000 redundant microbial genes present in at least 50% of individuals, which included genes involved in essential pathways, such as polysaccharide degradation, short‐chain fatty acid production, and amino acid and vitamin biosynthesis.1 Consequently, it has been proposed that there is a ‘core microbiome’ consisting of a set of universal metabolic functions, rather than common genera or species.20, 21
Metabolomic analysis has resulted in an emerging picture of the mammalian–microbial ‘co‐metabolome’.23 Gut microbes secrete a variety of health‐relevant metabolites that play a role in the etiology and prevention of complex diseases.24, 25 For instance, gut microbial conversion of choline to trimethylamine increases the risk of cardiovascular disease26 and gut microbes also transform dietary polyphenols into metabolically active antioxidative compounds.27 The microbiota strongly influences host metabolic phenotypes and treatment with antibiotics significantly alters the urine and fecal metabolome.28 Metabolites synthesized by the microbiota include compounds that can directly regulate and modulate host metabolism, such as neurotransmitters and hormones. In fact, the gut microbiota can be considered as an additional endocrine organ.29 The gut microbiota also metabolizes and transforms xenobiotics, including a variety of drugs, which must be taken into consideration in future drug development.30 Hence, the gut microbiota affects a variety of health‐relevant metabolic functions in human (Table 2).
Notably, despite the presence of a core set of metabolic functions, there is some interpersonal variety in the metabolic activity of the microbiota. For example, two distinct phenotypes in rats can be distinguished based on the gut microbial metabolism of phenylalanine, with a possible connection to biomarkers associated with autism.31 Moreover, changes in the gut microbial composition, e.g., through bariatric surgery, can alter the metabolic phenotype of the host.32, 33 Such findings may lead to the development of personalized medicine23 and personalized nutrition34 tailored to patients' individual genetic phenotypes, lifestyles, and microbiota compositions. The numerous factors affecting human health (e.g., genetic traits, environment, nutrition, and the microbiota composition) require an integrated systems biology approach.23 Such a systems biology framework could reveal nonintuitive relationships between these factors, identifying novel biomarkers.23, 35
SYSTEMS BIOLOGY APPROACHES FOR STUDYING HOST–MICROBE CO‐METABOLISM
Top‐Down and Bottom‐Up Systems Biology
The gut microbial ecosystem that co‐exists with the human host can be described as a superorganism whose metabolic potential far exceeds that of a human alone.3 High‐throughput methods, such as metagenomic, metatranscriptomic, and metabolomic analyses, have greatly increased our knowledge about the diversity and functionality of the human gut microbiota36 and the influence of the gut microbiota on metabolic phenotypes.37 To achieve a comprehensive understanding of host–microbe interactions, such ‘big data’ must be contextualized. One established method for placing multi‐omics data into context is overlaying these data with network reconstructions.38 There are a variety of network modeling techniques for high‐throughput data with different strengths and weaknesses, including constraint‐based modeling, kinetic modeling, and Bayesian approaches.39 The main methods discussed in this review include top‐down systems biology, topological models, and constraint‐based models, which are compared in Table 1.
Table 1.
Summary of the Main Modeling Methods Discussed in This Review
| Feature | Top‐Down Metabonomics | Topological Network Modeling | Constraint‐Based Modeling |
|---|---|---|---|
| Model system | Multivariate statistical model | Supra‐organism network | Genome‐scale reconstruction(s) |
| Scope | Metabolite profiles of the gut microbiota/host organs and biofluids | Microbiome‐wide | One or more target organisms (host or microbes) on the genome scale |
| Main inputs | Metabolomic measurements from biofluids or tissue | Metagenomic data | Target organism's genome sequence Optional inputs:
|
| Types of predictions |
|
|
|
| Advantages |
|
|
|
| Disadvantages |
|
|
|
A distinction can be made between top‐down approaches, in which network structures and conclusions are inferred through statistical analysis, and bottom‐up approaches that employ manually constructed and validated networks.40 In the top‐down approach, genome‐wide high‐throughput data are the starting point. Top‐down analysis can easily integrate metabolomic, transcriptomic, and proteomic data.41 From this view of the system as a whole, mechanisms closer to the bottom are inferred with the aim of biological discovery. Networks are inferred from experimental data. Rather than being knowledge‐based, top‐down systems biology aims to glean new knowledge from the correlations between data points.41 The strengths of top‐down approaches are that they are broad in scope, provide complete, genome‐wide views of the modeled organism(s), and do not require extensive manual curation efforts. A disadvantage of these methods is that they lack accuracy and mechanistic insight because network structures are merely inferred and not based on structural and biochemical knowledge.40 In contrast, the bottom‐up systems biology approach aims to be well structured and accurate. Pathways are assembled based on detailed information obtained from experimental studies and integrated into a large‐scale reconstruction.42 Bottom‐up reconstructions are mechanism based41 and predictive. A main weakness of the bottom‐up systems biology approach is the laborious network reconstruction involved. The genome‐scale reconstruction of a well‐studied microorganism can take up to 6 months.43 Moreover, owing to the need for manual curation and computational power, bottom‐up networks are limited in scope, which is especially true for network models of large microbial communities. However, automated reconstruction tools44, 45, 46, 47, 48, 49 and efficient algorithms50, 51 designed to partly overcome these weaknesses are available. The methods for bottom‐up network reconstruction of genome‐scale metabolic networks are well established,43 but this methodology has also been applied to nonmetabolic cellular processes, such as macromolecular synthesis52, 53 and signaling pathways.54, 55, 56
Top‐Down Host–Microbe Metabolomics
Top‐down systems biology combined with high‐throughput metabolomic data has provided valuable insight into the co‐metabolism of mammals and their microbiota. The metabotype, or the metabolic phenotype of a target organism as measured from its biofluids (e.g., blood, plasma, and urine), is commonly measured using nuclear magnetic resonance (NMR) spectroscopy or mass spectrometry (MS).23, 57 Metabolomic analyses are untargeted, resulting in the broad quantification of biofluid metabolites, or targeted, thus accurately quantifying selected metabolites of interest.57 Untargeted metabolomics may result in the discovery of novel biomarkers.57 The interpretation of NMR spectroscopy measurements via multivariate statistical analysis is deemed ‘metabonomics’.58, 59 Common multivariate statistical methods are principal component analysis (PCA) and partial least squares‐discriminant analysis (PLS‐DA), which result in a separation of data points into clusters according to different metabotypes.57 Statistical analyses may result in the discovery of biomarkers that explain the differences between control groups and clinical states.57 Metabolomic measurements can also be mapped onto pathways, for example, through metabolite set enrichment analysis (MSEA).57 MSEA compares metabolomic measurements with metabolic pathway maps or databases, which can identify upregulated pathways compared with the control condition.57
Top‐down systems biology approaches have been applied to a variety of human and animal studies. For example, the metabolic profiles of germfree mice colonized with human baby flora and conventional mice were compared.60 This analysis revealed significant differences in bile acid and lipid metabolism in the host.60 Another study investigated the effects of probiotic lactobacilli on germfree mice colonized with human by flora.61 Probiotic supplement modulated bile acid and energy metabolism and increased the levels of the microbial co‐metabolites indoleacetylglycine, phenylacetylglycine, and tryptamine.61 Probiotics, prebiotics, and synbiotics (the combined application of both) also modulated multiple host organs in another human baby flora mouse model.62 Zucker rats are established model organisms of obesity and type 2 diabetes. Different microbiome compositions and metabolic phenotypes have been found in lean and obese Zucker rats.63 Several studies have compared the metabolomes of germfree and conventional mice, revealing that the microbiota has systemic effects on whole‐body metabolism and on biofluid and tissue metabolite profiles.64, 65, 66, 67, 68 Similarly, antibiotic treatment of rats altered a wide range of urinary and fecal mammalian–microbial co‐metabolites demonstrating that the gut microbiota strongly impacts a variety of metabolic subsystems.28 Correlations between mammalian–microbial co‐metabolites and certain microbial species have been inferred through multivariate statistical analysis of metabolomic data. For instance, 10 bacteria were shown to be correlated with urinary metabolites (e.g., lactate, citrate, phenylacetylglutamine, and 4‐cresol sulfate) in humans.69 However, the mechanisms underlying such links between microbes and host metabolism remain poorly understood.
Topological Network Models
Topological network models of the human gut microbiome are constructed based on the microbiome‐wide metabolic gene content, typically with nodes representing metabolites and links representing reactions (Figure 1(a)). Metagenomic data serve as input for these models, which consider the gut microbiota as a single supra‐organism, where species boundaries and the species origin of genes are ignored. The resulting network reflects the metabolic potential of the entire gut microbiome and thus represents the gut ecosystem on a systems level.
Figure 1.
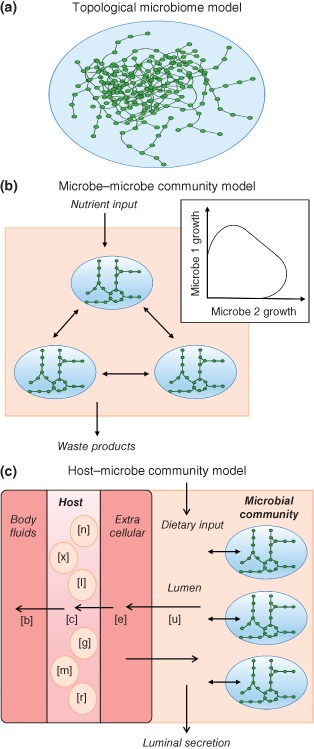
Schematic representation of the major network modeling approaches utilized in systems biology analyses of host–microbe interactions. (a) Topological microbiome model. In this approach (e.g., Ref 70), the gut microbiota is treated as a single supra‐organism without species–species boundaries, with nodes representing metabolites and links representing reactions. Topological features of the gut microbial metabolic network, e.g., betweenness centrality (defined as the proportion of shortest paths passing through a node) or neighborhood connectivity (average number of neighbors of a node's neighbors)70 can be elucidated. (b) Constraint‐based microbe–microbe model. In a constraint‐based multispecies model (e.g., Refs 71, 72), metabolic reconstructions targeting two or more individual species are joined in an organism‐resolved manner. Multispecies models allow the prediction of cross‐feeding and mutualistic, commensal, or competitive interactions between microbial species. The tradeoff between two simultaneously growing microbes can be computed (e.g., see Ref 72). (c) Constraint‐based host–microbe community interaction model. In a constraint‐based host–microbe model, a reconstruction of host metabolism is joined with one73 or more74 metabolic networks of representative gut microbes. The setup enables a tractable exchange of host and microbial metabolites and provides outlets for luminal secretion and host secretion into body fluids (e.g., blood and urine). Hence, the host biofluid metabolome can be predicted (see also Figure 2).
Greenblum et al. constructed one of the first topological network models by integrating shotgun metagenomic data from a human microbiome gene catalog assembled from 124 European subjects by Qin et al.1 into a metabolic network.70 This ‘metagenomic systems biology’ approach revealed topological network differences associated with obesity and inflammatory bowel disease. For instance, the betweenness centrality, which was defined as the proportion of shortest paths passing through a given node, was determined for each enzyme. Based on this feature, each enzyme in the network was classified as peripheral, intermediate, or central. Enzymes associated with obesity and inflammatory bowel disease tended to have low centrality and to be peripheral rather than central.70 Another approach based on metagenomic data (the HUMAnN method) was used to reconstruct microbial networks for seven body sites.75 The HUMAnN pipeline is based on short DNA sequence reads, from which the presence, absence, and abundance of microbial gene families and pathways are computed. From the resulting pathway coverage and pathway abundance data for the seven body sites, metabolic pathways that are enriched in certain habitats could be identified. For example, glycosaminoglycan degradation was unique to the gut microbiota.75 Yet another modeling framework used 5026 sequenced samples from 18 body sites and 239 individuals.76 The resulting network of 3005 co‐occurrence and co‐exclusion relationships between 197 microbial clades has been constructed using generalized boosted linear models and correlation and similarity measures (e.g., Pearson correlation). The interactions participated in were clade‐specific. For example, the known co‐exclusion of Streptococci and Porphyromonaceae in the subgingival plaque was captured. The Actinobacteria and Bacilli only formed co‐exclusion relationships with other clades, while pathogenic Treponema and Prevotella co‐occurred in the oral microbiome.76
To investigate the variable metabolic potential in human gut microbiomes, as well as links between gut microbes and human drug targets, another study mapped the gene catalog published by Qin et al.1 to the MetaCyc77 and the KEGG78 databases.79 The resulting metabolic network summarized the metabolic potential of the gut microbiota. Three distinct clusters of individuals with high, medium, or low metabolic potential were identified.79 Moreover, the microbial network was overlaid with Recon1, a manually curated reconstruction of human metabolism that covers the reactions occurring in any human cell.80 This strategy resulted in an interactome map of ‘non‐human’ microbial metabolites and the human proteome. The chemical similarities between drug molecules in DrugBank81 and metabolites in the interactome map were compared, predicting that 603 existing drugs could perturb 515 metabolic microbial reactions. Moreover, 18 microbial metabolites overlapped with known experimental drugs indicating that the gut microbiota acts as a natural pharmacy.79 Another supra‐organismal network82 was recently constructed from the KEGG database78 based on the genome annotations for selected bacterial strains reported to be present in the human gut.1 The resulting network, accounting for 3449 reactions, has been used to predict 49 amino acid biotransformation products, of which 26 were confirmed to be present in the cecal contents of mice. The majority of the detected metabolites were microbe‐derived and/or significantly reduced or absent in germfree mice,82 demonstrating the influence of the microbiota on shaping the luminal metabolome.
The seed set framework relies on genome‐scale reconstructions of individual microbes83 and a graph theory‐based algorithm to compute an organism's metabolic potential to extract metabolites from the environment. This ‘reverse ecology’ approach assumes that selection pressure from the environment and the presence of other species is reflected in the metabolic network of an organism.83 The seed set method was applied to 154 species from the human gut microbiota84 and the metabolic profiles, competition, and complementarity indices for each pair have been computed. The predicted interactions have been compared with co‐occurrence patterns for the 154 species based on their abundances from metagenomic data.1 Metabolic competition correlated positively with co‐occurrence suggesting that habitat filtering drives microbiome assembly.84
Taken together, these studies demonstrate the advantages of microbiome‐wide topological networks. Because species boundaries are not accounted for in most of these studies, topological networks are extensive in scope and present a global view of the gut microbial ecosystem and its metabolic capabilities. Moreover, they can be easily linked to high‐throughput data, such as those obtained through metagenomic or metabolomic analysis.85 One shortcoming of not accounting for species boundaries is that diffusion (or facilitated transport) of almost any compound in the supra‐organismal metabolic network is assumed, whereas, in reality, only a subset of metabolites can be exchanged between species, and their transport often requires energy, e.g., via ATP‐binding cassette transporters.86
Constraint‐Based Modeling
At the heart of the constraint‐based modeling and analysis (COBRA) approach lies well‐structured mathematical models that were constructed in a bottom‐up manner. Briefly, metabolic networks that describe the target organism on the genome scale are manually reconstructed based on the organism's genome sequence, biochemistry, and physiology. These genome‐scale reconstructions (GENREs) can be converted into predictive mathematical models.87 The process of generating GENREs has been divided into 96 steps.43 To speed up parts of this reconstruction process, tools, such as Model SEED,44 have been developed to generate automated draft reconstructions. Such draft reconstructions still require intensive manual curation and validation against the available literature to yield a high‐quality reconstruction that captures the metabolic traits of the target organism.43, 73, 88 This bottom‐up reconstruction process results in a biochemically, genetically, and genomically structured knowledge base for the target organism.43 More than 140 manually curated GENREs are now available,89 including many for microbes colonizing the human body.90
Flux balance analysis is one of the most broadly used methods for the interrogation of constraint‐based models.91 This method requires an objective function, such as the biomass objective function representing all molecular biomass precursors required to produce a new cell.92 The second key feature of flux balance analysis is the addition of constraints representing mass conservation, enzyme capacities, and environmental conditions (e.g., nutrient availability).92 It is assumed that the simulated biological system is in a steady state. Solving the set of linear equations representing the biochemical network and imposed linear inequality constraints results in the prediction of a phenotype.92 A variety of constraint‐based methods are available to contextualize high‐throughput data, such as metabolomic measurements.91, 93 For example, metabolomic measurements from physiologically normal and Leigh's syndrome fibroblasts were overlaid with a fibroblast reconstruction to study the metabolic differences between healthy and disease states.94 Additionally, metabolomic and transcriptional data were integrated with Recon1 to construct condition‐specific models for two cancer cell lines, which elucidated the distinct metabolism of the two cell types.95 The integration of metabolomic data into the metabolic network of Escherichia coli resulted in accurate prediction of aerobic and anaerobic growth.96 Moreover, tools have been developed to allow the simultaneous contextualization of quantitative metabolomic and proteomic measurements97 or of transcriptomic and metabolomic datasets,95, 98 yielding condition‐specific models. Another established method for the contextualization of metabolomic data is 13C flux analysis, in which labeled carbon substrates are measured.99 Because constraint‐based modeling does not directly capture metabolite concentrations, fluxes are mathematically inferred from time‐dependent changes in concentrations.99 The integration of metabolomic data requires adequate information for the metabolites, e.g., InChI strings.100, 101
Constraint‐Based Multispecies Interaction Models
A growing number of constraint‐based modeling efforts have been devoted to developing multispecies models. Unlike the supra‐organism approach employed in topological network models, constraint‐based models combine multiple species by setting well‐defined species boundaries.90, 102 Another advantage is that they account for the genomic and biochemical traits of each included species. Typically, the species are reconstructed separately and then joined through an appropriate in silico scheme90 (Figure 1(b) and (c)). Moreover, a community objective function must be defined to allow the optimization of multispecies growth. In a first effort to model microbe–microbe interactions, the interaction between a sulfate reducer and a methanogen was simulated using a small‐scale model that allowed nutrient exchange through an additional shared compartment.103 Simultaneous growth was simulated by fixing the ratios between the two microbes.103 In a more complex modeling scheme, Klitgord and Segre joined seven microbe reconstructions pairwise in a shared in silico environment, where each species retained its separate extracellular space.104 To optimize simultaneous growth, minimal growth of each species was assumed. The interactions between the pairs were then systematically investigated by simulating different compositions of minimal media as well as genetic perturbations. Three types of synthetic interactions (mutualism, neutralism, and commensalism) were distinguished.104 Taffs et al. developed three modeling approaches integrating microbial groups as guilds into a consortium.105 This approach was applied for modeling thermophilic, phototrophic natural communities.105 Using an approach combining metabolic modeling and in vitro culture, Wintermute and Silver showed that E. coli mutants that are auxotrophic for amino acids can complement each other's growth.106 In yet another study, Freilich et al. predicted the metabolic interactions between 6903 bacterial pairs derived from 118 automated metabolic models.107 Each pair was grown on an in silico competition‐inducing medium. Based on the predicted growth rates, ‘winners’ that grew faster in co‐culture and ‘losers’ that grew slower were identified. Furthermore, give–take interactions in samples from 59 ecological niches were predicted.107 In a first effort to link high‐throughput data with metabolic modeling of the gut microbiota, Shoaie et al. reconstructed three gut microbe species: Eubacterium rectale (Firmicutes), Bacteroides thetaiotaomicron (Bacteroidetes), and Methanobrevibacter smithii (Archaea).71 A co‐growth model of the three species was then constructed and tailored to be condition specific based on transcriptomic data from E. rectale and B. thetaiotaomicron grown in ex‐germfree mice.108 Another study investigated the metabolic interactions between 11 gut microbes spanning three phyla.72 The pairwise tradeoffs between microbes (Figure 1(b)) were investigated on 12 varying nutrient environments. Anoxic conditions were predicted to induce mutualistic interactions between certain microbes, which were abolished in the presence of oxygen.72
Various strategies have been applied to overcome the challenge of predicting realistic simultaneous growth in a microbial consortium. For instance, the dynamic multispecies metabolic modeling (DMMM) framework integrates existing metabolic reconstructions and uses dynamic flux balance analysis to predict time‐dependent growth.109 Multiple microbes can interact through transfer of metabolic products from one species through the other. However, DMMM cannot directly predict simultaneous community growth as the biomass objective functions of the included species are independent from each other. Using this method, Zhuang et al. modeled the competition between two Fe(III)‐reducing bacteria in a soil community.109 Recently, dynamic flux balance analysis was also implemented into a lattice where a separate optimization problem is solved in each box.110 This approach, deemed COMETS (Computation of Microbial Ecosystems in Time and Space), enables the simulation of spatiotemporal dynamics of a multispecies microbial community.110 A community flux balance analysis approach solves a nonlinear optimization problem to simulate steady‐state balanced growth of a microbial community, resulting in the prediction of the individual species abundances at optimal total community growth.111 Yet another modeling framework (OptCom) optimizes multiple objective functions on the community and single species level,112 thereby predicting community growth. Different types of interactions, such as commensalism, competition, parasitism, and mutualism, can be modeled with OptCom. As a downside, OptCom is computationally intensive compared with other frameworks, such as DMMM. OptCom was applied to predicting the interactions within a phototrophic microbial community.112 An extension of this framework (dynamic OptCom) allows for dynamic modeling and the inclusion of substrate uptake kinetics.113 Recently, OptCom was also applied to modeling the interaction between two gut microbes, the Firmicutes representative Faecalibacterium prausnitzii and the Actinobacterium representative Bifidobacterium adolescentis. 114 The existing studies demonstrate that constraint‐based modeling accurately captures the behavior of individual species and natural interaction patterns of commensalism, mutualism, and competition. Moreover, unexpected, nonintuitive species–species interactions can be predicted. A disadvantage of constraint‐based modeling is the intensive manual curation effort required to construct such models, which has caused most studies performed to date to be small in scope and to include only a limited number of species.
Constraint‐Based Modeling of Host–Microbe Interactions
While microbe–microbe interaction models are well established, few constraint‐based studies have been conducted to model host–microbe interactions. In a first effort, Bordbar et al. constructed a host–pathogen model simulating the infection of the human alveolar macrophage with Mycobacterium tuberculosis. The model was constructed by placing a reconstruction of M. tuberculosis into an in silico compartment simulating the intracellular phagosome.115 A constraint‐based model of a host and a commensal microbe73 linked a published mouse reconstruction116 with a reconstruction of the prominent human gut symbiont B. thetaiotaomicron. The two species could consume simulated dietary inputs and exchange nutrients with each other through an in silico compartment simulating the intestinal lumen. The mouse could secrete into a separate outlet representing mouse biofluids, e.g., blood or urine. This setup was modeled after a gnotobiotic mouse mono‐associated with B. thetaiotaomicron, which is a well‐established animal model used in gut microbiome research.117 To enable simultaneous growth, the tradeoff between host and microbe biomass production was computed. The in silico model captured known traits of the in vivo model, including B. thetaiotaomicron's foraging on dietary and host glycans as well as the production of short‐chain fatty acids,117 which were then consumed by the host. Moreover, the model predicted that lethal mouse gene deletion phenotypes would be rescued by the presence of B. thetaiotaomicron and vice versa. Finally, the implementation of the separate host outlet enabled the prediction of the mouse biofluid metabolome, which was validated against experimental metabolomic data.73
Moving from these initial efforts to a comprehensive, predictive model of human–microbe co‐metabolism requires a well‐curated, extensive network of human metabolism. The global human reconstruction Recon2 was curated through a community effort118 and is far more comprehensive than its predecessor, Recon1.80 Importantly, it captures the majority of known exometabolites, which are extracellular metabolites found in biofluids, such as blood and plasma,118 making it an excellent tool for prediction of the human metabolome and the contextualization of metabolomic data. Most health‐relevant metabolites and pathways known to be influenced by the gut microbiota are present in Recon2 (Table 2). Taking advantage of these characteristics, Recon2 was used to predict the global effects of a representative gut microbe community on the host's metabolome.74 The community accounted for 11 published, manually curated gut microbe reconstructions spanning three phyla (Bacteroidetes, Firmicutes, and Proteobacteria), including commensal, probiotic, and pathogenic bacteria.90 The microbial reconstructions were linked to Recon2 using a previously established framework,73 which enables dietary input and metabolic exchange between host and microbes (Figure 1(c)). The human global metabolome was systematically predicted in the presence of the 11 microbes and in their absence (‘germfree’ human). Recon2 secreted a total of 342 metabolites. Surprisingly, only 11 of these metabolites could not be secreted by the ‘germfree’ human, but the quantitative secretion flux of 52 metabolites was at least fivefold higher in the presence of the microbe community (Figure 2). Notably, the secretion of known mammalian–microbial co‐metabolites, such as phenylacetylglutamine and 4‐hydroxyphenylacetate,69 as well as several hormones and neurotransmitters, was up to 160‐fold higher in the presence of the 11 microbes (Figure 2). Moreover, the microbes' potential to secrete luminal metabolites was systematically predicted, including a variety of phenolic compounds as well as short‐chain fatty acids, lactate, formate, and ethanol.74 These metabolites are known to have beneficial or detrimental effects on host health.120 Thanks to the bottom‐up systems biology approach, underlying mechanisms for the observed microbial effects on host metabolite secretion could be proposed. For instance, the microbes increased glutathione and leukotriene production by the host through secreting the synthesis‐limiting precursor l‐cysteine.74 In summary, this systems‐level framework predicted that the microbes profoundly affected host metabolism, in agreement with the emerging view of the microbiota as an additional organ.29 In future efforts, more comprehensive gut microbial communities could be considered, which would be a prerequisite for the integrative analysis of metagenomic and metatranscriptomic data. Such applications will be valuable for gaining mechanistic insight into human–microbe co‐metabolism.
Table 2.
Metabolites Relevant for Human Health and Disease, and Associated References
| Metabolite | Health Implications | References | HMDB ID | Recon2 ID | |
|---|---|---|---|---|---|
| Short‐chain fatty acids produced by gut microbiota | Acetate | Serves as carbon source in the liver, affects immune system by binding GPR receptors, may have a role in adipogenesis, and stimulates colonic function. | 10, 119 | HMDB00042 | ac |
| Propionate | Serves as carbon source in the liver, has anti‐inflammatory properties, lowers blood cholesterol, stimulates satiety, and alters brain function in rats. | 10, 119 | HMDB00237 | ppa | |
| Butyrate | Serves as the main energy source for colonocytes, has anti‐inflammatory properties, prevents oxidative stress, and may protect against colonic cancer. | 119, 120 | HMDB00039 | but | |
| Microbial fermentation products | Lactate | Certain circumstances in the gut may lead to toxic levels of d‐lactate in blood, causing d‐lactic acidosis. Positively correlated with Faecalibacterium prausnitzii. | 69, 119 | HMDB01311 | lac_D |
| HMDB00190 | lac_L | ||||
| Formate | Decreased levels were found in urine of IBD patients. Formate is associated with blood pressure. | 121, 122 | HMDB00142 | for | |
| Ethanol | Produced by many gut bacteria, disrupts the intestinal epithelial barrier. Ethanol consumption can promote intestinal overgrowth. | 123, 124 | HMDB00108 | etoh | |
| Acetaldehyde | Produced from ethanol by gut microbiota, toxic and carcinogenic. Disrupts the intestinal epithelial barrier. | 24, 123 | HMDB00990 | acald | |
| Succinate | Decreased levels were found in urine of IBD patients and in a rat model after bariatric surgery. | 32, 121 | HMDB00254 | succ | |
| Lipid metabolism | Choline | Choline deficiency and disrupted choline metabolism are associated with NAFLD. Choline has also been linked to CVD risk. | 25, 26, 125 | HMDB00097 | chol |
| Betaine | Phosphatidylcholine‐derived betaine has been linked to CVD risk. | 26 | HMDB00043 | glyb | |
| Ethanolamine | Host‐derived ethanolamine can be exploited as a carbon source by pathogenic Salmonella typhimurium during inflammation. | 126 | HMDB00149 | etha | |
| Phosphatidylcholine | Biomarker of disrupted fatty acid metabolism, related to obesity and resistance to insulin. Phosphatidylcholine metabolites are linked to CVD. | 26, 57 | pchol_hs | ||
| Phosphatidyl‐ethanolamine | Altered in plasma of individuals with type 2 diabetes. | 127 | HMDB60501 | pe_hs | |
| l‐Carnitine | Higher l‐carnitine levels were found in obese subjects. Dietary l‐carnitine is converted to TMA by gut microbiota, linking it to CVD risk. | 128, 129 | HMDB00062 | crn | |
| Acylcarnitines | Biomarkers of disrupted fatty acid metabolism after high‐fat feeding. Acylcarnitine pools are altered in obese subjects. | 57, 128 | e.g., acrn, c4crn, and c8crn | ||
| Dimethylamine | Increased urinary levels in NAFLD. Negatively correlated with F. prausnitzii. | 69, 125 | HMDB00087 | ||
| Trimethylamine (TMA) | It is formed from choline and l‐carnitine by gut bacteria, and has been linked to CVD. Increased urinary levels in NAFLD. | 26, 125, 129 | HMDB00906 | ||
| Bile acids | Cholate | Gut microbes transform bile acids to secondary bile acids, permitting their reabsorption via the colonic epithelium. The gut microbiota may contribute to obesity, type 2 diabetes, inflammation, and cancer by controlling bile acid pools. | 10, 124, 130 | HMDB00619 | cholate |
| Lithocholate | HMDB00761 | C03990 | |||
| Glycocholate | HMDB00138 | gchola | |||
| Taurocholate | HMDB00036 | tchola | |||
| Deoxycholate | HMDB00626 | C04483 | |||
| Glycochenodeoxycholate | HMDB00637 | dgchol | |||
| Taurochenodeoxycholate | HMDB00951 | tdchola | |||
| Amino acids | Branched‐chain amino acids | High levels of leucine, isoleucine, and valine are associated with obesity and type 2 diabetes. | 128, 131, 132, 133 | HMDB00172 | ile_L |
| HMDB00687 | leu_L | ||||
| HMDB00883 | val_L | ||||
| Aromatic amino acids | High levels of tyrosine, phenylalanine, and tryptophan are associated with type 2 diabetes. | 128, 132, 133 | HMDB00159 | phe_L | |
| HMDB00158 | tyr_L | ||||
| HMDB00929 | trp_L | ||||
| Glutamine and glutamate | High glutamate/glutamine ratio is associated with type 2 diabetes. | 132 | HMDB00641 | gln_L | |
| HMDB00148 | glu_L | ||||
| Taurine | Elevated levels were found in fecal samples of UC patients. Associated with certain gut bacterial species. | 69, 134 | HMDB00251 | taur | |
| Phenolic compounds | Phenylacetate | Increased production by gut bacteria on a high‐protein diet, may give rise to toxic products. Elevated in colorectal cancer. | 25, 135 | HMDB00209 | pac |
| Phenylpropionate | Can be converted to benzoic acid by gut bacteria and give rise to hippurate, which is implicated in a variety of disease states. | 136 | HMDB11743 | ||
| 3‐Hydroxyphenylpropionate (3‐HPPA) | Can be converted to benzoic acid by gut bacteria and give rise to hippurate, which is implicated in a variety of disease states. | 136 | HMDB00375 | 3hhpa1 | |
| Phenylacetylglutamine | Decreased excretion in autism. Lower levels in obese subjects. Positively associated with age in humans. Associated with certain gut bacterial species. | 31, 69, 128, 137 | HMDB06344 | pheacgln | |
| Benzoic acid | Converted into hippurate by the host, which is implicated in a variety of disease states. | 136 | HMDB01870 | bz | |
| 4‐Hydroxyphenylacetate | Elevated in colorectal cancer. Negatively correlated with Subdoligranulum variable. | 25, 69 | HMDB00020 | 4hphac | |
| Hippurate (N‐benzoylglycinate) | Related to a variety of health and disease states. Hippurate levels in urine are decreased in CD patients due to altered gut microbiota. Diminished excretion was also found in obese individuals as well as patients with schizophrenia and autism. Associated with certain gut bacterial species. | 31, 69, 136, 138, 139 | HMDB00714 | bgly | |
| p‐Cresol | Produced by gut bacteria. May be a significant factor in autism, is elevated in colorectal cancer, and has been linked to inflammatory bowel disease. Also linked to cardiovascular disease in chronic kidney disease patients. Interferes with the sulfonation of acetaminophen (paracetamol). | 25, 31, 140, 141 | HMDB01858 | pcresol1 | |
| p‐Cresyl sulfate | Formed from gut bacteria‐derived p‐cresol, and is directly linked to progression of chronic kidney disease. Elevated in the urine of children with autism. Positively associated with age in humans. Associated with certain gut bacterial species. | 25, 69, 137, 142 | HMDB11635 | pcs1 | |
| Indolic compounds | Indole | Produced by gut bacteria, converted to uremic toxin indoxyl sulfate by the host. | 143 | HMDB00738 | indole1 |
| Indoxyl sulfate | Formed from bacteria‐derived indole, and is directly linked to progression of chronic kidney disease. | 142 | HMDB00682 | inds1 | |
| Polyamines | Cadaverine | Produced by gut bacteria. Increased levels were found in fecal samples of UC patients. | 134 | HMDB02322 | |
| Putrescine | Produced by gut bacteria and in human tissues. Polyamines are toxic at high concentrations and associated with cancer. Increased fecal putrescine was found in a rat model after bariatric surgery. | 24, 32 | HMDB01414 | ptrc | |
| Spermine | HMDB01256 | sprm | |||
| Spermidine | HMDB01257 | spmd | |||
| Hormones/Precursors | GABA (γ‐aminobutyric acid) | Directly produced by gut bacteria with implications for the gut–brain axis. Increased urinary levels were found in a rat model after bariatric surgery. | 29, 32 | HMDB00112 | 4abut |
| Dopamine | Directly produced by gut bacteria with implications for the gut–brain axis. | 29 | HMDB00073 | dopa | |
| Norepinephrine | Directly produced by gut bacteria with implications for the gut–brain axis. | 29 | HMDB00216 | nrpphr | |
| Serotonin | Directly produced by gut bacteria. The microbiota also regulates tryptophan availability, affecting serotonin biosynthesis in the CNS and thus brain and behavior. | 29 | HMDB00259 | srtn | |
| Histamine | Histamine and N‐methylhistamine levels are increased in IBD patients. Histamine is also directly produced by gut bacteria. | 29, 144 | HMDB00870 | hista | |
| N‐Methylhistamine | HMDB00898 | mhista | |||
| l‐Dopa | l‐Dopa is dehydroxylated by the gut microbiota. Manipulating the microbiota has been proposed as an approach to control l‐Dopa bioavailability in PD treatment. | 29, 145, 146 | HMDB00181 | 34dhphe | |
| Other | Ammonia | Produced by many gut bacteria. Inhibits the mitochondrial oxygen condition and short‐chain fatty acid oxidation in colonocytes. | 147 | HMDB00051 | nh4 |
| Hydrogen sulfide | Produced by some gut bacteria, e.g., Escherichia coli. Toxic to colonocytes, disturbs their metabolism. Found to be increased in stool samples of colonic cancer patients. Implicated in IBD. | 24, 147, 148 | HMDB00598 | ||
| Glutathione | Glutathione levels in the liver were lower in mice colonized with human baby flora than in conventional mice. Increased glutathione synthesis as stress response was also found in NAFLD. | 60, 149 | HMDB00125 | gthrd |
CD, Crohn's disease; CNS, central nervous system; CVD, cardiovascular disease; IBD, inflammatory bowel disease; NAFLD, nonalcoholic fatty liver disease; PD, Parkinson's disease; UC, ulcerative colitis.
If available, HMDB150 (http://www.hmdb.ca/) and Recon2118 (http://humanmetabolism.org) IDs are shown.
Will be included in subsequent releases of Recon2.
Figure 2.
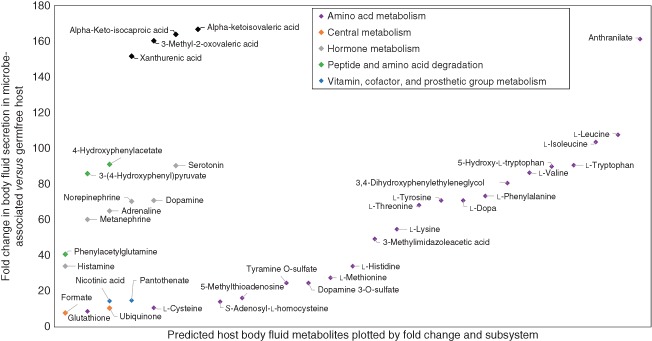
Prediction of health‐relevant host body fluid secretion using a constraint‐based modeling framework. Using a constraint‐based modeling framework (Figure 1(c)), the maximal quantitative metabolite secretion was predicted in the presence and in the absence of a model community of 11 microbes. A dietary regime approximating the amounts of protein, carbohydrate, and fat consumed by a typical Western citizen (http://www.ars.usda.gov/) was simulated. Shown are examples for metabolites for which the secretion flux was at least fivefold increased in the presence of the microbe community compared with the ‘germfree’ condition. The complete data analysis is available in Ref 74.
FUTURE PERSPECTIVES
Elucidating the Unknown Metabolic Potential of the Microbiota
Despite the recent advances in the field of human gut microbiome research, our understanding of the mechanisms through which the microbiota affects host health is still incomplete. Two worldwide research initiatives, the Human Microbiome Project and MetaHIT, have resulted in enormous amounts of data, including more than 2800 reference sequences for microbes from various sites in the body (http://www.hmpdacc.org/). However, many of these strains are uncultured and uncharacterized, and the genome sequence alone does not offer complete insight into the metabolic capabilities of specific bacteria.151 As a result, it is difficult to identify keystone species through metagenomic approaches alone.151 Ultimately, only cultivation can fully elucidate the biochemical and physiological traits of a strain151; yet cultivating the mostly anaerobic gut microbiota is difficult, time‐consuming, and impossible to be performed for thousands of species. Constraint‐based modeling could bridge the gap between genome sequences and in vitro culture by predicting a species' metabolic potential based on its genome sequence.88, 152, 153 Such predictions could identify potential keystone species, whose metabolic properties (e.g., nutrient requirements) could be predicted and subsequently experimentally validated. For instance, a combined in silico/in vitro approach has yielded a chemically defined growth medium for the oxygen‐sensitive gut symbiont F. prausnitzii.88 Such an iterative approach could be readily applied to the other poorly characterized or uncharacterized species in the gut.
Predicting Mechanisms by Combining Top‐Down and Bottom‐Up Approaches
In addition to the characterization of individual species in the gut, it is of utmost importance to identify links between gut microbial metabolism and human phenotypes. Metagenomic datasets have provided valuable insights, for example, giving rise to the enterotype concept.9 However, many of these studies used top‐down approaches, which identified patterns rather than mechanisms.154 Moreover, the causality between phenotypes and the microbiome composition (i.e., whether the host's phenotype drives changes in the gut microbiome composition or vice versa) remains unclear.154 The combination of top‐down approaches revealing patterns and bottom‐up methods elucidating the mechanisms underlying these patterns is a key future challenge.154 Supporting experimental bottom‐up approaches with computational methods will require well‐structured, accurate networks. Constraint‐based reconstructions are ideal tools to provide insight into phenotype‐gut microbiome causality because they can predict mechanisms at the biochemical level.39 Initial efforts could demonstrate that constraint‐based modeling can provide mechanistic hypotheses on how microbes affect the host's potential to synthesize health‐relevant metabolites.73, 74 A host–microbe community model could also be combined with top‐down methods to contextualize high‐throughput data (Figure 3). For instance, in a second phase of the Human Microbiome Project, a multi‐omic analysis of the microbiotas of mothers and neonates will be performed, including metagenomic, metatranscriptomic, metabolomic, and metaproteomic analyses.155 In addition, host lipidomes and cytokines will be analyzed.155 These datasets will be accessible through public databases155 and could be readily mapped onto the human metabolic reconstruction Recon2,118 which accounts for many lipid derivates, or be used in a gut microbiota community model. Moreover, top‐down statistical approaches have previously inferred associations between metabolites and specific microbes from metabolomic data. For instance, F. prausnitzii has been shown to correlate with the presence of eight human urinary metabolites.69 Integrating top‐down inference models with bottom‐up reconstructions could elucidate the mechanisms underlying such patterns. A proposed pipeline for using constraint‐based modeling to contextualize high‐throughput data is shown in Figure 3.
Figure 3.
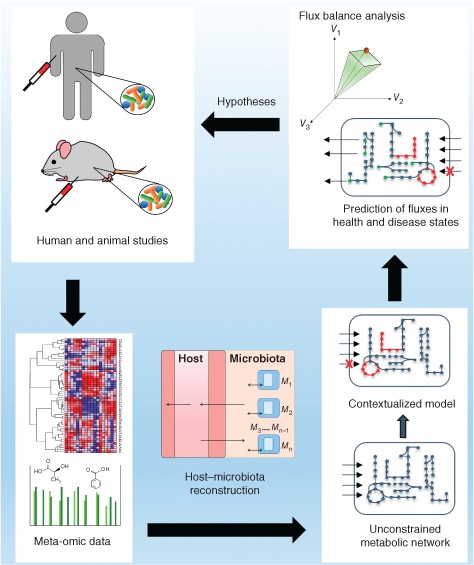
Schematic overview of a pipeline for using constraint‐based models to contextualize high‐throughput metagenomic, metatranscriptomic, metaproteomic, and metabolomic data from human and animal studies.
Toward a Predictive Bottom‐Up Host–Microbe Community Model: Challenges
Currently, the application of constraint‐based modeling to gut microbiome research is limited by the availability of high‐quality reconstructions. While the number of high‐quality, manually curated metabolic reconstructions is steadily growing, the thousands of species inhabiting the human gut are still poorly represented. A number of bacteria colonizing the human gut have been reconstructed71, 88, 90, 114; however, they do not represent the gut microbiome composition well. While Proteobacteria are overrepresented, the important Bacteroides and Clostridium groups are only represented by one reconstructed species, and reconstructions for minor phyla, such as Verrucomicrobia, are lacking. Moreover, the currently available reconstructions lack standardization in terms of nomenclature and the reconstruction structure, which hampers their integration onto a community model.74, 101, 156 To overcome this challenge, MetaNetX, a tool that allows the mapping of metabolic reconstructions from different sources into a common namespace, has been developed.157 Using automated reconstruction tools, such as Model SEED44 or Pathway Tools,47 one could easily construct a community model of hundreds of species from the genome sequences archived by the Human Microbiome Project.6 The resultant loss of accuracy would be compensated by a dramatic increase in scope. Considering that most gut microbes are both uncultivated and uncharacterized, automated draft reconstructions are a reasonable first approximation of their metabolism. A challenge in this context is that the quality of a draft reconstruction depends mostly on the genome sequence. Many published sequences are incomplete. Additionally, gene annotations are lacking or unspecific and/or the associated pathways are not accounted for by the automated reconstruction pipelines, resulting in a less predictive draft reconstruction. Moreover, even universal pathways vary in the enzymes carrying out individual steps.158 As a result, annotation platforms often fail to correctly reconstruct these variant pathways.158 Several recent comparative genomics studies have resulted in the improvement of genome annotations in gut microbial genomes.159, 160, 161 Such efforts will ultimately result in an improved predictive potential of automated reconstructions derived from the genome sequence.
Peripheral and species‐specific pathways are generally lacking in automated reconstructions.44 For instance, the dietary glycan degradation by gut microbes is highly specialized and species‐specific162 and the inclusion of the associated pathways into metabolic reconstructions currently requires intensive manual curation.73 Recently, a computational pipeline has been developed to predict the glycan degradation potential of gut bacterial species163 and will facilitate the incorporation of species‐specific carbohydrate degradation pathways into metabolic reconstructions. Xenobiotic transformations by gut microbes are currently not well captured in draft metabolic reconstructions. The ongoing advances in metagenomic analyses will further elucidate drug transformations performed by gut bacteria,145 which could then be included into reconstruction pipelines.
Constraint‐Based Modeling of Host–Microbe Co‐Metabolism: Future Applications
The field of human gut microbiome research has exploded during the last decade and will undoubtedly grow even more in the upcoming years. The prevalence of lifestyle diseases directly linked to the gut microbiota is estimated to increase exponentially in the next decades. For instance, worldwide obesity has nearly doubled since 1980; there are currently 1.4 billion overweight people, including 500 million obese individuals.164 The prevalence of type 2 diabetes in developed and developing countries is predicted to double between 2000 and 2030.165 Thus, the interest in the relationship between the human microbiota and host health is ever growing. In this section, we propose key contributions that constraint‐based host–microbe modeling could make.
A key application area of systems biology research on host–microbe interactions is drug development. It is well established that the gut microbiota affects drug metabolism.30 In fact, at least 30 drugs have been shown to be co‐metabolized by the microbiota.30 For instance, the well‐studied drug acetaminophen (paracetamol) is differentially metabolized in individuals due to gut microbial activity.140 Microbiota‐derived p‐cresol competes over human sulfo‐transferase 1 with acetaminophen, resulting in lower acetaminophen sulfonation capacity in individuals with high bacterial p‐cresol production. As, many xenobiotics are substrates for sulfo‐transferase 1 and sulfonation alters the physical properties of molecules, this finding has implications for the metabolism and toxicity of various drugs.140 The cardiac drug digoxin has been shown to be inactivated by the gut bacterium Eggerthella lenta. 166 Using a computational framework, the transformation of drugs and xenobiotics (e.g., antibiotics) can be predicted and linked to specific species. Recon2 accounts for 1290 drugs included in DrugBank,81 mapped to 308 enzymes and enzymatic complexes,118 making it an excellent tool for modeling drug metabolism. As constraint‐based modeling can predict drug effects at a mechanistic level,35 and its applications for drug discovery are promising (reviewed in more detail in Refs 35, 167, 168). For instance, metabolic modeling has led to the prediction and subsequent validation of a novel cancer drug.169, 170 Recently, Sahoo et al. expanded Recon2 with a manually curated drug metabolism module for the five most highly prescribed drug groups, including acetaminophen.171 This expanded human network was then used to predict the effects of dietary intake and inherited metabolic disorders on drug metabolism. The flux through sulfo‐transferase 1, which carries out the sulfonation of acetaminophen, has been predicted to be lower under a vegetarian diet compared with a Western or balanced diet.171 Interestingly, the same reaction was found to be affected by high gut microbial production of p‐cresol, which competes with acetaminophen for sulfo‐transferase 1.140 Currently, such studies are using the human metabolic network as the sole modeling environment and are therefore not accounting for the well‐known drug transformations performed by the microbiota. By using a host–microbe community framework,74 drug co‐metabolism by humans and microbiota depending on the diet could be investigated.
A logical next step after modeling the co‐metabolism of drugs and xenobiotics is the prediction of individual‐specific drug metabolism. The global human reconstruction can be tailored to be both cell‐type and individual specific, e.g., by overlaying it with transcriptomic or metabolomic data,172 and it has been applied successfully to the contextualization of high‐throughput data from pathological states, such as type 2 diabetes173 and cancer.95, 174 Recently, Agren et al. constructed personalized genome‐scale models for carcinoma patients and used these models to predict cancer drug targets.175 Moreover, Yizhak et al. built personalized cancer cell models for more than 700 breast and lung cancer patients.176 Low growth rates in silico were found to be correlated with longer patient survival.176 Personalized human cell models could also be constructed based on intestinal metabolomic measurements (e.g., condition‐specific models based on measured drug degradation products). One could even envision a personalized gut microbiota model based on an individual's gut microbiome composition. Such knowledge‐based, bottom‐up network models would valuably complement or be combined with the existing top‐down inference models, such as the pharmacometabonomics approach developed by Nicholson et al.177
Biomarker discovery is another important application of systems biology. For instance, constraint‐based modeling‐based analyses have predicted biomarkers of inborn errors in human metabolism,118, 178 with 77% accuracy in the case of Recon2.118 The human reconstruction was also applied to predicting novel biomarkers of type 2 diabetes173, 179 and Alzheimer's disease.180 Linking Recon2 with a microbial community74 could be applied to predicting the effects of the microbes on disease‐associated biomarkers in humans.
Yet another useful application is modeling the interplay between the diet, microbiota, and host energy metabolism. Several constraint‐based approaches have been employed to model energy metabolism (reviewed in Ref 181). Obesity can generally be considered to be the result of an imbalance between energy uptake and consumed energy. The gut microbiota has long been known to contribute approximately 10% of the host energy intake182 and to directly modulate host energy metabolism.3 Systems biology models have been proposed as a tool for modeling the thermodynamic properties of the gut microbiota and its costs from the perspective of the host.183 There are established methods for integrating thermodynamic constraints into genome‐scale models,184, 185, 186, 187, 188, 189 which could be used for this type of application. Multiple studies have investigated adipocyte metabolism using cell‐type‐specific models.190, 191, 192 Such genome‐scale adipocyte models would be combined with a microbe community model to elucidate the effect of the presence of microbes on host fat storage and energy metabolism.
Finally, while this review focuses on human–gut microbiota interactions, the described systems biology approach is not limited to human hosts. For instance, we73 and others82 have shown that mouse–gut microbiota interactions can also be computationally modeled. Ex‐germfree mice colonized with a defined or conventional microbiota are important animal models employed in gut microbiome research,193 especially in metabolomics analyses.65, 67 Computational models can be useful in complementing or possibly partially replacing such animal studies. Other mammals could be reconstructed using the human reconstruction as a template,172 and these reconstructions could subsequently be combined with a microbe community model. Moreover, host–microbe modeling can be carried out for organisms other than mammals. For example, the survival of the endangered honeybee may be improved by manipulating its gut microbiota,194 which could be predicted in silico. Another potential application of constraint‐based multispecies modeling is the investigation of plant–microbe interactions. There are several plant genome‐scale reconstructions195, 196 as well as tissue‐specific reconstructions of Arabidopsis thaliana, 197 and multiorgan models of maize198 and barley,199 which could be applied to modeling the interactions between plants and their associated symbiotic or pathogenic microbes.
ACKNOWLEDGMENT
This work was funded by an ATTRACT program grant from the Luxembourg National Research Fund (FNR) to I.T. (FNR/A12/01).
REFERENCES
- 1. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hooper LV, Midtvedt T, Gordon JI. How host‐microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr 2002, 22:283–307. [DOI] [PubMed] [Google Scholar]
- 3. Sommer F, Backhed F. The gut microbiota—masters of host development and physiology. Nat Rev Microbiol 2013, 11:227–238. [DOI] [PubMed] [Google Scholar]
- 4. Kaiko GE, Stappenbeck TS. Host‐microbe interactions shaping the gastrointestinal environment. Trends Immunol 2014, 35:538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lawley TD, Walker AW. Intestinal colonization resistance. Immunology 2013, 138:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Human Microbiome Project Consortium . Structure, function and diversity of the healthy human microbiome. Nature 2012, 486:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006, 444:1022–1023. [DOI] [PubMed] [Google Scholar]
- 8. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science 2005, 308:1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. Enterotypes of the human gut microbiome. Nature 2011, 473:174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489:242–249. [DOI] [PubMed] [Google Scholar]
- 11. Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 2005, 102:11070–11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Finegold SM, Downes J, Summanen PH. Microbiology of regressive autism. Anaerobe 2012, 18:260–262. [DOI] [PubMed] [Google Scholar]
- 13. Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, Haapaniemi E, Kaakkola S, Eerola‐Rautio J, Pohja M, et al. Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov Disord 2015, 30:350–358. [DOI] [PubMed] [Google Scholar]
- 14. Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, Benezra A, DeStefano J, Meier MF, Muegge BD, et al. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature 2014, 510:417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sonnenburg ED, Sonnenburg JL. Starving our microbial self: the deleterious consequences of a diet deficient in microbiota‐accessible carbohydrates. Cell Metab 2014, 20:779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500:541–546. [DOI] [PubMed] [Google Scholar]
- 17. Ze X, Le Mougen F, Duncan SH, Louis P, Flint HJ. Some are more equal than others: the role of “keystone” species in the degradation of recalcitrant substrates. Gut Microbes 2013, 4:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez‐Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti‐inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA 2008, 105:16731–16736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 2010, 8:292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shafquat A, Joice R, Simmons SL, Huttenhower C. Functional and phylogenetic assembly of microbial communities in the human microbiome. Trends Microbiol 2014, 22:261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature 2009, 457:480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nicholson JK, Holmes E, Wilson ID. Gut microorganisms, mammalian metabolism and personalized health care. Nat Rev Microbiol 2005, 3:431–438. [DOI] [PubMed] [Google Scholar]
- 24. Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 2014, 12:661–672. [DOI] [PubMed] [Google Scholar]
- 25. Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host‐gut microbiota metabolic interactions. Science 2012, 336:1262–1267. [DOI] [PubMed] [Google Scholar]
- 26. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moco S, Martin FP, Rezzi S. Metabolomics view on gut microbiome modulation by polyphenol‐rich foods. J Proteome Res 2012, 11:4781–4790. [DOI] [PubMed] [Google Scholar]
- 28. Zheng X, Xie G, Zhao A, Zhao L, Yao C, Chiu NH, Zhou Z, Bao Y, Jia W, Nicholson JK, et al. The footprints of gut microbial‐mammalian co‐metabolism. J Proteome Res 2011, 10:5512–5522. [DOI] [PubMed] [Google Scholar]
- 29. Clarke G, Stilling RM, Kennedy PJ, Stanton C, Cryan JF, Dinan TG. Gut microbiota: the neglected endocrine organ. Mol Endocrinol 2014, 28:1221–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li H, Jia W. Cometabolism of microbes and host: implications for drug metabolism and drug‐induced toxicity. Clin Pharmacol Ther 2013, 94:574–581. [DOI] [PubMed] [Google Scholar]
- 31. Clayton TA. Metabolic differences underlying two distinct rat urinary phenotypes, a suggested role for gut microbial metabolism of phenylalanine and a possible connection to autism. FEBS Lett 2012, 586:956–961. [DOI] [PubMed] [Google Scholar]
- 32. Li JV, Ashrafian H, Bueter M, Kinross J, Sands C, le Roux CW, Bloom SR, Darzi A, Athanasiou T, Marchesi JR, et al. Metabolic surgery profoundly influences gut microbial‐host metabolic cross‐talk. Gut 2011, 60:1214–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seyfried F, Li JV, Miras AD, Cluny NL, Lannoo M, Fenske WK, Sharkey KA, Nicholson JK, le Roux CW, Holmes E. Urinary phenotyping indicates weight loss‐independent metabolic effects of Roux‐en‐Y gastric bypass in mice. J Proteome Res 2013, 12:1245–1253. [DOI] [PubMed] [Google Scholar]
- 34. Xie G, Li X, Li H, Jia W. Toward personalized nutrition: comprehensive phytoprofiling and metabotyping. J Proteome Res 2013, 12:1547–1559. [DOI] [PubMed] [Google Scholar]
- 35. Schmidt BJ, Papin JA, Musante CJ. Mechanistic systems modeling to guide drug discovery and development. Drug Discov Today 2013, 18:116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Baarlen P, Kleerebezem M, Wells JM. Omics approaches to study host‐microbiota interactions. Curr Opin Microbiol 2013, 16:270–277. [DOI] [PubMed] [Google Scholar]
- 37. Martin FP, Collino S, Rezzi S, Kochhar S. Metabolomic applications to decipher gut microbial metabolic influence in health and disease. Front Physiol 2012, 3:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hyduke DR, Lewis NE, Palsson BO. Analysis of omics data with genome‐scale models of metabolism. Mol Biosyst 2013, 9:167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bordbar A, Monk JM, King ZA, Palsson BO. Constraint‐based models predict metabolic and associated cellular functions. Nat Rev Genet 2014, 15:107–120. [DOI] [PubMed] [Google Scholar]
- 40. Oberhardt MA, Palsson BO, Papin JA. Applications of genome‐scale metabolic reconstructions. Mol Syst Biol 2009, 5:320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bruggeman FJ, Westerhoff HV. The nature of systems biology. Trends Microbiol 2007, 15:45–50. [DOI] [PubMed] [Google Scholar]
- 42. Reed JL, Famili I, Thiele I, Palsson BO. Towards multidimensional genome annotation. Nat Rev Genet 2006, 7:130–141. [DOI] [PubMed] [Google Scholar]
- 43. Thiele I, Palsson BO. A protocol for generating a high‐quality genome‐scale metabolic reconstruction. Nat Protoc 2010, 5:93–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. High‐throughput generation, optimization and analysis of genome‐scale metabolic models. Nat Biotechnol 2010, 28:977–982. [DOI] [PubMed] [Google Scholar]
- 45. Boele J, Olivier BG, Teusink B. FAME, the flux analysis and modeling environment. BMC Syst Biol 2012, 6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Agren R, Liu L, Shoaie S, Vongsangnak W, Nookaew I, Nielsen J. The RAVEN toolbox and its use for generating a genome‐scale metabolic model for Penicillium chrysogenum . PLoS Comput Biol 2013, 9:e1002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karp PD, Paley SM, Krummenacker M, Latendresse M, Dale JM, Lee TJ, Kaipa P, Gilham F, Spaulding A, Popescu L, et al. Pathway tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief Bioinform 2010, 11:40–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Swainston N, Smallbone K, Mendes P, Kell D, Paton N. The SuBliMinaL toolbox: automating steps in the reconstruction of metabolic networks. J Integr Bioinform 2011, 8:186. [DOI] [PubMed] [Google Scholar]
- 49. Feng X, Xu Y, Chen Y, Tang YJ. MicrobesFlux: a web platform for drafting metabolic models from the KEGG database. BMC Syst Biol 2012, 6:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vlassis N, Pacheco MP, Sauter T. Fast reconstruction of compact context‐specific metabolic network models. PLoS Comput Biol 2014, 10:e1003424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thiele I, Vlassis N, Fleming RM. fastGapFill: efficient gap filling in metabolic networks. Bioinformatics 2014, 30:2529–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thiele I, Fleming RM, Que R, Bordbar A, Diep D, Palsson BO. Multiscale modeling of metabolism and macromolecular synthesis in E. coli and its application to the evolution of codon usage. PLoS One 2012, 7:e45635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thiele I, Jamshidi N, Fleming RM, Palsson BO. Genome‐scale reconstruction of Escherichia coli's transcriptional and translational machinery: a knowledge base, its mathematical formulation, and its functional characterization. PLoS Comput Biol 2009, 5:e1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li F, Thiele I, Jamshidi N, Palsson BO. Identification of potential pathway mediation targets in Toll‐like receptor signaling. PLoS Comput Biol 2009, 5:e1000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aurich MK, Thiele I. Contextualization procedure and modeling of monocyte specific TLR signaling. PLoS One 2012, 7:e49978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Papin JA, Palsson BO. The JAK‐STAT signaling network in the human B‐cell: an extreme signaling pathway analysis. Biophys J 2004, 87:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dumas ME, Kinross J, Nicholson JK. Metabolic phenotyping and systems biology approaches to understanding metabolic syndrome and fatty liver disease. Gastroenterology 2014, 146:46–62. [DOI] [PubMed] [Google Scholar]
- 58. Nicholson JK, Lindon JC, Holmes E. ‘Metabonomics:’ understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29:1181–1189. [DOI] [PubMed] [Google Scholar]
- 59. Martin FP, Collino S, Rezzi S. 1H NMR‐based metabonomic applications to decipher gut microbial metabolic influence on mammalian health. Magn Reson Chem 2011, 49(suppl 1):S47–S54. [DOI] [PubMed] [Google Scholar]
- 60. Martin FP, Dumas ME, Wang Y, Legido‐Quigley C, Yap IK, Tang H, Zirah S, Murphy GM, Cloarec O, Lindon JC, et al. A top‐down systems biology view of microbiome‐mammalian metabolic interactions in a mouse model. Mol Syst Biol 2007, 3:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Martin FP, Wang Y, Sprenger N, Yap IK, Lundstedt T, Lek P, Rezzi S, Ramadan Z, van Bladeren P, Fay LB, et al. Probiotic modulation of symbiotic gut microbial‐host metabolic interactions in a humanized microbiome mouse model. Mol Syst Biol 2008, 4:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Martin FP, Sprenger N, Yap IK, Wang Y, Bibiloni R, Rochat F, Rezzi S, Cherbut C, Kochhar S, Lindon JC, et al. Panorganismal gut microbiome‐host metabolic crosstalk. J Proteome Res 2009, 8:2090–2105. [DOI] [PubMed] [Google Scholar]
- 63. Waldram A, Holmes E, Wang Y, Rantalainen M, Wilson ID, Tuohy KM, McCartney AL, Gibson GR, Nicholson JK. Top‐down systems biology modeling of host metabotype‐microbiome associations in obese rodents. J Proteome Res 2009, 8:2361–2375. [DOI] [PubMed] [Google Scholar]
- 64. Claus SP, Ellero SL, Berger B, Krause L, Bruttin A, Molina J, Paris A, Want EJ, de Waziers I, Cloarec O, et al. Colonization‐induced host‐gut microbial metabolic interaction. MBio 2011, 2:e00271–10. Available at: http://mbio.asm.org/content/2/2/e00271‐10.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Claus SP, Tsang TM, Wang Y, Cloarec O, Skordi E, Martin FP, Rezzi S, Ross A, Kochhar S, Holmes E, et al. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol Syst Biol 2008, 4:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Velagapudi VR, Hezaveh R, Reigstad CS, Gopalacharyulu P, Yetukuri L, Islam S, Felin J, Perkins R, Boren J, Oresic M, et al. The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res 2010, 51:1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA 2009, 106:3698–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mestdagh R, Dumas ME, Rezzi S, Kochhar S, Holmes E, Claus SP, Nicholson JK. Gut microbiota modulate the metabolism of brown adipose tissue in mice. J Proteome Res 2012, 11:620–630. [DOI] [PubMed] [Google Scholar]
- 69. Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H, Zhang Y, Shen J, Pang X, Zhang M, et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci USA 2008, 105:2117–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci USA 2012, 109:594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shoaie S, Karlsson F, Mardinoglu A, Nookaew I, Bordel S, Nielsen J. Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci Rep 2013, 3:2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Heinken A, Thiele I. Anoxic conditions promote species‐specific mutualism between gut microbes in silico. Appl Environ Microbiol 2015. doi:10.1128/AEM.00101-15. Available at: http://aem.asm.org/content/early/2015/03/30/AEM.00101‐15.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Heinken A, Sahoo S, Fleming RM, Thiele I. Systems‐level characterization of a host‐microbe metabolic symbiosis in the mammalian gut. Gut Microbes 2013, 4:28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Heinken A, Thiele I. Systematic prediction of health‐relevant human‐microbial co‐metabolism through a computational framework. Gut Microbes 2015, 6:120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez‐Mueller B, Zucker J, Thiagarajan M, Henrissat B, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 2012, 8:e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, Huttenhower C. Microbial co‐occurrence relationships in the human microbiome. PLoS Comput Biol 2012, 8:e1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Caspi R, Altman T, Dale JM, Dreher K, Fulcher CA, Gilham F, Kaipa P, Karthikeyan AS, Kothari A, Krummenacker M, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 2010, 38:D473–D479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res 2008, 36:D480–D484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jacobsen UP, Nielsen HB, Hildebrand F, Raes J, Sicheritz‐Ponten T, Kouskoumvekaki I, Panagiotou G. The chemical interactome space between the human host and the genetically defined gut metabotypes. ISME J 2013, 7:730–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R, Palsson BO. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci USA 2007, 104:1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V, et al. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res 2011, 39:D1035–D1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sridharan GV, Choi K, Klemashevich C, Wu C, Prabakaran D, Pan LB, Steinmeyer S, Mueller C, Yousofshahi M, Alaniz RC, et al. Prediction and quantification of bioactive microbiota metabolites in the mouse gut. Nat Commun 2014, 5:5492. [DOI] [PubMed] [Google Scholar]
- 83. Borenstein E, Kupiec M, Feldman MW, Ruppin E. Large‐scale reconstruction and phylogenetic analysis of metabolic environments. Proc Natl Acad Sci USA 2008, 105:14482–14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Levy R, Borenstein E. Metabolic modeling of species interaction in the human microbiome elucidates community‐level assembly rules. Proc Natl Acad Sci USA 2013, 110:12804–12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Roume H, Muller EE, Cordes T, Renaut J, Hiller K, Wilmes P. A biomolecular isolation framework for eco‐systems biology. ISME J 2013, 7:110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sahoo S, Aurich MK, Jonsson JJ, Thiele I. Membrane transporters in a human genome‐scale metabolic knowledgebase and their implications for disease. Front Physiol 2014, 5:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Palsson B. Systems Biology: Properties of Reconstructed Networks. Cambridge: Cambridge University Press; 2006. [Google Scholar]
- 88. Heinken A, Khan MT, Paglia G, Rodionov DA, Harmsen HJ, Thiele I. A functional metabolic map of Faecalibacterium prausnitzii, a beneficial human gut microbe. J Bacteriol 2014, 196:3289–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Monk J, Nogales J, Palsson BO. Optimizing genome‐scale network reconstructions. Nat Biotechnol 2014, 32:447–452. [DOI] [PubMed] [Google Scholar]
- 90. Thiele I, Heinken A, Fleming RM. A systems biology approach to studying the role of microbes in human health. Curr Opin Biotechnol 2013, 24:4–12. [DOI] [PubMed] [Google Scholar]
- 91. Lewis NE, Nagarajan H, Palsson BO. Constraining the metabolic genotype‐phenotype relationship using a phylogeny of in silico methods. Nat Rev Microbiol 2012, 10:291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Orth JD, Thiele I, Palsson BO. What is flux balance analysis? Nat Biotechnol 2010, 28:245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Schellenberger J, Que R, Fleming RM, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, et al. Quantitative prediction of cellular metabolism with constraint‐based models: the COBRA toolbox v2.0. Nat Protoc 2011, 6:1290–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Vo TD, Paul Lee WN, Palsson BO. Systems analysis of energy metabolism elucidates the affected respiratory chain complex in Leigh's syndrome. Mol Genet Metab 2007, 91:15–22. [DOI] [PubMed] [Google Scholar]
- 95. Aurich MK, Paglia G, Rolfsson O, Hrafnsdóttir S, Magnúsdóttir M, Stefaniak MM, Palsson BO, Fleming RMT, Thiele I. Prediction of intracellular metabolic states from extracellular metabolomic data. Metabolomics 2015. Available at: http://link.springer.com/article/10.1007%2Fs11306‐014‐0721‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. McCloskey D, Gangoiti JA, King ZA, Naviaux RK, Barshop BA, Palsson BO, Feist AM. A model‐driven quantitative metabolomics analysis of aerobic and anaerobic metabolism in E. coli K‐12 MG1655 that is biochemically and thermodynamically consistent. Biotechnol Bioeng 2014, 111:803–815. [DOI] [PubMed] [Google Scholar]
- 97. Yizhak K, Benyamini T, Liebermeister W, Ruppin E, Shlomi T. Integrating quantitative proteomics and metabolomics with a genome‐scale metabolic network model. Bioinformatics 2010, 26:i255–i260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Schmidt BJ, Ebrahim A, Metz TO, Adkins JN, Palsson BO, Hyduke DR. GIM3E: condition‐specific models of cellular metabolism developed from metabolomics and expression data. Bioinformatics 2013, 29:2900–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zamboni N, Sauer U. Novel biological insights through metabolomics and 13C‐flux analysis. Curr Opin Microbiol 2009, 12:553–558. [DOI] [PubMed] [Google Scholar]
- 100. Haraldsdottir HS, Thiele I, Fleming RM. Comparative evaluation of open source software for mapping between metabolite identifiers in metabolic network reconstructions: application to Recon 2. J Cheminform 2014, 6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Thiele I, Palsson BO. Reconstruction annotation jamborees: a community approach to systems biology. Mol Syst Biol 2010, 6:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Greenblum S, Chiu HC, Levy R, Carr R, Borenstein E. Towards a predictive systems‐level model of the human microbiome: progress, challenges, and opportunities. Curr Opin Biotechnol 2013, 24:810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Stolyar S, Van Dien S, Hillesland KL, Pinel N, Lie TJ, Leigh JA, Stahl DA. Metabolic modeling of a mutualistic microbial community. Mol Syst Biol 2007, 3:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Klitgord N, Segre D. Environments that induce synthetic microbial ecosystems. PLoS Comput Biol 2010, 6:e1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Taffs R, Aston JE, Brileya K, Jay Z, Klatt CG, McGlynn S, Mallette N, Montross S, Gerlach R, Inskeep WP, et al. In silico approaches to study mass and energy flows in microbial consortia: a syntrophic case study. BMC Syst Biol 2009, 3:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wintermute EH, Silver PA. Emergent cooperation in microbial metabolism. Mol Syst Biol 2010, 6:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Freilich S, Zarecki R, Eilam O, Segal ES, Henry CS, Kupiec M, Gophna U, Sharan R, Ruppin E. Competitive and cooperative metabolic interactions in bacterial communities. Nat Commun 2011, 2:589. [DOI] [PubMed] [Google Scholar]
- 108. Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, Shah N, Wang CY, Magrini V, Wilson RK, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci USA 2009, 106:5859–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zhuang K, Izallalen M, Mouser P, Richter H, Risso C, Mahadevan R, Lovley DR. Genome‐scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J 2011, 5:305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Harcombe WR, Riehl WJ, Dukovski I, Granger BR, Betts A, Lang AH, Bonilla G, Kar A, Leiby N, Mehta P, et al. Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep 2014, 7:1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Khandelwal RA, Olivier BG, Roling WF, Teusink B, Bruggeman FJ. Community flux balance analysis for microbial consortia at balanced growth. PLoS One 2013, 8:e64567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zomorrodi AR, Maranas CD. OptCom: a multi‐level optimization framework for the metabolic modeling and analysis of microbial communities. PLoS Comput Biol 2012, 8:e1002363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zomorrodi AR, Islam MM, Maranas CD. d‐OptCom: dynamic multi‐level and multi‐objective metabolic modeling of microbial communities. ACS Synth Biol 2014, 3:247–257. [DOI] [PubMed] [Google Scholar]
- 114. El‐Semman IE, Karlsson FH, Shoaie S, Nookaew I, Soliman TH, Nielsen J. Genome‐scale metabolic reconstructions of Bifidobacterium adolescentis L2‐32 and Faecalibacterium prausnitzii A2‐165 and their interaction. BMC Syst Biol 2014, 8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bordbar A, Lewis NE, Schellenberger J, Palsson BO, Jamshidi N. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol Syst Biol 2010, 6:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sigurdsson MI, Jamshidi N, Steingrimsson E, Thiele I, Palsson BO. A detailed genome‐wide reconstruction of mouse metabolism based on human Recon 1. BMC Syst Biol 2010, 4:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD, Gordon JI. Glycan foraging in vivo by an intestine‐adapted bacterial symbiont. Science 2005, 307:1955–1959. [DOI] [PubMed] [Google Scholar]
- 118. Thiele I, Swainston N, Fleming RM, Hoppe A, Sahoo S, Aurich MK, Haraldsdottir H, Mo ML, Rolfsson O, Stobbe MD, et al. A community‐driven global reconstruction of human metabolism. Nat Biotechnol 2013, 31:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Macfarlane GT, Macfarlane S. Bacteria, colonic fermentation, and gastrointestinal health. J AOAC Int 2012, 95:50–60. [DOI] [PubMed] [Google Scholar]
- 120. Russell WR, Hoyles L, Flint HJ, Dumas ME. Colonic bacterial metabolites and human health. Curr Opin Microbiol 2013, 16:246–254. [DOI] [PubMed] [Google Scholar]
- 121. Stephens NS, Siffledeen J, Su X, Murdoch TB, Fedorak RN, Slupsky CM. Urinary NMR metabolomic profiles discriminate inflammatory bowel disease from healthy. J Crohns Colitis 2013, 7:e42–e48. [DOI] [PubMed] [Google Scholar]
- 122. Holmes E, Loo RL, Stamler J, Bictash M, Yap IK, Chan Q, Ebbels T, De Iorio M, Brown IJ, Veselkov KA, et al. Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 2008, 453:396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Elamin EE, Masclee AA, Dekker J, Jonkers DM. Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr Rev 2013, 71:483–499. [DOI] [PubMed] [Google Scholar]
- 124. Joyce SA, Gahan CG. The gut microbiota and the metabolic health of the host. Curr Opin Gastroenterol 2014, 30:120–127. [DOI] [PubMed] [Google Scholar]
- 125. Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, Tatoud R, Blanc V, Lindon JC, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin‐resistant mice. Proc Natl Acad Sci USA 2006, 103:12511–12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, Baumler AJ. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci USA 2011, 108:17480–17485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wang C, Kong H, Guan Y, Yang J, Gu J, Yang S, Xu G. Plasma phospholipid metabolic profiling and biomarkers of type 2 diabetes mellitus based on high‐performance liquid chromatography/electrospray mass spectrometry and multivariate statistical analysis. Anal Chem 2005, 77:4108–4116. [DOI] [PubMed] [Google Scholar]
- 128. Kim JY, Park JY, Kim OY, Ham BM, Kim HJ, Kwon DY, Jang Y, Lee JH. Metabolic profiling of plasma in overweight/obese and lean men using ultra performance liquid chromatography and Q‐TOF mass spectrometry (UPLC‐Q‐TOF MS). J Proteome Res 2010, 9:4368–4375. [DOI] [PubMed] [Google Scholar]
- 129. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, et al. Intestinal microbiota metabolism of L‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 2013, 19:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res 2006, 47:241–259. [DOI] [PubMed] [Google Scholar]
- 131. Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. A branched‐chain amino acid‐related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 2009, 9:311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Cheng S, Rhee EP, Larson MG, Lewis GD, McCabe EL, Shen D, Palma MJ, Roberts LD, Dejam A, Souza AL, et al. Metabolite profiling identifies pathways associated with metabolic risk in humans. Circulation 2012, 125:2222–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, et al. Metabolite profiles and the risk of developing diabetes. Nat Med 2011, 17:448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Le Gall G, Noor SO, Ridgway K, Scovell L, Jamieson C, Johnson IT, Colquhoun IJ, Kemsley EK, Narbad A. Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J Proteome Res 2011, 10:4208–4218. [DOI] [PubMed] [Google Scholar]
- 135. Russell WR, Gratz SW, Duncan SH, Holtrop G, Ince J, Scobbie L, Duncan G, Johnstone AM, Lobley GE, Wallace RJ, et al. High‐protein, reduced‐carbohydrate weight‐loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr 2011, 93:1062–1072. [DOI] [PubMed] [Google Scholar]
- 136. Lees HJ, Swann JR, Wilson ID, Nicholson JK, Holmes E. Hippurate: the natural history of a mammalian‐microbial cometabolite. J Proteome Res 2013, 12:1527–1546. [DOI] [PubMed] [Google Scholar]
- 137. Swann JR, Spagou K, Lewis M, Nicholson JK, Glei DA, Seeman TE, Coe CL, Goldman N, Ryff CD, Weinstein M, et al. Microbial‐mammalian cometabolites dominate the age‐associated urinary metabolic phenotype in Taiwanese and American populations. J Proteome Res 2013, 12:3166–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Williams HR, Cox IJ, Walker DG, Cobbold JF, Taylor‐Robinson SD, Marshall SE, Orchard TR. Differences in gut microbial metabolism are responsible for reduced hippurate synthesis in Crohn's disease. BMC Gastroenterol 2010, 10:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Williams HR, Cox IJ, Walker DG, North BV, Patel VM, Marshall SE, Jewell DP, Ghosh S, Thomas HJ, Teare JP, et al. Characterization of inflammatory bowel disease with urinary metabolic profiling. Am J Gastroenterol 2009, 104:1435–1444. [DOI] [PubMed] [Google Scholar]
- 140. Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host‐microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci USA 2009, 106:14728–14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Poesen R, Viaene L, Verbeke K, Augustijns P, Bammens B, Claes K, Kuypers D, Evenepoel P, Meijers B. Cardiovascular disease relates to intestinal uptake of p‐cresol in patients with chronic kidney disease. BMC Nephrol 2014, 15:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Wu IW, Hsu KH, Lee CC, Sun CY, Hsu HJ, Tsai CJ, Tzen CY, Wang YC, Lin CY, Wu MS. p‐Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol Dial Transplant 2011, 26:938–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Meijers BK, Evenepoel P. The gut‐kidney axis: indoxyl sulfate, p‐cresyl sulfate and CKD progression. Nephrol Dial Transplant 2011, 26:759–761. [DOI] [PubMed] [Google Scholar]
- 144. Winterkamp S, Weidenhiller M, Otte P, Stolper J, Schwab D, Hahn EG, Raithel M. Urinary excretion of N‐methylhistamine as a marker of disease activity in inflammatory bowel disease. Am J Gastroenterol 2002, 97:3071–3077. [DOI] [PubMed] [Google Scholar]
- 145. Haiser HJ, Turnbaugh PJ. Developing a metagenomic view of xenobiotic metabolism. Pharmacol Res 2013, 69:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Sousa T, Paterson R, Moore V, Carlsson A, Abrahamsson B, Basit AW. The gastrointestinal microbiota as a site for the biotransformation of drugs. Int J Pharm 2008, 363:1–25. [DOI] [PubMed] [Google Scholar]
- 147. Davila AM, Blachier F, Gotteland M, Andriamihaja M, Benetti PH, Sanz Y, Tome D. Re‐print of “Intestinal luminal nitrogen metabolism: role of the gut microbiota and consequences for the host.” Pharmacol Res 2013, 69:114–126. [DOI] [PubMed] [Google Scholar]
- 148. Hughes R, Magee EA, Bingham S. Protein degradation in the large intestine: relevance to colorectal cancer. Curr Issues Intest Microbiol 2000, 1:51–58. [PubMed] [Google Scholar]
- 149. Soga T, Sugimoto M, Honma M, Mori M, Igarashi K, Kashikura K, Ikeda S, Hirayama A, Yamamoto T, Yoshida H, et al. Serum metabolomics reveals γ‐glutamyl dipeptides as biomarkers for discrimination among different forms of liver disease. J Hepatol 2011, 55:896–905. [DOI] [PubMed] [Google Scholar]
- 150. Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S, et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res 2009, 37:D603–D610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Allen‐Vercoe E. Bringing the gut microbiota into focus through microbial culture: recent progress and future perspective. Curr Opin Microbiol 2013, 16:625–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Reed JL, Patel TR, Chen KH, Joyce AR, Applebee MK, Herring CD, Bui OT, Knight EM, Fong SS, Palsson BO. Systems approach to refining genome annotation. Proc Natl Acad Sci USA 2006, 103:17480–17484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Manichaikul A, Ghamsari L, Hom EF, Lin C, Murray RR, Chang RL, Balaji S, Hao T, Shen Y, Chavali AK, et al. Metabolic network analysis integrated with transcript verification for sequenced genomes. Nat Methods 2009, 6:589–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Huttenhower C, Knight R, Brown CT, Caporaso JG, Clemente JC, Gevers D, Franzosa EA, Kelley ST, Knights D, Ley RE, et al. Advancing the microbiome research community. Cell 2014, 159:227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Integrative HMPRNC . The Integrative Human Microbiome Project: dynamic analysis of microbiome‐host omics profiles during periods of human health and disease. Cell Host Microbe 2014, 16:276–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Ravikrishnan A, Raman K. Critical assessment of genome‐scale metabolic networks: the need for a unified standard. Brief Bioinform 2015. doi: 10.1093/bib/bbv003. [DOI] [PubMed] [Google Scholar]
- 157. Ganter M, Bernard T, Moretti S, Stelling J, Pagni M. MetaNetX.org: a website and repository for accessing, analysing and manipulating metabolic networks. Bioinformatics 2013, 29:815–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. de Crecy‐Lagard V. Variations in metabolic pathways create challenges for automated metabolic reconstructions: examples from the tetrahydrofolate synthesis pathway. Comput Struct Biotechnol J 2014, 10:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ravcheev DA, Thiele I. Systematic genomic analysis reveals the complementary aerobic and anaerobic respiration capacities of the human gut microbiota. Front Microbiol 2014, 5:674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Magnusdottir S, Ravcheev DA, de Crecy‐Lagard V, Thiele I. Systematic genome assessment of B‐vitamin biosynthesis suggests co‐operation among gut microbes. Front Genet 2015, 6:148. doi: 10.3389/fgene.2015.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Ravcheev DA, Godzik A, Osterman AL, Rodionov DA. Polysaccharides utilization in human gut bacterium Bacteroides thetaiotaomicron: comparative genomics reconstruction of metabolic and regulatory networks. BMC Genomics 2013, 14:873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Martens EC, Kelly AG, Tauzin AS, Brumer H. The devil lies in the details: how variations in polysaccharide fine‐structure impact the physiology and evolution of gut microbes. J Mol Biol 2014, 426:3851–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Eilam O, Zarecki R, Oberhardt M, Ursell LK, Kupiec M, Knight R, Gophna U, Ruppin E. Glycan degradation (GlyDeR) analysis predicts mammalian gut microbiota abundance and host diet‐specific adaptations. MBio 2014, 5:e01526–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. WHO . Fact sheet on obesity and overweight. Available at: http://www.who.int/mediacentre/factsheets/fs311/en/. (Accessed November 30, 2014).
- 165. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27:1047–1053. [DOI] [PubMed] [Google Scholar]
- 166. Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta . Science 2013, 341:295–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Kell DB, Goodacre R. Metabolomics and systems pharmacology: why and how to model the human metabolic network for drug discovery. Drug Discov Today 2014, 19:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Oberhardt MA, Yizhak K, Ruppin E. Metabolically re‐modeling the drug pipeline. Curr Opin Pharmacol 2013, 13:778–785. [DOI] [PubMed] [Google Scholar]
- 169. Folger O, Jerby L, Frezza C, Gottlieb E, Ruppin E, Shlomi T. Predicting selective drug targets in cancer through metabolic networks. Mol Syst Biol 2011, 7:501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Frezza C, Zheng L, Folger O, Rajagopalan KN, MacKenzie ED, Jerby L, Micaroni M, Chaneton B, Adam J, Hedley A, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477:225–228. [DOI] [PubMed] [Google Scholar]
- 171. Sahoo S, Haraldsdottir HS, Fleming RM, Thiele I. Modeling the effects of commonly used drugs on human metabolism. FEBS J 2015, 282:297–317. [DOI] [PubMed] [Google Scholar]
- 172. Bordbar A, Palsson BO. Using the reconstructed genome‐scale human metabolic network to study physiology and pathology. J Intern Med 2012, 271:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Zelezniak A, Pers TH, Soares S, Patti ME, Patil KR. Metabolic network topology reveals transcriptional regulatory signatures of type 2 diabetes. PLoS Comput Biol 2010, 6:e1000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Li L, Zhou X, Ching WK, Wang P. Predicting enzyme targets for cancer drugs by profiling human metabolic reactions in NCI‐60 cell lines. BMC Bioinformatics 2010, 11:501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Agren R, Mardinoglu A, Asplund A, Kampf C, Uhlen M, Nielsen J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome‐scale metabolic modeling. Mol Syst Biol 2014, 10:721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Yizhak K, Gaude E, Le Devedec S, Waldman YY, Stein GY, van de Water B, Frezza C, Ruppin E. Phenotype‐based cell‐specific metabolic modeling reveals metabolic liabilities of cancer. eLIFE 2014, 3:e03641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Nicholson JK, Wilson ID, Lindon JC. Pharmacometabonomics as an effector for personalized medicine. Pharmacogenomics 2011, 12:103–111. [DOI] [PubMed] [Google Scholar]
- 178. Shlomi T, Cabili MN, Ruppin E. Predicting metabolic biomarkers of human inborn errors of metabolism. Mol Syst Biol 2009, 5:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Li L, Jiang H, Qiu Y, Ching WK, Vassiliadis VS. Discovery of metabolite biomarkers: flux analysis and reaction‐reaction network approach. BMC Syst Biol 2013, 7(suppl 2):S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Stempler S, Yizhak K, Ruppin E. Integrating transcriptomics with metabolic modeling predicts biomarkers and drug targets for Alzheimer's disease. PLoS One 2014, 9:e105383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Shestov AA, Barker B, Gu Z, Locasale JW. Computational approaches for understanding energy metabolism. WIREs Syst Biol Med 2013, 5:733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. McNeil NI. The contribution of the large intestine to energy supplies in man. Am J Clin Nutr 1984, 39:338–342. [DOI] [PubMed] [Google Scholar]
- 183. Gordon JI. Honor thy gut symbionts redux. Science 2012, 336:1251–1253. [DOI] [PubMed] [Google Scholar]
- 184. Noor E, Haraldsdottir HS, Milo R, Fleming RM. Consistent estimation of Gibbs energy using component contributions. PLoS Comput Biol 2013, 9:e1003098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Henry CS, Broadbelt LJ, Hatzimanikatis V. Thermodynamics‐based metabolic flux analysis. Biophys J 2007, 92:1792–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Kummel A, Panke S, Heinemann M. Systematic assignment of thermodynamic constraints in metabolic network models. BMC Bioinformatics 2006, 7:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Fleming RM, Thiele I, Provan G, Nasheuer HP. Integrated stoichiometric, thermodynamic and kinetic modelling of steady state metabolism. J Theor Biol 2010, 264:683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Haraldsdottir HS, Thiele I, Fleming RM. Quantitative assignment of reaction directionality in a multicompartmental human metabolic reconstruction. Biophys J 2012, 102:1703–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Fleming RM, Thiele I. von Bertalanffy 1.0: a COBRA toolbox extension to thermodynamically constrain metabolic models. Bioinformatics 2011, 27:142–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Bordbar A, Feist AM, Usaite‐Black R, Woodcock J, Palsson BO, Famili I. A multi‐tissue type genome‐scale metabolic network for analysis of whole‐body systems physiology. BMC Syst Biol 2011, 5:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Galhardo M, Sinkkonen L, Berninger P, Lin J, Sauter T, Heinaniemi M. Integrated analysis of transcript‐level regulation of metabolism reveals disease‐relevant nodes of the human metabolic network. Nucleic Acids Res 2014, 42:1474–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Mardinoglu A, Agren R, Kampf C, Asplund A, Nookaew I, Jacobson P, Walley AJ, Froguel P, Carlsson LM, Uhlen M, et al. Integration of clinical data with a genome‐scale metabolic model of the human adipocyte. Mol Syst Biol 2013, 9:649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol 2007, 19:59–69. [DOI] [PubMed] [Google Scholar]
- 194. Crotti E, Sansonno L, Prosdocimi EM, Vacchini V, Hamdi C, Cherif A, Gonella E, Marzorati M, Balloi A. Microbial symbionts of honeybees: a promising tool to improve honeybee health. N Biotechnol 2013, 30:716–722. [DOI] [PubMed] [Google Scholar]
- 195. de Oliveira Dal'Molin CG, Quek LE, Palfreyman RW, Brumbley SM, Nielsen LK. AraGEM, a genome‐scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiol 2010, 152:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Saha R, Suthers PF, Maranas CD. Zea mays iRS1563: a comprehensive genome‐scale metabolic reconstruction of maize metabolism. PLoS One 2011, 6:e21784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Mintz‐Oron S, Meir S, Malitsky S, Ruppin E, Aharoni A, Shlomi T. Reconstruction of Arabidopsis metabolic network models accounting for subcellular compartmentalization and tissue‐specificity. Proc Natl Acad Sci USA 2012, 109:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Simons M, Saha R, Amiour N, Kumar A, Guillard L, Clement G, Miquel M, Li Z, Mouille G, Lea PJ, et al. Assessing the metabolic impact of nitrogen availability using a compartmentalized maize leaf genome‐scale model. Plant Physiol 2014, 166:1659–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Grafahrend‐Belau E, Junker A, Eschenroder A, Muller J, Schreiber F, Junker BH. Multiscale metabolic modeling: dynamic flux balance analysis on a whole‐plant scale. Plant Physiol 2013, 163:637–647. [DOI] [PMC free article] [PubMed] [Google Scholar]