

Abstract
An abnormally high number of macrophages are present in human brain arteriovenous malformations (bAVM) with or without evidence of prior hemorrhage, causing unresolved inflammation that may enhance abnormal vascular remodeling and exacerbate the bAVM phenotype. The reasons for macrophage accumulation at the bAVM sites are not known. We tested the hypothesis that persistent infiltration and pro-inflammatory differentiation of monocytes in angiogenic tissues increase the macrophage burden in bAVM using two mouse models and human monocytes. Mouse bAVM was induced through deletion of AVM causative genes, Endoglin (Eng) globally or Alk1 focally, plus brain focal angiogenic stimulation. An endothelial cell and vascular smooth muscle cell co-culture system was used to analyze monocyte differentiation in the angiogenic niche. After angiogenic stimulation, the Eng-deleted mice had fewer CD68+ cells at 2 weeks (P=0.02), similar numbers at 4 weeks (P=0.97), and more at 8 weeks (P=0.01) in the brain angiogenic region compared with wild-type (WT) mice. Alk1-deficient mice also had a trend towards more macrophages/microglia 8 weeks (P=0.064) after angiogenic stimulation and more RFP+ bone marrow-derived macrophages than WT mice (P=0.01). More CD34+ cells isolated from peripheral blood of patients with ENG or ALK1 gene mutation differentiated into macrophages than those from healthy controls (P<0.001). These data indicate that persistent infiltration and pro-inflammatory differentiation of monocytes might contribute to macrophage accumulation in bAVM. Blocking macrophage homing to bAVM lesions should be tested as a strategy to reduce the severity of bAVM.
Keywords: Angiogenesis, Animal models, Arteriovenous malformations, Cerebrovascular disease, Macrophages, Microglia
Introduction
Arteriovenous malformations (AVMs) are active angiogenic lesions consisting of tangles of abnormal vessels shunting blood directly from arteries to veins without a true capillary bed [1]. An abnormally high number of macrophages are present in and around vascular walls in human brain AVM (bAVM) specimens, with or without hemorrhage, suggesting that macrophage accumulation is not simply a response to hemorrhage [2-6]. Polymorphisms in inflammatory cytokines and elevated expressions of inflammation-related genes in AVM patients further suggest their active roles in bAVM pathogenesis [7-9]. Macrophage accumulation in bAVM can cause unresolved inflammation, which then enhances abnormal vascular remodeling and the severity of the bAVM phenotype. We have also found increased macrophage burden in bAVM in mouse models generated through conditional deletion of hereditary hemorrhagic telangiectasia (HHT) causative genes, endoglin (Eng) or activin-like kinase 1 (Alk1, ACVLR1), in combination with focal angiogenic stimulation [10-13]. HHT is one of the known familial cases with high prevalence of AVMs in multiple organs including the brain [1,14]. The mechanism of macrophage accumulation in bAVM has not been fully explored [6]. Understanding the mechanism could lead to the development of a new therapy for treating bAVM patients.
It has been shown that Eng-deficiency impairs monocyte homing to the injury site [15,16]. Recruitment of monocytes to the infarcted heart and subsequent vessel formation are severely impaired in Eng heterozygous (Eng+/−) mice [17]. Decreased homing is linked to the inability of monocytes to respond to stromal cell-derived factor 1α (SDF-1α). It has also been shown that Eng-deficiency in endothelial cells (ECs) reduces leukocyte adhesion and transmigration [18], and impairs the EC-autonomous capacity to upregulate SDF-1 expression in response to ischemic injury in a hind-limb ischemic injury model [19]. We found that there is about 10% reduction in monocyte homing to the brain angiogenic focus in mice with Eng+/− bone marrow (BM) compared to mice with wild-type (WT) BM two weeks after AAV-VEGF injection [20]. In addition, we have shown that Eng+/− mice have fewer CD68+ cells than WT mice in the peri-infarct area 3 days after ischemic stroke [21]. Taken together, Eng-deficiency appears to impair monocyte adhesion and migration. There is currently no explanation regarding how Eng-deficient macrophages accumulate in bAVM and whether loss of Alk1 affects these processes.
In this study, we used two mouse models to test if persistent infiltration causes macrophage accumulation and unresolved inflammation in bAVM through quantification of BM-derived macrophages, local microglia and CD68+ cells in bAVM lesions. It has been shown that HHT patients have more CD34+ cells in peripheral blood; however, when cultured in EC growth medium, fewer CD34+ cells display EC phenotypes [22]. Therefore, we also tested the differentiation of HHT CD34+ monocytes in angiogenic niches.
Materials and methods
Ethical standards
All protocols involving human samples were approved by the Research Ethics Board of St. Michael's Hospital, University of Toronto, and the Committee on Human Research of the University of California, San Francisco (UCSF), in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki). The experimental protocols involving animal usage were approved by the Institutional Animal Care and Use Committee (IACUC) of UCSF and conformed to National Institutes of Health (NIH) guidelines. The staff of the Animal Core Facility and the IACUC of UCSF provided animal husbandry under the guidance of supervisors who are certified animal technologists, and IACUC faculty members and veterinary residents located on the San Francisco General Hospital campus provided veterinary care.
Animals
Mice were fed standard rodent food and water ad libitum, and were housed (less than five per cage) in 421 × 316 cm2 sawdust-lined cages in an air-conditioned environment with 12-h light/dark cycles.
Mouse Model 1 was induced using R26CreER/+;Eng2f/2f mouse line that has a Rosa promoter driving and estrogen-inducible cre recombinase expression, and an Eng gene with exons 5 and 6 flanked by loxP sites [23]. Adeno-associated viral vectors (AAV) with cytomegalovirus (CMV) promoter driving VEGF packaged in AAV1 capsid (AAV1-VEGF) were injected into the brain of 8-week-old R26CreER/+;Eng2f/2f mice to induce bAVM as previously described [24]. Briefly, mice were anesthetized with isoflurane inhalation and placed in a stereotactic frame (David Kopf Instruments, Tujunga, CA). A hole was drilled in the pericranium, 1 mm posterior to the coronal suture and 2 mm lateral to the sagittal suture. AAV1-VEGF (2 μl) viral suspension containing 2×109 viral genome (vg) was stereotactically injected into the right basal ganglia 3 mm below the cortex. Control mice received 2 μl of AAV1-LacZ (2×109 vg). Tamoxifen (TM, in corn oil, 2.5 mg/25 g body weight, Sigma-Aldrich, Carlsbad, CA) or corn oil (control) was injected intraperitoneally for 3 consecutive days starting from the day of viral injection to globally delete the Eng gene.
The mice were randomly assigned to four experimental groups treated with (1) corn oil plus AAV1-LacZ, (2) corn oil plus AAV1-VEGF, (3) TM plus AAV1-LacZ, and (4) TM plus AAV1-VEGF. Brain samples were collected 2, 4 and 8 weeks after viral injection (Fig. 1a).
Fig. 1. Animal models.

a Model 1: bAVMs were in induced in R26CreER/+;Eng2f/2f mouse line that has a Rose promoter driving and estrogen inducible cre recombinase and an Eng gene with exons 5 and 6 flanked by loxP sites [23] by injection of AAV1-VEGF stereotactically into the basal ganglia to induce brain focal angiogenesis and i.p. injection of 3 doses of tamoxifen (TM) on 3 consecutive days to globally delete the Eng gene. Brain samples were collected 2, 4 and 8 weeks after AAV1-VEGF injection. b Model 2: bAVMs were induced in Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice that have Alk1 gene deleted in one allele and floxed in the other allele [25], RFP gene knocked into one allele of Ccr2 gene and GFP knocked into one allele of Cx3cr1 gene [26] through co-injection of Ad-Cre and AAV1-VEGF stereotactically into the cortex to induce brain focal deletion of Alk1 floxed allele and angiogenesis. Brain samples were collected 8 weeks after the vector injection.
Mouse Model 2 was induced in Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice that have the Alk1 gene deleted in one allele and floxed in the other allele (exons 4 to 6 flanked by loxP sites) [25], red fluorescent protein (RFP) gene knocked into one allele of Ccr2 gene, and green fluorescent protein gene (GFP) knocked into one allele of Cx3cr1 gene [26]. The macrophages in these mice expressed RFP, and the microglia, GFP. Adenoviral vector carrying CMV promoter driving Cre recombinase expression (Ad-Cre, 2×107 plaque forming unit) and AAV1-VEGF (2×109 vg) were co-injected stereotactically into the cortex of 8-week-old Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice to induce bAVM as described previously [11]. AAV1-LacZ was used as vector control. Briefly, the needle was inserted 1 mm into the cortex and advanced 2 mm parallel to the surface, and the vector was then injected. Exons 4 to 6 of the Alk1-floxed allele were deleted in the brain through injection of Ad-Cre, leading to loss of Alk1 gene function. In this model, bAVM developed 8 weeks after vector injection.
The three experimental groups were: (1) Alk1+/+(WT);Ccr2RFP/+/Cx3cr1GFP/+ mice injected with Ad-Cre/AAV1-VEGF; (2) Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice injected with Ad-Cre/AAV1-VEGF; and (3) Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice injected with Ad-Cre/AAV1-LacZ. Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice were randomly assigned to either group 2 or group 3. Brain samples were collected 8 weeks after vector injection (Fig. 1b).
Immunohistochemistry
Model 1
Brain samples were collected 2, 4, and 8 weeks after induction. After being anesthetized with isoflurane inhalation, mice in the 8-week group were perfused through the left cardiac ventricle with heparinized PBS to remove the intravascular blood before brain sample collection. Mice in the 2- and 4-week groups were not perfused. Brain samples were frozen in dry ice, and cut into 20-μm-thick coronal sections, which were then co-stained with antibodies against CD31 (an EC marker, 1:50, Abcam, Burlingame, CA) and CD68 (a macrophage marker, 1:500; AbD Serotec, Raleigh, NC). Positive stains were visualized by incubating the sections with fluorescent-labeled secondary antibodies: Alexa Fluor 488 goat anti-rabbit IgG (1:500, Invitrogen, South San Francisco, CA) for CD 31, and Alexa Fluor 594 goat anti-rat IgG (1:500, Invitrogen) for CD68.
Model 2
Mice were anesthetized with isoflurane inhalation and perfused with PBS through the left cardiac ventricle followed by 4% paraformaldehyde. Brain samples were collected, incubated in 4% paraformaldehyde containing 20% sucrose for 2 days and frozen in dry ice, and then sectioned into 20-μm-thick sections. RFP+ and GFP+ cells were directly detected under a fluorescent microscope (Keyence BZ-9000, Itasca, IL).
Macrophage quantification
Two coronal sections per brain sample, 0.5 mm rostral and 0.5 mm caudal to the viral injection site, were selected. Images from three areas (to the right and left of and below the injection site) per section were taken under a 200× magnification. CD68+ cells in Model 1 and RFP+ and GFP+ cells in Model 2 were quantified using NIH Image 1.63 software by three investigators who were blinded to the experimental groups.
ELISA
To examine the VEGF expression in the AAV1-VEGF-injected brain, brain tissue around the vector injection site was collected from R26CreER/+;Eng2f/2f mice and lysed in a cell lysis buffer (Tris-buffered saline, protease inhibitors, 0.1% NP-40). Human VEGF ELISA was performed using an ELISA kit (R&D systems, Minneapolis, MN) according to the manufacturer's instructions.
Latex casting
Mice were anesthetized using isoflurane inhalation, and the thoracic cavities were opened. After cutting off the left and right atria, latex dye (Blue latex, Connecticut Valley Biological Supply Co., Southampton, MA) was injected into the left ventricle using a 22-gauge needle with a 3-ml syringe. Brain samples were collected, fixed with 10% formalin overnight, dehydrated using methanol series, and clarified with an organic solvent (benzyl alcohol:benzyl benzoate, 1:1; Sigma-Aldrich) [27].
Informed consent
Informed written consent for study participation was obtained from all patients and healthy volunteers.
Differentiation of CD34+ cells in endothelial cell (EC) and vascular smooth muscle cell (vSMC) co-culture transwell
A total of 8 clinically diagnosed HHT patients, 3 with known ENG mutations (HHT1), 3 with known ALK1 mutations (HHT2), and 2 unidentified were used in this study. Healthy age- and gender-matched volunteers (n=7) served as controls.
Peripheral blood mononuclear cells (PBMNCs) were isolated from 50 ml of venous blood by Ficoll (StemCell Technology, Vancouver, Canada) density gradient centrifugation. CD34+ cells were isolated using CD34-antibody-coated Dynabeads (Life Technologies, South San Francisco, CA) according to the manufacturer's instructions.
An EC and vSMC co-culture system [28] was used to mimic the angiogenic niche (Online resource Supp. Figure 1). The membrane of the insert and the bottom of the well were pre-coated with human fibronectin (30 mg/ml). Human umbilical vein ECs (HUVECs, Lonza, Allendale, NJ) were plated onto the membrane of the insert (5×105 cells/cm2), and human aortic SMCs (HASMC, Lonza) were seeded to the well (2×105 cells/cm2). HUVECs on the insert were cultured in M199 medium containing 20% FBS, while the SMCs were cultured in F12k medium containing 10% FBS. Upon confluence, the insert and bottom well were assembled together. The HUVECs and HASMCs were cultured for 24 hours in a M199 medium containing 2% FBS. CD34+ cells (105) labeled with Green CMFDA (5-chloromethylfluorescein diacetate) were applied onto the HUVEC layer and cultured for 7 days. The CD34+ cells either incorporated with HUVECs (adhesive cell with an EC phenotype) or migrated to the bottom chamber (migrated cells with a macrophage phenotype) [28]. The EC phenotype of adhesive cells and macrophage phenotype of migrated cells were confirmed by fluorescent-activated cell sorting (FACS) using EC markers CD31 and CD146, and macrophage markers CD11b and CD14 (Online resource Supp. Figure 2). Monoclonal antibodies used in the FACS were anti CD31-V450 (1:100), anti CD146-PE (1:50), anti CD14-PE-Cy7 (1:100, BD Biosciences, San Jose, CA), and anti-CD11b-V450 (1:100, Biolegend, San Diego, CA). BD CompBead Anti-Mouse Ig Kappa (BD Biosciences) incubated with each antibody was used to optimize fluorescence compensation settings for multicolor flow cytometric analysis. The samples were analyzed on a FACS LSR II (BD Biosciences), and percentage of CMFDA cells was calculated using DIVA V 6.1.3 software.
ENG and ALK1 expressions were analyzed using quantitative real-time-PCR in monocytes isolated from 11 controls, 7 HHT1 patients and 6 HHT2 patients. Total RNA was extracted from CD34+ monocytes using RNAzol@RT (Molecular Research Center, Cincinnati, OH) and reverse-transcribed into cDNA using SuperScript* III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Real-time PCR was performed using TaqMan Fast Advanced Master Mix (Applied Biosystems, Foster City, CA). Gene-specific primers and probes purchased from Applied Biosystems were used: ALK1 (ACVRL1, Hs00953798_m1), ENG (Hs00923996_m1) and GAPDH (Hs02758991_g1). All samples were run in triplicate, and relative gene expression was calculated using the comparative threshold cycle (CT) and normalized to GAPDH (ΔCT). Results are exhibited as fold-changes compared to cells isolated from controls.
Statistical analysis
Data are represented as mean ± SD. Prism 6 (GraphPad Software Inc., La Jolla, CA) was used for all the statistical analyses in this study. Sample sizes are shown in figure legends and were determined based on our previous study [11].
For animal studies, one-way analysis of variance (ANOVA) was used to determine statistical significance among groups, followed by Tukey's post-hoc test. Student's t-test was performed when two groups were compared. A p value of <0.05 was considered statistically significant.
Two-way ANOVA was used to compare the differentiation of CD34+ cells in the transwell co-culture system.
Results
Eng-deficient mice (Model 1) had fewer CD68+ cells at the early stage and more CD68+ cells at the later stage of angiogenic stimulation
Injection of AAV1-VEGF expressing human VEGF165 protein into the brain resulted in expression of human VEGF in the injection site (66.6±31.1 pg/mg of brain tissue). VEGF in the AAV-LacZ injection site (background) was 1.7±2.6pg/mg (Figure 2a).
Fig. 2. VEGF expression and chaotically assembled vessels in the angiogenic region of Eng-deficient mice (Model 1).
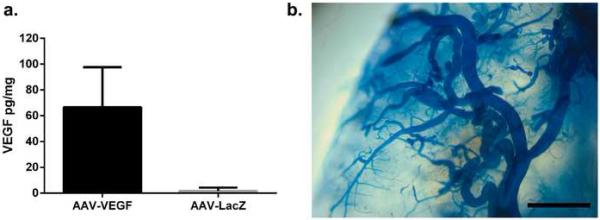
a Quantification of human VEGF levels in the AAV1-VEGF-injected region. N=6. b Representative image of chaotically assembled bAVM vessels in AAV1-VEGF injection site. Scale bar: 1 mm.
Similar to our previous findings [13], R26CreER/+;Eng2f/2f mice developed bAVM 8 weeks after TM-induced global Eng deletion and intra-brain injection of AAV1-VEGF (Figure 2b). There were more abnormal vessels (dysplasia index, DI: number of vessels larger than 15 μm/200 vessels) in TM/AAV1-VEGF-treated mice than those treated with TM/AAV1-LacZ, corn oil/AAV1-VEGF or corn oil/AAV-LacZ (P<0.0001, Online resource Supp. Figure 3). Therefore the bAVM model was successfully induced.
There were more macrophages (544±168/mm2) in the bAVM lesion of TM/AAV1-VEGF-treated mice 8 weeks after angiogenic stimulation than in the brain angiogenic region of those treated with corn oil/AAV1-VEGF (345±79/mm2, P=0.01), corn oil/AAV1-LacZ (208±75/mm2, P<0.0001) and TM/AAV1-LacZ (212±15/mm2, P<0.0001, Fig. 3), consistent with our previous data showing that bAVMs increase macrophage burden [11-13].
Fig. 3. More CD68+ cells in bAVM lesion of Eng-deficient brain (Model 1) 8-weeks after AAV1-VEGF injection.

a Representative images of sections stained with antibodies specific to CD68 (red, macrophages) and CD31 (green, brain microvasculatures). Nuclei were counterstained with DAPI (blue). The animals were perfused with heparinized PBS before sample collection. Arrows indicate CD68+ cells outside vessels or on vessel wall. Scale bar: 100 μm. b Bar graph shows quantifications. *: P<0.05; N=6
To investigate the dynamic of macrophage homing to the brain angiogenic region of Eng-deficient mice, we quantified CD68+ cells at 2 and 4 weeks after angiogenic induction. At 2 weeks, TM/AAV1-VEGF-treated mice had fewer CD68+ cells (463±97/mm2) in the angiogenic region than those treated with corn oil/AAV1-VEGF (607±136/mm2, P=0.02) (Fig. 4a & c). At 4 weeks, mice treated with TM/AAV1-VEGF and corn oil/AAV1-VEGF had similar numbers of CD68+ cells in the brain angiogenic region (499±83/mm2 vs. 496±144/mm2, P=0.97, Fig. 4b & c). These data confirm previous observations that the ability of Eng-deficient monocyte homing to angiogenic/injury tissue is impaired [17,29].
Fig. 4. Slow onset and persistent CD68+ cells homed to the brain angiogenic region of Eng-deficient mice (Model 1).

a, b Representative images of sections stained with antibodies specific to CD68 (red, macrophages) and CD31 (green, brain microvasculature). Nuclei were counterstained with DAPI (blue). Scale bar: 100 μm c Bar graph shows quantifications. *: P<0.05; N=6
Alk1-deficient brain (Model 2) had more BM-derived macrophages in bAVM
The influence of Alk1 deficiency on monocytes has not been thoroughly studied. We have shown in our previous studies that bAVM in Alk1-deficient mice also have CD68+ cell accumulation [11,12]. To investigate if BM-derived macrophages with one functional Alk1 allele also accumulate in the bAVM lesion, we used Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice that have the Alk1 gene deleted in one allele and exons 4 to 6 floxed in the other allele [25], RFP gene knocked into one allele of Ccr2 gene, and GFP knocked into one allele of the Cx3cr1 gene [26]. In these mice, the BM-derived macrophages expressed RFP, and the microglia, GFP. Brain focal deletion of Alk1 exon 4 to 6 from the floxed allele was achieved by stereotactic injection of Ad-Cre. The brain AVM phenotype was induced by co-injecting Ad-Cre and AAV1-VEGF [10]. The BM-derived macrophages in this model were Alk1+/− (heterozygous). Eight weeks after Ad-Cre/AAV1-VEGF-injection, more dysplastic vessels (DI: 1.7±0.5) were detected in the brain of Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice than in the brain of WT;Ccr2RFP/+/Cx3cr1GFP/+ mice (no Alk1 mutation; DI: 0.8±0.4, P=0.048) or in the brain of Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice injected with Ad-Cre/AAV1-LacZ (no angiogenic stimulation; DI: 0.5±0.5, P=0.007) (Online resource Supp. Figure 4). No additional abnormality was found in these mice. These data are similar to our previous finding [10] that homozygous deletion of Alk1 gene plus angiogenic stimulation induce bAVMs in Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice.
The Ad-Cre/AAV1-VEGF-injected brain of Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice had more RFP+ BM-derived macrophages (51±32/mm2) in the angiogenic region than the brain of WT;Ccr2RFP/+/Cx3cr1GFP/+ mice (7±10/mm2, P=0.01) or Ad-Cre/AAV-LacZ injected Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice (7±8/mm2, P=0.01) (Fig. 5a & b). The numbers of GFP+ microglia were similar among the groups; however, activated GFP+ microglia (larger cell-body) were clustered around the abnormal vessels in the bAVM region of the Ad-Cre/AAV1-VEGF-injected Alk11f/2f;Ccr2RFP/+/Cx3cr1GFP/+ mice (Fig. 5c & d).
Fig. 5. BM-derived macrophages and microglia clustered around bAVM vessels in the Alk1-deficient brain 8 weeks after angiogenic stimulation (Model 2).

a Representative images show RFP+ BM-derived macrophage. b Quantification of RFP+ cells. * P<0.05 c Representative images show GFP+ microglia. d Quantification of GFP+ microglia. Scale bar: 100 μm. Nuclei were counterstained with DAPI. N=5
More HHT CD34+ cells differentiated into macrophages in the angiogenic niche
In the angiogenic niche mimicked by the EC and vSMC co-culture system, CD34+ cells isolated from peripheral blood can differentiate into ECs or macrophages [28]. It has been reported that HHT patients have more CD34+ cells in the peripheral blood than healthy controls, but few CD34+ cells displayed EC phenotypes in culture, raising the issue of whether HHT CD34+ cells have an increased propensity to differentiate into macrophages [22]. To examine the differentiation potential of HHT CD34+ cells in an angiogenic niche, we used the EC/vSMC co-culture system [28]. The viabilities of normal and HHT CD34+ cells were similar in the co-culture system (P=0.15, Fig. 6a). Compared to normal controls (64±12%), more HHT CD34+ cells (80±14%) differentiated towards the macrophage lineage (migrating to the bottom chamber) in the culture system (p<0.001, Fig. 6b). We also analyzed ENG and ALK1 expression in HHT monocytes using cells isolated from different control and HHT patient cohorts. We found that compared to control monocytes, ENG expression decreased in both HHT1 and HHT2 monocytes (P<0.001), and ALK1 expression decreased only in HHT2 monocytes (P=0.017) (Fig. 7). Therefore, HHT CD34+ cells are more likely to differentiate into macrophages in angiogenic niches.
Fig. 6. More HHT CD34+ cells differentiated into macrophages.

a Quantification of surviving cells. b Percentage of cells that migrated to bottom chamber. *: P=0.001
Fig. 7. ENG and ALK1 expression in HHT monocytes.

a ENG expression decreased in both HHT1 and HHT2 monocytes. ***: P<0.001 vs. control. b ALK1 expression decreased only in HHT2 monocytes. *: P=0.017 vs. control.
Discussion
We showed in this study that in Eng-deleted mice, fewer macrophages homed to the brain angiogenic region at the early stage (2 weeks) after angiogenic stimulation and more in the later stage (8 weeks) when bAVM had formed. This is consistent with our previous finding that the homing and clearance of Eng-deficient macrophages in injury tissue (angiogenic region) are impaired [29]. The accumulation of macrophages in the angiogenic region of Eng-deficient mice could be due to persistent infiltration and delayed clearance of BM-derived macrophages. Increased vascular permeability and occurrence of microhemorrhage could also cause persistent macrophage accumulation in bAVM. We have also shown that Alk1+/− BM-derived macrophages accumulate in Alk1-deleted bAVM, and that CD34+ HHT monocytes are more likely to differentiate into macrophages in the angiogenic niche. Together with previous findings [17,24,29], these data suggest that deletion of one allele of Eng or perhaps Alk1 is sufficient to alter macrophage function, and that the abnormal macrophage function contributes to the accumulation of macrophages in bAVM.
Although Eng deletion in macrophages alone is insufficient for bAVM development [13], the macrophage burden is increased in bAVM lesions in both Eng- and Alk1-deficient mouse models [11,13]. Combined with clinical evidence that macrophages are present in human bAVM specimens [2-4], these data suggest that macrophages play an active role in bAVM pathogenesis.
We previously reported that in WT mice, the infiltration of BM-derived macrophages in the angiogenic focus begins at week 1, peaks at week 2, decreases thereafter and returns to normal level at 6-8 weeks [30]. Systemic deletion of Eng leads to a temporal difference in macrophage responses. Eng+/− mice have fewer CD68+ cells in the peri-infarct area at 3 days but more at 60 days after permanent occlusion of a distal middle cerebral artery [29]. Similarly, compared to WT mice, Eng-deficient mice (Model 1) had fewer macrophages in the brain angiogenic region at the early stage of angiogenesis. The number of macrophages remained stable with a slight increase thereafter, resulting in unresolved inflammation. Using a brain focal Alk1-deficient model (Model 2), we showed that both microglia and BM-derived macrophages were present in the lesion. Since the mice in the 8-week-old groups of Model 1 and Model 2 were intravascularly perfused with heparinized PBS before sample collection, the macrophages detected in the bAVM lesions in these mice were either on the vessel wall or in the brain parenchyma.
This study could not rule out the possibility that macrophage infiltration was due to the response to the increased permeability and hemorrhage of the dysplastic AVM vessels [2,11]. In contrast to the WT brain, where the number of macrophages decreased between 2 and 4 weeks after angiogenic stimulation, the number of macrophages was unchanged or increased slightly during this period in the Eng-deficient brain, suggesting that the accumulation of macrophages in the brain angiogenic region started between 2 and 4 weeks after angiogenic stimulation, at the time when bAVM had not formed [10]. In addition, we could not determine if the homing ability of Eng-deficient monocytes to angiogenic/injury tissue is impaired or just altered. However, Post et al. have reported that PBMNCs from HHT1 patients (ENG deficiency) have a reduced ability to home to the infarcted mouse myocardium, and also demonstrated that the decreased homing of HHT1-MNCs is caused by the cells’ impaired ability to respond to SDF-1α [15]. Therefore, the delayed and persistent infiltration of monocytes in the Eng-deficient mouse brain is very likely due to the impairment of their homing ability. However, future studies are needed to determine whether their homing ability toward other chemotactic agents, such as VEGF, is also impaired.
Excessively high levels of VEGF are known to cause inflammation. Therefore, we injected the same dose of AAV-VEGF into the brain of WT mice as control. The CD68+ cell loads in the brain angiogenic foci of R26CreER/+;Eng2f/2f and WT mice were compared in parallel (Fig. 3 & 4). The CD68+ cells in the angiogenic foci of WT mice (345±79/mm2) were significantly fewer than those in the AVM lesion (544±168/mm2, P=0.01) 8 weeks after the vector injection.
Macrophages were also found in human bAVM specimens without iron deposits [5,31]. Therefore, macrophage accumulation cannot simply be explained as a response to hemorrhage in bAVM pathogenesis. The unresolved inflammation caused by macrophage accumulation can enhance abnormal vascular remodeling [32] leading to the instability of bAVM vessels. We would like to caution that the etiologies of sporadic bAVM and HHT bAVM may not be exactly the same. The data we obtained from studying HHT bAVM mouse models cannot be applied directly to sporadic bAVM. Further studies are needed to verify if macrophages play the same roles in sporadic bAVM and HHT bAVM.
It should be noted that the GFP-labeled Cx3cr1-positive cells not only represent active microglia but also inactive resident microglia. Hence, the increase in active microglia in the bAVM region could be underestimated by simply quantifying the number of GFP+ cells. It has also been reported that BM-derived macrophages can trans-differentiate into microglia under disease conditions [26], and that the monocytes may also express a low level of Cx3cr1 [33]. Therefore, a portion of microglia we detected in Alk1-deficient brain samples could have actually originated from BM-derived macrophages.
We tried to evaluate the role of macrophages by depleting them with clodronate treatment. This was tested successfully in a stroke-plus-tibia-fracture mouse model using one injection of clodronate [34]. However, since our bAVM model took 8 weeks to develop, multiple doses of clodronate were needed to inhibit bAVM formation, consequently causing high mortality. New agents that can deplete microglia or block myeloid-derived macrophage infiltration are currently being tested.
The percentage of CD34+ cells in the peripheral blood mononuclear cells (PBMNCs) from HHT patients was significantly higher than those from healthy controls. There were no differences in the fraction of CD133+ or CD34+/VEGFR2+ cells between HHT and control PBMNCs. However, the circulating angiogenic cells (CAC) derived from HHT patients not only showed a significant reduction in EC-selective surface markers following a 7-day culture, but also a significant increase in the rate of apoptosis and blunted migration in response to VEGF and SDF-1 [22]. We showed that HHT CD34+ peripheral blood cells are more likely to differentiate into macrophages than EC in an angiogenic niche compared to normal controls. It is not surprising that more CD34+ cells isolated from both normal controls and HHT patients differentiated into macrophage-like cells, because only small portions of CD34+ monocytes are endothelial progenitor cells or angiogenic precursor cells. Majority of them will adopt the macrophage phenotype once they enter tissues. Here we showed that compared to normal CD34+ cells, fewer HHT CD34+ cells were able to become ECs. It could have been due to the reduced ability of HHT CD34+ cells to differentiate into the EC phenotype in the angiogenic niche, or perhaps there were fewer endothelial progenitor cells or CACs in HHT CD34+ cells than in normal CD34+ cells. Although reduced expression of ENG has been confirmed in HHT1 and HHT2 CD34+ cells and reduced expression of ALK1 in HHT2 CD34+ cells, future studies are needed to understand how the reduction of ENG or ALK1 expressions alters monocyte EC differentiation. However, the vascular niches composed of HUVEC and HASMC cannot completely resemble the vascular niche in the mouse brain. Our ultimate goal is to uncover the pathogenesis of human bAVM. We will determine in a future research study if the data can be reproduced in vascular niches composed of human brain microvascular ECs and human brain SMCs.
In summary, using two mouse models and human CD34+ cells isolated from peripheral blood of HHT patients, our data suggest that persistent macrophage infiltration and the pro-inflammation phenotype of HHT mononuclear cells drive unresolved inflammation in bAVM and lesion progression. We will further investigate and analyze if blocking macrophage infiltration can reduce abnormal vascular remodeling and lesion progression in bAVM.
Supplementary Material
Acknowledgements
This study was supported by grants to H. Su from the National Institutes of Health (R01 NS027713, R01 HL122774 and R21 NS083788), and from the Michael Ryan Zodda Foundation and the UCSF Research Evaluation and Allocation Committee (REAC). We thank members of the UCSF BAVM Study Project (http://avm.ucsf.edu) for their support, and Voltaire Gungab for assistance with manuscript preparation.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution at which the studies were conducted.
References
- 1.Kim H, Su H, Weinsheimer S, Pawlikowska L, Young WL. Brain arteriovenous malformation pathogenesis: a response-to-injury paradigm. Acta Neurochir Suppl. 2011;111:83–92. doi: 10.1007/978-3-7091-0693-8_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Y, Zhu W, Bollen AW, Lawton MT, Barbaro NM, Dowd CF, Hashimoto T, Yang GY, Young WL. Evidence of inflammatory cell involvement in brain arteriovenous malformations. Neurosurgery. 2008;62:1340–1349. doi: 10.1227/01.neu.0000333306.64683.b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y, Pawlikowska L, Yao JS, Shen F, Zhai W, Achrol AS, Lawton MT, Kwok PY, Yang GY, Young WL. Interleukin-6 involvement in brain arteriovenous malformations. Ann Neurol. 2006;59:72–80. doi: 10.1002/ana.20697. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y, Fan Y, Poon KY, Achrol AS, Lawton MT, Zhu Y, McCulloch CE, Hashimoto T, Lee C, Barbaro NM, Bollen AW, Yang GY, Young WL. MMP-9 expression is associated with leukocytic but not endothelial markers in brain arteriovenous malformations. Front Biosci. 2006;11:3121–3128. doi: 10.2741/2037. [DOI] [PubMed] [Google Scholar]
- 5.Guo Y, Tihan T, Lawton MT, Kim H, Young WL, Zhao Y, Su H. Distinctive distribution of lymphocytes in unruptured and previously untreated brain arterviovenous malformation. Neuroimmunol Neuroinflamm. 2014;1:147–152. doi: 10.4103/2347-8659.143674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma L, Guo Y, Zhao YL, Su H. The role of macrophage in the pathogenesis of brain arteriovenous malformation. Int J Hematol Res. 2015;1:52–56. doi: 10.17554/j.issn.2409-3548.2015.01.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Achrol AS, Pawlikowska L, McCulloch CE, Poon KY, Ha C, Zaroff JG, Johnston SC, Lee C, Lawton MT, Sidney S, Marchuk D, Kwok PY, Young WL. Tumor necrosis factor-alpha-238G>A promoter polymorphism is associated with increased risk of new hemorrhage in the natural course of patients with brain arteriovenous malformations. Stroke. 2006;37:231–234. doi: 10.1161/01.STR.0000195133.98378.4b. [DOI] [PubMed] [Google Scholar]
- 8.Pawlikowska L, Tran MN, Achrol AS, McCulloch CE, Ha C, Lind DL, Hashimoto T, Zaroff J, Lawton MT, Marchuk DA, Kwok PY, Young WL. Polymorphisms in genes involved in inflammatory and angiogenic pathways and the risk of hemorrhagic presentation of brain arteriovenous malformations. Stroke. 2004;35:2294–2300. doi: 10.1161/01.STR.0000141932.44613.b1. [DOI] [PubMed] [Google Scholar]
- 9.Hashimoto T, Lawton MT, Wen G, Yang GY, Chaly T, Jr., Stewart CL, Dressman HK, Barbaro NM, Marchuk DA, Young WL. Gene microarray analysis of human brain arteriovenous malformations. Neurosurgery. 2004;54:410–423. doi: 10.1227/01.neu.0000103421.35266.71. discussion 423-425. [DOI] [PubMed] [Google Scholar]
- 10.Walker EJ, Su H, Shen F, Choi EJ, Oh SP, Chen G, Lawton MT, Kim H, Chen Y, Chen W, Young WL. Arteriovenous malformation in the adult mouse brain resembling the human disease. Ann Neurol. 2011;69:954–962. doi: 10.1002/ana.22348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W, Guo Y, Walker EJ, Shen F, Jun K, Oh SP, Degos V, Lawton MT, Tihan T, Davalos D, Akassoglou K, Nelson J, Pile-Spellman J, Su H, Young WL. Reduced mural cell coverage and impaired vessel integrity after angiogenic stimulation in the Alk1-deficient brain. Arterioscler Thromb Vasc Biol. 2013;33:305–310. doi: 10.1161/ATVBAHA.112.300485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen W, Sun Z, Han Z, Jun K, Camus M, Wankhede M, Mao L, Arnold T, Young WL, Su H. De novo cerebrovascular malformation in the adult mouse after endothelial Alk1 deletion and angiogenic stimulation. Stroke. 2014;45:900–902. doi: 10.1161/STROKEAHA.113.003655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi EJ, Chen W, Jun K, Arthur HM, Young WL, Su H. Novel brain arteriovenous malformation mouse models for type 1 hereditary hemorrhagic telangiectasia. PLoS One. 2014;9:e88511. doi: 10.1371/journal.pone.0088511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bharatha A, Faughnan ME, Kim H, Pourmohamad T, Krings T, Bayrak-Toydemir P, Pawlikowska L, McCulloch CE, Lawton MT, Dowd CF, Young WL, Terbrugge KG. Brain arteriovenous malformation multiplicity predicts the diagnosis of hereditary hemorrhagic telangiectasia: quantitative assessment. Stroke. 2012;43:72–78. doi: 10.1161/STROKEAHA.111.629865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Post S, Smits AM, van den Broek AJ, Sluijter JP, Hoefer IE, Janssen BJ, Snijder RJ, Mager JJ, Pasterkamp G, Mummery CL, Doevendans PA, Goumans MJ. Impaired recruitment of HHT-1 mononuclear cells to the ischaemic heart is due to an altered CXCR4/CD26 balance. Cardiovasc Res. 2010;85:494–502. doi: 10.1093/cvr/cvp313. [DOI] [PubMed] [Google Scholar]
- 16.Dingenouts CK, Goumans MJ, Bakker W. Mononuclear cells and vascular repair in HHT. Front Genet. 2015;6:114. doi: 10.3389/fgene.2015.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Laake LW, van den Driesche S, Post S, Feijen A, Jansen MA, Driessens MH, Mager JJ, Snijder RJ, Westermann CJ, Doevendans PA, van Echteld CJ, ten Dijke P, Arthur HM, Goumans MJ, Lebrin F, Mummery CL. Endoglin has a crucial role in blood cell-mediated vascular repair. Circulation. 2006;114:2288–2297. doi: 10.1161/CIRCULATIONAHA.106.639161. [DOI] [PubMed] [Google Scholar]
- 18.Rossi E, Sanz-Rodriguez F, Eleno N, Duwell A, Blanco FJ, Langa C, Botella LM, Cabanas C, Lopez-Novoa JM, Bernabeu C. Endothelial endoglin is involved in inflammation: role in leukocyte adhesion and transmigration. Blood. 2013;121:403–415. doi: 10.1182/blood-2012-06-435347. [DOI] [PubMed] [Google Scholar]
- 19.Young K, Conley B, Romero D, Tweedie E, O'Neill C, Pinz I, Brogan L, Lindner V, Liaw L, Vary CP. BMP9 regulates endoglin-dependent chemokine responses in endothelial cells. Blood. 2012;120:4263–4273. doi: 10.1182/blood-2012-07-440784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi EJ, Walker EJ, Jun K, Kuo R, Su H, Young WL. Endoglin deficiency in bone marrow is sufficient to cause vascular dysplasia in the adult mouse brain after VEGF stimulation [Abstract]. Stroke. 2012;43:A3153. doi: 10.1161/STROKEAHA.112.671974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen F, Degos V, Han Z, Choi EJ, Young WL, Su H. Endoglin deficiency exacerbates ischemic brain injury [Abstract]. Stroke. 2013;44:ATMP69. [Google Scholar]
- 22.Zucco L, Zhang Q, Kuliszewski MA, Kandic I, Faughnan ME, Stewart DJ, Kutryk MJ. Circulating angiogenic cell dysfunction in patients with hereditary hemorrhagic telangiectasia. PLoS One. 2014;9:e89927. doi: 10.1371/journal.pone.0089927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allinson KR, Carvalho RL, van den Brink S, Mummery CL, Arthur HM. Generation of a floxed allele of the mouse Endoglin gene. Genesis. 2007;45:391–395. doi: 10.1002/dvg.20284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi EJ, Walker EJ, Degos V, Jun K, Kuo R, Su H, Young WL. Endoglin deficiency in bone marrow is sufficient to cause cerebrovascular dysplasia in the adult mouse after vascular endothelial growth factor stimulation. Stroke. 2013;44:795–798. doi: 10.1161/STROKEAHA.112.671974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, Park A, Wu X, Kaartinen V, Roman BL, Oh SP. ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2 (HHT2). Blood. 2008;111:633–642. doi: 10.1182/blood-2007-08-107359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, Charo IF. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One. 2010;5:e13693. doi: 10.1371/journal.pone.0013693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen W, Young WL, Su H. Induction of brain arteriovenous malformation in the adult mouse. Methods Mol Biol. 2014;1135:309–316. doi: 10.1007/978-1-4939-0320-7_25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shih YT, Wang MC, Yang TL, Zhou J, Lee DY, Lee PL, Yet SF, Chiu JJ. beta2-Integrin and Notch-1 differentially regulate CD34+CD31+ cell plasticity in vascular niches. Cardiovas Res. 2012;96:296–307. doi: 10.1093/cvr/cvs256. [DOI] [PubMed] [Google Scholar]
- 29.Shen F, Degos V, Chu PL, Han Z, Westbroek EM, Choi EJ, Marchuk D, Kim H, Lawton MT, Maze M, Young WL, Su H. Endoglin deficiency impairs stroke recovery. Stroke. 2014;45:2101–2106. doi: 10.1161/STROKEAHA.114.005115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao Q, Liu J, Pappu R, Su H, Rola R, Gabriel RA, Lee CZ, Young WL, Yang GY. Contribution of bone marrow-derived cells associated with brain angiogenesis is primarily through leucocytes and macrophages. Arterioscler Thromb Vasc Biol. 2008;28:2151–2157. doi: 10.1161/ATVBAHA.108.176297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo Y, Saunders T, Su H, Kim H, Akkoc D, Saloner DA, Hetts SW, Hess C, Lawton MT, Bollen AW, Pourmohamad T, McCulloch CE, Tihan T, Young WL. Silent intralesional microhemorrhage as a risk factor for brain arteriovenous malformation rupture. Stroke. 2012;43:1240–1246. doi: 10.1161/STROKEAHA.111.647263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aihara K, Mogi M, Shibata R, Bishop-Bailey D, Reilly MP. Inflammation and vascular remodeling. Int J Vasc Med. 2012;2012:596796. doi: 10.1155/2012/596796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 34.Degos V, Maze M, Vacas S, Hirsch J, Guo Y, Shen F, Jun K, van Rooijen N, Gressens P, Young WL, Su H. Bone fracture exacerbates murine ischemic cerebral injury. Anesthesiology. 2013;118:1362–1372. doi: 10.1097/ALN.0b013e31828c23f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.