

Abstract
Streptococcus pneumoniae (Sp) strain TIGR4 is a virulent, encapsulated serotype that causes bacteremia, otitis media, meningitis and pneumonia. Increased bacterial resistance and limited efficacy of the available vaccine to some serotypes complicate the treatment of diseases associated to this microorganism. Flavodoxins are bacterial proteins involved in several important metabolic pathways. The Sp flavodoxin (Spfld) gene was recently reported to be essential for the establishment of meningitis in a rat model, which makes SpFld a potential drug target. To facilitate future pharmacological studies, we have cloned and expressed SpFld in E. coli and we have performed an extensive structural and biochemical characterization of both the apo form and its active complex with the FMN cofactor. SpFld is a short-chain flavodoxin containing 146 residues. Unlike the well-characterized long-chain apoflavodoxins, the Sp apoprotein displays a simple two-state thermal unfolding equilibrium and binds FMN with moderate affinity. The X-ray structures of the apo and holo forms of SpFld differ at the FMN binding site, where substantial rearrangement of residues at the 91–100 loop occurs to permit cofactor binding. This work will set up the basis for future studies aiming at discovering new potential drugs to treat S. pneumoniae diseases through the inhibition of SpFld.
Introduction
Streptococcus pneumoniae is a gram-positive bacterium that causes pneumonia, meningitis, otitis media, acute sinusitis, septicemia, and conjunctivitis, among several other diseases [1–4]. The World Health Organization (WHO) considers Streptococcus pneumoniae a major health problem due to the elevated mortality of some of the diseases associated to this pathogen. It is estimated that close to 500,000 children under 5 years died in 2008 as a consequence of pneumococcal infections [5]. More specifically, pneumococcal meningitis causes death in around 25% of cases, plus neurological diseases in half of the survivors [6]. The emerging resistance of S. pneumoniae to established antibiotics [7], the fact that current polysaccharide-related vaccines are only protective to certain infective serotypes, and the fluctuation of the prevalence of some pneumococcal serotypes over time due to selection forces (i.e.: 7vPCV vaccine) or serotype replacement [8] advice for the identification of new S. pneumoniae targets, common to the different serotypes, that could be used to develop novel antibiotics to specifically combat S. pneumoniae infections.
In a recent work [9], several Sp TIGR4 genes [10] were identified as essential for S. pneumoniae infection during experimental meningitis in rats. One of them is the gene encoding flavodoxin (Spfld), a protein that has been shown to be essential for several other bacteria (http://tubic.tju.edu.cn/deg/) [11], including Escherichia coli [12, 13], Salmonella enterica [14], Haemophilus influenzae [15], Vibrio cholerae [16], and Helicobacter pylori [17]. Flavodoxins are electron transfer proteins that participate in a variety of reactions in prokaryotes and in some unicellular algae [18]. In Azobacter vinelandii, Fld participates in the reduction of nitrate [19], in E. coli it mediates the activation of different enzymes such as anaerobic ribonucleotide reductase [20], cobalamin-dependent methionine synthase [21], biotin synthase [22] and pyruvate formate-lyase [23]. In some organisms, such as S. pneumoniae, the importance of flavodoxin has been tentatively associated to its participation in methionine synthesis [9]. The identification of flavodoxin as an essential protein for S. pneumoniae infectivity and the fact that flavodoxin is not present in humans open the possibility of using it as a target for the development of novel and specific antimicrobials against S. pneumoniae. Related to this potential use of flavodoxins as drug targets [24] the flavodoxin from Helicobacter pylori (HpFld) has been subjected to detailed functional [25], structural [26] and biophysical [27–30] analyses that have facilitated the discovery of novel specific inhibitors with bactericidal properties [31, 32]. In contrast to this wealth of information on HpFld, very little is known of SpFld. According to its gene sequence translation [9], it contains 147 residues and very likely belongs to the so-called short-chain flavodoxins [18] characterized by the lack of a 20-residue loop that, in the long-chain flavodoxins, appears intercalated in the fifth β-strand. Hence, SpFld shows a higher sequence identity with other short-chain flavodoxins, such as Streptococcus pyrogenes (76%) or Desulfovibrio desulfuricans (45,3%), than with long-chain flavodoxins, such as those from Helicobacter pylori (25,7%), Anabaena PCC7119 (22,4%) or Escherichia coli (25,7%). The more detailed flavodoxin molecular and structural studies have been devoted to a few long-chain flavodoxins, especially those from Anabaena PCC7119 [33–35], Azobobacter vinelandii [36] and Helicobacter pylori [17, 28]. The lack of information about SpFld can hardly be alleviated by resourcing to the limited data available for other short-chain flavodoxins. In this work we provide a detailed biophysical characterization of SpFld and report the x-ray structures of the apo and holo forms, the conformational stability of the protein and the thermodynamic characterization of the cofactor binding equilibrium. These studies provide the target characterization needed to pursue the rational discovery of specific inhibitors against SpFld. In addition, they will contribute to understand better the specific structural and thermodynamic characteristics of short-chain flavodoxins.
Results and Discussion
Spectroscopic properties of the holo and apo SpFld forms
The UV-visible absorption spectrum of purified SpFld (Fig 1a) shows the typical hallmarks of flavin binding proteins, with two sizable absorption peaks at 371 and 456 nm, a pronounced shoulder at 480 nm, and no absorption beyond 550 nm, indicative of a completely oxidized flavin cofactor. The very low 271 nm/456 nm absorbance ratio of 3.3 is consistent with the absence of tryptophan residues and the presence of only five tyrosine residues in its sequence. The UV-visible spectrum of apo SpFld shows a single peak with maximal absorbance at 277 nm (Fig 1a) and a completely flat spectrum beyond 350 nm, indicative of both a full removal of the cofactor and a lack of protein aggregation phenomena. The apoflavodoxin molar extinction coefficient at 277 nm in 150 mM NaCl, 100 mM MES, pH 6.5, determined as described [37], was 7.2 mM-1cm-1. The extinction coefficient at 456 nm (λmax) of the fully oxidized holoprotein in the same buffer, determined as described [38], was 9.1 mM-1cm-1. To observe SpFld reduced species, a partial FMN reduction in pseudo-anaerobic conditions was done with sodium dithionite (Fig 1b). The stabilized semiquinone exhibits a maximum at 592 nm. The low accumulation of semiquinone observed suggests (assuming a typical FMN semiquinone extinction coefficient around 5.0 mM-1 cm-1 at that wavelength) that the Eox/sq redox potential could be slightly more negative than the Esq/hq one. Most of the flavin signal (95% at λmax) is lost in the reduced form. The fluorescence emission spectra of holo SpFld (Fig 1c) displays characteristic peaks of tyrosine residues and of the FMN cofactor. On the other hand, the apoprotein shows a single maximum of emission at 310 nm, as expected from its amino acid composition.
Fig 1. S. pneumoniae flavodoxin absorption and fluorescence spectra.
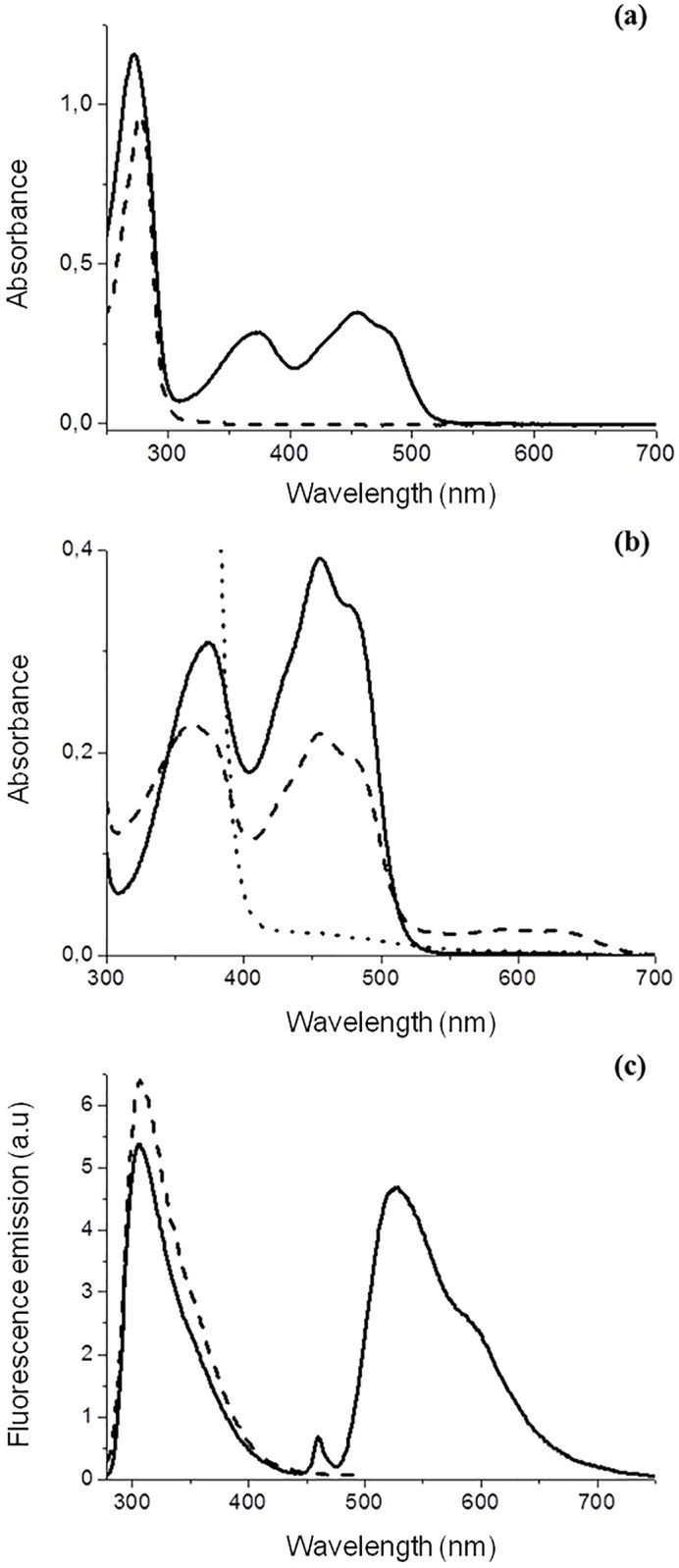
(a) UV/VIS absorbance of 39 μM holo (solid line) and 134 μM apoflavodoxin (dashed line) in 150 mM NaCl, 100 mM MES, pH 6.5. (b) Spectra related to three redox states of 43 μM SpFld in 150 mM NaCl, 100 mM MES, pH 6.5 at 10°C: Oxidized (solid line), semiquinone (dashed line) with absorption maxima at 592 nm and reduced (dotted line). (c) Fluorescence emission spectra of 20 μM SpFld holoprotein (solid line) and 20 μM apoprotein (dashed line), in 150 mM NaCl, 100 mM MES, pH 6.5, 10°C.
The far-UV CD spectra of the holo and apoproteins (Fig 2a) indicate that, in solution, the secondary structure content of the apo form is lower than that of the holoprotein. In contrast, in the crystal structures (see below) the two forms appear to display very similar secondary structure contents. The near-UV CD spectra of apo SpFld (Fig 2b) is characterized by a single negative peak at 280 nm that must arise from the tyrosine residues in the protein. The presence of a peak in this spectral region, which is characteristic of well-folded proteins [39], indicates that, in solution, at least one tyrosine residue is located in a structurally well-defined region. On the other hand, the near-UV CD spectrum of holoprotein (Fig 2b and 2c) is more complex and reveals several contributions of the FMN cofactor that also arise as a consequence of its interaction with a well-structured protein moiety [40]. The FMN-protein interaction gives rise to prominent peaks in the visible region at the wavelengths of maximal absorbance of the FMN cofactor. The CD data together suggests that both the apo and holo forms display well defined tertiary structures in solution, but suggests that the apo form overall secondary structure content may be lower than that observed in the crystal structure (see below). The near-UV CD spectra and the fluorescence emission spectra of apo SpFld are different from those of Anabaena and Hp flavodoxins, which are dominated by positive peaks in the near-UV CD and by longer fluorescence emission wavelengths characteristic of tryptophan containing proteins. SpFld thus displays a specific spectroscopic identity that allows it to be distinguished from the aforementioned long-chain flavodoxins based on simple fluorescence and CD spectra.
Fig 2. CD spectra of holo and apo SpFld in 150 mM NaCl, 100 mM MES, pH 6.5, 4°C.
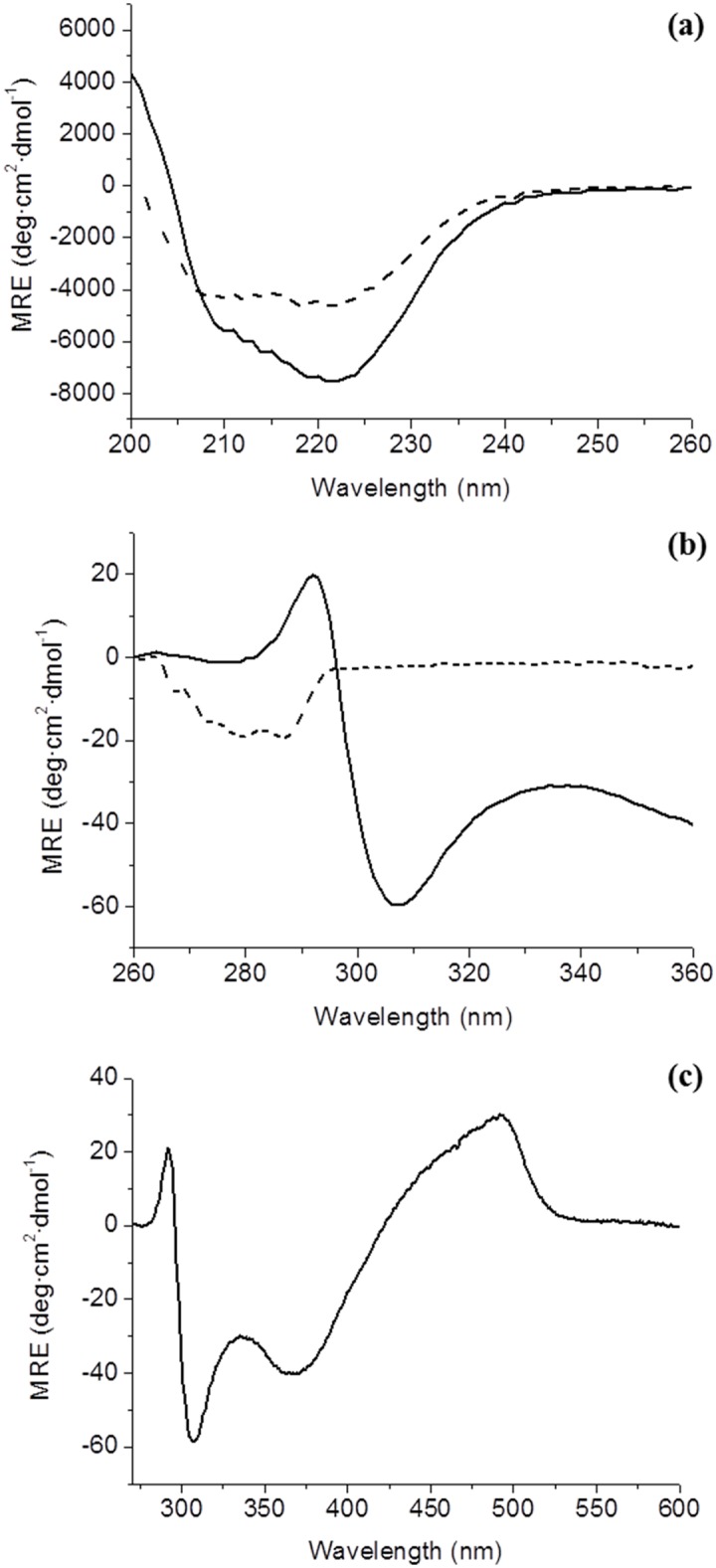
(a) Far-CD spectra of 80 μM of holo and apo SpFld (continuous and dashed lines respectively) (b) Near-CD spectra of 80 μM of holo (continuous line) and apo (dashed line) SpFld. (c) Visible-CD spectrum of 80 μM of holoprotein.
SpFld thermal stability
The stability of holo and apo SpFld has been determined from thermal unfolding curves. The far UV-CD and near-UV CD thermal unfolding curves of the apo SpFld, roughly normalized to signal spans from approximately 0 to 1, are shown in Fig 3a. Their fair superposition suggests that the apoprotein follows a two-state thermal unfolding mechanism. The individual fits of the two curves to two-state equations (eqs 1 and 2) give similar temperatures of mid denaturation (around 30°C) and similar enthalpy changes (around 50 kcal mol-1). Their global analysis confirms the two-state model and provides a Tm of 30.8 ± 2°C and an enthalpy change of 41 ± 5 kcal mol-1 (Table 1).
Fig 3. Thermal unfolding curves monitored spectroscopically in 150 mM NaCl, 100 mM MES pH 6.5.

(a) (80 μM) holo SpFld by Fluorescence (open triangles, gray line), far-UV CD (black circles, black line) and near-UV CD (open squares, gray line). (b) (80 μM) apo SpFld by far-UV CD (black circles, black line) and near-UV CD (open squares, gray line).
Table 1. Thermal stability parameters for holo and apo SpFld.
| Holo Flavodoxina | Apo Flavodoxina | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Far-UV CD |
Near-UV CD |
Fluoresc. | Global fitb | DSC | Far-UV CD |
Near-UV CD |
Global fitb | DSC | |
| Tm | 44.7±0.4 | 43.7±1 | 44.5±0.3 | 44.6±0.2 | 46.7±0.5 | 30.3±0.1 | 30.4±0.8 | 30.8±2 | 34.2±0.5 |
| (°C) | |||||||||
| ΔHm | 52±2 | 38±3 | 65.8±0.1 | 51±1 | 61±2 | 51±1 | 50.5±0.6 | 41±5 | 55±2 |
| (kcal mol-1) | |||||||||
a In 150 mM NaCl, 100 mM MES, pH 6.5.
b Parameters obtained through global analysis of the spectroscopic curves using a two-state unfolding model.
The thermal unfolding of holo SpFld was monitored by far-UV CD, near-UV CD and FMN fluorescence. The unfolding curves, roughly normalized to signal spans from zero to one (Fig 3b), can also be superimposed, suggesting a simple two-state unfolding equilibrium for the holoprotein [41]. Their individual fitting to a two-state equation provide Tm values around 44.5°C and enthalpy changes around 50 kcal mol-1. The global analysis of the three curves gave rise to a Tm of 44.6 ± 0.2°C and an enthalpy change of 51±1 kcal mol-1 (Table 1), similar to the values obtained in the individual fits. The fact that the Tm of the holoflavodoxin is higher than that of the apoprotein is the expected behavior for any protein whose native conformation binds a ligand, in this case the FMN cofactor, as it has been discussed before in the context of the apoflavodoxin/FMN equilibrium in other species [28, 30, 42].
The thermal unfolding of the holo and apo SpFld has also been investigated by DSC (Fig 4). Analysis of the thermograms of either form of the protein confirms that the unfolding equilibrium is two-state with only one cooperative transition. The calorimetric Tm and ΔH values are similar to those derived from the spectroscopic analysis (Table 1) and confirm that the holoprotein is more stable than the apoprotein.
Fig 4. DSC analysis in the absence of FMN (open squares) or in the presence of 40 μM of FMN (open circles) in 150 mM NaCl, 100 mM MES, pH 6.5.

The continuous line is the non-linear fitting curve obtained by using a two-state model for the unfolding of apoFld.
Although little is known about the stability of short-chain flavodoxins, the stability of long-chain ones has been studied in detail [18], specially using Anabaena PCC 7119 flavodoxin, as a model. In long-chain flavodoxins, the thermal unfolding of the apoform is at least three-state (four-state in H. pylori [28]) because a partly unfolded intermediate conformation accumulates as the temperature is raised before the protein becomes fully unfolded. The structure of this major thermal intermediate has been determined at atomic resolution [43]. Roughly, two-thirds of it displays a native conformation while the other third, encompassing the long loop of long-chain flavodoxins and neighboring residues, is unfolded. In addition, protein engineering experiments have shown that removal of the long loop gives rise to a shortened well-folded Anabaena apoflavodoxin, which behaves as two-state towards thermal unfolding [44]. Therefore, it is well established that the equilibrium thermal intermediate of long-chain flavodoxins is associated to the autonomous unfolding of the long loop. Our spectroscopic and calorimetric analysis of the short-chain Sp apoflavodoxin thermal unfolding indicates that no thermal intermediate accumulates, which suggests that short-chain apoflavodoxins may generally follow a simple two-state equilibrium unfolding mechanism.
Unlike in previous studies in our group on the stability of other flavodoxins, we have used here a slightly acidic buffer and a higher ionic strength because, otherwise, the conformational stability of Sp apoflavodoxin is significantly lower, and it is difficult to determine accurately. In fact, at the conditions typically used to characterize Anabaena or Hp apoflavodoxins (i.e. pH 7 and around 20 mM ionic strength), Sp apoflavodoxin appears to be unfolded (not shown). This lower stability is unlikely related to the absence of a long loop in the short-chain SpFld. Our previous analysis of the influence of the Anabaena PCC 7119 long loop in the conformational stability of that long-chain flavodoxin [44] showed that removal of the long loop hardly modified the conformational stability of the protein. The low stability of SpFld in low ionic strength conditions is more likely related to its peculiar amino acid composition. In fact, the progressively decreasing conformational stability of the H. pylori, Anabaena PCC 7119 and Sp apoflavodoxins at neutral pH and low ionic strength correlates with their increasing relative content of acidic residues (27/164, 31/169 and 38/148, respectively) that gives rise to an increasingly large excess of negative charge at pH 7 (approximately -12, -17 and -28, respectively). This interpretation is in agreement with the fact that, in the Anabaena protein, more stable variants have been engineered by reducing the net negative charge of the apoprotein [45, 46].
SpFld interaction with FMN
The higher thermostability of Sp holoflavodoxin compared to that of the apo form is a consequence of the favorable interactions established between the apoprotein and the cofactor. To quantitate this effect, the interaction between the apoprotein and the FMN cofactor was studied by isothermal titration calorimetry at 15, 20 and 25°C (Fig 5 and Table 2). In addition, buffer-independent binding parameters were determined by using buffers with different ionization enthalpies (Table 2). As observed for other flavodoxins, FMN binding is markedly exothermic. The overall binding process is enthalpically driven, with a small unfavorable entropic contribution. This suggests a considerable conformational entropy loss that is not compensated by the desolvation entropy. A large part of the hydrophobic FMN moiety (isoalloxazine ring) does not become buried inside the binding site; moreover, the polar region of FMN strongly interacts with amino acid residues in the active site and becomes completely buried upon binding. The dissociation constant at 25°C is 83 nM, not far from that of Helicobacter pylori flavodoxin (27 nM), but two orders of magnitude higher than that of Anabaena flavodoxin (0.26 nM). As previously observed for other flavodoxins, there is no significant exchange of protons coupled to FMN binding (Table 2). The binding data obtained at the three temperatures indicated allows estimating a binding heat capacity change of -0.5 kcal K-1·mol-1, similar to that of other flavodoxins.
Fig 5. Titration of 20 μM Apo SpFld with 300 μM FMN at 25°C in 150 mM NaCl, 100 mM MES, pH 6.5.

The upper panel shows the thermogram (thermal power as function of time) and the lower panel shows the binding isotherm (normalized heat as a function of the molar ratio). The continuous line is the non-linear fitting curve obtained using a model considering a single ligand binding site. The inset in the upper panel shows a comparison of the thermodynamic binding profiles obtained by ITC for FMN interacting with apoflavodoxins from S. pneumoniae, Helicobacter pylori, and Anabaena PCC7119: ΔG (black bars) ΔH (gray bars) -TΔS (white bars). Data for the H. pylori and Anabaena complexes come from reference [28]
Table 2. Apoflavodoxin/FMN binding energetics.
X-ray structures of the apo and holo forms of SpFld
To understand the molecular basis of the interaction of the FMN cofactor with the protein moiety and to provide a structural frame for the identification and characterization of SpFld inhibitors, we have solved the crystal structures of the SpFld in the apo form (Fig 6a) and in complex with FMN (Fig 6b and S1 Fig), at 2.18 and 1.69 Å, respectively (Table 3). Either structure shows the archetypical α/β sandwich flavodoxin fold (5 α-helices that pack against a five-stranded β-sheet with a 21345 topology, forming an α/β/α sandwich). It has been reported that some short-chain flavodoxins contain a bulge in the fifth beta strand that has been interpreted as reminiscent of an evolutionary event where the long loop was lost from long-chain flavodoxins to give rise to short-chain flavodoxins [44]. This unique bulge is also found in the fifth beta strand of Sp flavodoxin, unlike in any of the other four strands (Fig 7). Overall, the holo (note that the holo form contains selenomethionine (SeMet) residues) and apo SpFld structures are similar with an r.m.s.d of 0.8 Å. The main differences between the two structures are traced to two of the three FMN binding loops encompassing residues 10–14 (phosphate loop), 57–66 (50’ loop) and 92–100 (90’ loop), the latter two sandwiching the isoalloxazine moiety. A further noticeable difference is the presence of a small 310 helix stretch at residues 38–40 in the holoprotein that is absent in the apo form (Fig 6c).
Fig 6.

Ribbon structure representation of apo SpFl in blue (a) and holo SpFld in cyan (b). Changes in the key FMN binding loops indicated in (b) can be observed in the structural superimposition of the holo (cyan) and apo (blue) flavodoxins shown in (c). Also in (c), a red arrow points to a short 310 helix formed in the holoprotein. The FMN-SpFld hydrogen bonding interactions are highlighted in (d).
Table 3. Data collection and refinement statistics of holo and apo SpFld crystallographic structures.
| SpFld-apo | SpFld-FMN | |
|---|---|---|
| (SeMet form) | ||
| Space group | P212121 | H3 |
| Wavelength (Å) | 0.92 | 0.976 |
| Resolution (Å) | 20–1.60 | 37.00–2.07 |
| (1.69–1.60)a | (2.18–2.07) | |
| N. of molecules in Asymmetric unit | 1 | 2 |
| Cell dimensions (Å) | a = 29.87 | a = 101.68 |
| b = 60.71 | b = 101.68 | |
| c = 61.80 | c = 68.28 | |
| Unique reflections | 15310 | 15961 |
| Completeness | 99.2 (99.5) | 99.2 (99.6) |
| Rsym | 0.102 (0.703) | 0.094 (0.421) |
| I/σ(I) | 13.9 (2.7) | 18.9 (6.7) |
| Mn(I) half-set correlation CC(1/2) | 0.998 (0.810) | 0.999 (0.934) |
| Redundancy | 9.9 (9.6) | 16.0 (16.1) |
| Alpha_twin | 0.478 | |
| Rwork/Rfree | 0.204/0.228 | 0.187/0.228 |
| RMSD from ideal geometry, bonds (Å) | 0.007 | 0.017 |
| RMSD from ideal geometry, angles (°) | 1.448 | 2.047 |
| <B> overall (Å2) | 15.32 | 39.22 |
| <B> FMN (Å2) | - | 32.90 |
| <B> solvent (Å2) | 36.28 | 36.58 |
| <B> phosphate (Å2) | 11.97 | - |
| Ramachandran plot: | ||
| Most favoured (%) | 97.90 | 96.49 |
| Additionally allowed (%) | 2.10 | 3.51 |
| Generously allowed (%) |
a Values in parentheses refer to the highest resolution shell. Ramachandran plot statistics were determined with PROCHECK, ref. [63].
Fig 7. Stick representation of the 5-stranded β-sheet of SpFld showing a characteristic bulge in strand 5, which has been attributed [44] to originate in evolutionary splicing of two preexisting short strands, 5a and 5b that, in long-chain flavodoxins, are connected by their characteristic long loop.

Similar secondary structure induction due to FMN interaction is also observed in the crystal structures of holo and apo forms of short-chain flavodoxin MioC of E. coli [47]. The extent of the structural changes brought about by FMN binding in other flavodoxins of known structure has been analysed using DaliLite [48]. The larger r.m.s.d. deviation between the holo and apo forms was found for MioC Flavodoxin (2,6 Å), followed by Anabaena and Sp (0.8 Å), and then H. pylori (0.5 Å). Those data show a high preservation of the backbone structure upon FMN binding and indicate that segmental movements are essentially confined to the known dynamic loops of the protein; 50´s-Loop and 90´s-Loop. Smaller changes are observed in the phosphate binding loop upon FMN binding because, in the different apoproteins studied, this loop always carries bound anions: a phosphate anion in Sp apoflavodoxin, a chloride anion in Helicobacter pylori [26], or a sulfate anion in Anabaena [49] (S2 Fig).
Accommodation of the FMN into the Sp apoprotein takes place with changes in the dihedral angles of several residues of the isoalloxazine binding loops. In the 57–66 loop, the main changes are observed in residues G60, D61, G62 and E63, while in the 92–100 loop they occur in residues D92, Y95 and D96. No significant changes are noticed in the FMN phosphate binding loop, possibly reflecting that the conformation adopted by this loop in the apoprotein is highly influenced by the phosphate anion bound, which mimics the equivalent moiety in the FMN of the holoprotein. In its bound conformation, the FMN phosphate group forms hydrogen bonds with S10 (sc), M11 (N), T12 (sc and N), N14 (N) and T15 (sc and N) (Fig 6d). As previously reported for the apo/holo flavodoxin pairs in Anabaena PCC7119 and H. pylori [17, 26, 35, 49], the entrance of the FMN cofactor makes the two isoalloxazine binding loops to move apart. In this reorganization, existing interactions between the two loops in the apoform (Y95/G60 and E97/G62) are broken (Fig 8) and the protein expands: the holoprotein surface area is 7745 Å compared to 7135 Å for the apoprotein (S3 Fig). In the holoprotein, FMN is embedded by Y59, D92, E97, Y95, C99 and G60. The isoalloxazine ring of FMN establishes π-π interactions with Y59 and Y95 (Fig 9). While the presence of a FMN binding tyrosine residue is most common in the 90´s-Loop of flavodoxins, in the 50´s-Loop several other residues have been reported (e.g. W, A, M or L).
Fig 8. Stick representation of the FMN binding site in the Sp apo and holoflavodoxins indicating two key glycine residues (G60 and G62) that close the gap left by FMN in the apoprotein by interacting with residues of the 90’ loop: Y95 and E97.

Fig 9. Stick representation of the aromatic/aromatic pairs established by the FMN sandwiching Y59 and Y95 residues with neighboring Y57 and F94, respectively.

Interestingly, both Y59 and Y95 establish T-shape interactions with neighboring Y57 and F94, respectively (Fig 9). These aromatic pairs are not present in the apo form and it is possible that they contribute to stabilize the conformation of the FMN-sandwiching residues. No such aromatic pairs have been previously observed in the FMN binding sites of the other short-chain flavodoxins of known structure (e.g. Desulfovibrio desulfuricans, D. vulgaris, D. gigas, Clostridium Beijerinckii, Escherichia coli YqcA flavodoxin or Megasphaera elsdenii).
The flavodoxin from Desulfovibrio vulgaris (Dv) is the short-chain flavodoxin for which more detailed structural studies are available. Its sequence shows a 44% identity with that of Sp flavodoxin. The structures of the two flavodoxins (Dv and Sp), superimposed in Fig 10, reveal their high similarity (r.m.s.d. 1.2 Å), the main difference being noticed in the 50´s-Loop, which is longer in Dv flavodoxin. While in the 90´s-Loop both flavodoxins use tyrosine residues with essentially identical orientations on the si face of FMN, in the 50´s-Loop the sandwiching aromatic residues in the two flavodoxins are different as are their orientations on the re face. The interaction of the FMN phosphate at its binding loop is almost identical in the two flavodoxins, as expected from the high conservation of this loop sequence. Finally, the hydrogen bonding pattern of the polar atoms of the isoalloxazine rings (see Fig 6d for the Sp flavodoxin pattern) is not identical in the two flavodoxins, but it is clearly analogous specially for the nitrogen atoms (N3 and N10).
Fig 10. Structural comparison of the short flavodoxins from Desulfovibrio vulgaris (pdb id: 1j8q; ribbon and residues in green) and from Streptococcus pneumoniae (this work, pdb id: 5lji; ribbon and residues in cyan).

The FMN cofactors of the two flavodoxins and the corresponding sandwiching aromatic residues (Y98 and W60 in Dv flavodoxin, and Y59 and Y95 in Sp flavodoxin) are shown in a stick representation.
Suitability of SpFld as a novel drug target to combat Streptococcus pneumoniae infections
Streptococcus pneumoniae infections cause more than one million deaths every year, affecting principally young children and older people in developing countries. Appearance and propagation of resistant strains, the relative ineffectiveness of some vaccines for some serotypes, and virulence serotype changing, points towards an urgent search for novel Sp drug targets [50]. Antibiotics being the main line of therapy to pneumococcal infections, the main issue in pneumococcal infections management is the rising frequency of antibiotic resistance, principally to penicillin, macrolides and also to fluoroquinolones [51]. In this context, the identification of novel Sp-specific antibiotics is an important goal. Interestingly, a genome-wide identification of essential genes in S. pneumoniae TIGR 4 strain in a murine meningitis model identified three genes: adenylosuccinate synthetase (PurA), ABC transporter, and flavodoxin [9]. Although the specific role of flavodoxin in S. pneumoniae has not been established, it is clear that Sp infectiveness in this experimental meningitis model is severely attenuated in a SpFld knockout mutant [9]. Taking as a precedent our own discovery of specific inhibitors of the essential flavodoxin from the pathogen Helicobacter pylori, which are currently under preclinical evaluation [31, 32], we propose that novel Sp-specific antimicrobials can be developed based on inhibiting the activity of Sp flavodoxin. It is not obvious that the Hp inhibitors previously discovered [31, 32] should be effective against Sp because SpFld lacks the pocket near the FMN that is present in Hp flavodoxin where an alanine residue replaces the usual aromatic residue at the 50´s-Loop. Therefore, to facilitate the development of Sp specific flavodoxin inhibitors, we have cloned, expressed Sp flavodoxin, and solved the x-ray structure of both the apoprotein and the holo-form in complex with the FMN cofactor. Besides, we have characterized its thermal stability, a convenient reporting property than can be used to identify potential inhibitors by detecting their stabilizing effects against thermal denaturation exerted upon binding to the target protein. This work provides therefore the missing structural and thermodynamic knowledge of the Sp flavodoxin that will facilitate the search for novel antimicrobials to inhibit SpFld function as a means to control the Streptococcus pneumoniae infection.
Materials and Methods
Cloning
The flavodoxin coding sequence was amplified from Streptococcus pneumoniae TIRGR4 genomic DNA using the following primers:
5´-CGGAATTCGGATCCGGCAGCAGCCATCATCATCATCATCATCATCATGGCAGCGGCCTGGAAGTTCTGTTCCAGGGGCCCATGGCATTAGCAAAAATTG-3´ and 5’-CGGAATTCGTCGACTTAACCCACTTTCGCTGCCAGTTCTTC-3’, which additionally served to introduce 8 His residues at the N-terminal of the expressed protein. The resulting product from amplification was digested and cloned into the pMALC2X using BamHI and SalI restriction sites (underlined). The plasmid was then introduced into DH5alpha E. coli cells (Life Technologies, Inc., Rockville, MD, USA) and transformed cells were selected on Luria Bertani plates or in liquid medium (LB) containing 100 μg ml-1 ampicillin (Sigma, St. Louis, MO, USA). Positive clones identified by colony–PCR (BIORON) were sequenced and shown to contain the DNA construct encoding the full length SpFld plus 8 additional histidine residues at the N-terminus.
Sp flavodoxin expression, purification and apoflavodoxin preparation
The BL21 (DE3) strain (Novagen), cultured in 2XTY media, was used for recombinant protein overexpression. E. coli BL21 (DE3) cells were grown to an optical density of 0.6–0.8 at 37°C, followed by induction with 1 mM IPTG (Sigma Aldrich). After 10 h of induction at 20°C, the cells were collected by centrifugation at 11295 g for 20 min. The resulting pellet was suspended in 160 ml of binding buffer (150 mM NaCl, 10 mM imidazole, 20 mM sodium phosphate buffer, pH 7.4) per L of culture, and sonicated in ice in the presence of excess FMN, 5 μl of benzonase (Sigma Aldrich, stock solution at 250 units μl-1) per 25 ml of solution, lysozyme (Sigma Aldrich, >95% pure) final concentration 1 mg ml-1 and 500 μL protease inhibitors cocktail stock solution (1 M PMSF + 10 mM benzamidine + 0.5 mM leupeptin) per 25 ml of cell volume, using 10 cycles, each consisting of a 30 s pulse plus a 30 s cooling step. The lysate was centrifuged at 39086 g for 20 min. The cell pellet and its initial lysate showed a characteristic blue color indicating the flavodoxin FMN cofactor was in the semiquinone redox state [2]. The color changed to yellow (corresponding to oxidized FMN) as the purification progressed.
The supernatant was filtered through a 0.45 μm membrane and loaded onto a Co2+HiTrap TALON crude column (GE Healthcare) equilibrated with binding buffer. The column was washed with binding buffer till negative Bradford reaction of the eluate, and the bound protein was eluted using a 150 mM NaCl, 500 mM imidazole, sodium phosphate buffer, pH 7.4 (elution buffer). The eluted solution containing recombinant SpFld was dialyzed against 150 mM NaCl, 25 mM Tris buffer pH 7.5 to remove the imidazole, and incubated overnight at 4°C with 0.5U of PreScission protease in order to remove the pMAL-(His)8 tail. The mixture was loaded onto coupled GST and histidine columns (GE Healthcare) equilibrated in the dialysis buffer, and SpFld without affinity tags was collected as the unbinding fraction, dialyzed against 25 mM Tris pH 7.0, loaded into a 5 ml HiTrap Q FF column (GE Healthcare) equilibrated with 5 column volumes in the same buffer, and eluted with 5–10 column volumes with a linear NaCl gradient from 0 to 1M in Tris 25 mM pH 7.0. Pure fractions according to SDS-PAGE were concentrated to 2.5 ml by ultrafiltration using 10 kDa Centricons (Millipore) and loaded onto a gel filtration column (Superdex 200 GE Healthcare) equilibrated with 150 mM NaCl, 25 mM Tris, pH 7.5 from where a single peak corresponding to the monomer was collected. The purity of the obtained SpFld was estimated to be >95% by SDS/PAGE (S4 Fig). A single band of 16 kDa was observed, in agreement with the expected molecular mass of 16025 Da. SpFld was dialyzed against 150 mM NaCl, 100 mM MES pH 6.5, and concentrated to around 1 mM. Aliquots were fast frozen in liquid nitrogen and stored at -80°C for further use.
For the crystallization of FMN-bound flavodoxin, selenomethionine substituted SpFld was similarly expressed in E. coli BL21 (DE3) strain, using the SelenoMethionine media from Molecular Dimensions. The protein was purified following the same protocol used for the native protein (see above). MALDI-TOF mass spectrometry, carried out in the Proteomics facility from UAB, showed that three out of four possible selenomethionine residues were incorporated into the protein. Sp apoflavodoxin (deprived from the FMN cofactor) was obtained by precipitation with trichloroacetic acid (TCA) [52]. The protein pellet was washed five times with 3% w/v TCA and 1 mM DTT, resuspended in 1 M MES pH 6.5, 15 mM NaCl, and subsequently dialyzed in 150 mM NaCl, 100 mM MES, pH 6.5 and stored as the holoprotein.
UV-Visible absorption, circular dichroism and fluorescence emission spectra
The UV-visible absorption spectrum of holo and apo SpFld in 150 mM NaCl, 100 mM MES, pH 6.5, was collected at 25°C using a Cary 100Biospectrometer (Varian). To estimate SpFld apoflavodoxin molar extinction coefficient in 150 mM NaCl, 100 mM MES, pH 6.5 (working buffer), the Gill von Hippel method was used [37]. The extinction coefficient of the denatured protein in 6 M guanidine-HCl, 20 mM phosphate sodium, pH 6.5, was first calculated from its content of cysteines and tyrosines. Then, the ratio of absorbances at the wavelength selected of protein solutions or equal concentration prepared in either denaturing buffer or in working buffer equaled the ratio of the calculated extinction coefficients in denaturing buffer over that in working buffer. All measures were done by triplicate. To determine the holoflavodoxin extinction coefficient, the Mayhew and Massey method was used [38].
Far-UV, near-UV and visible-UV circular dichroism (CD) spectra of the holo and apoproteins in 150 mM NaCl, 100 mM MES, pH 6.5, were acquired at 4°C in a Chirascan spectropolarimeter (Applied-Photophysics). For near-UV and visible spectra, 80 μM protein solutions in a 1-cm path-length cuvette were used, while for far-UV, 80 μM protein solutions were used in a 0.01-cm path-length cuvette. To accurately determine the concentration of protein in the CD samples, small volumes of concentrated stock solutions of either apo or holoflavodoxin were initially diluted to prepare solutions that provided an approximation to the actual concentrations. Based on that, protein samples around 80 μM were prepared and used to acquire the CD spectra. After having recorded the CD spectra, the actual concentrations of those samples were accurately determined by recording their full absorbance spectra. For apoflavodoxin quantification the near-UV peak was used, and for holoflavodoxin quantification the major visible peak was used. The absorbances of either of those peaks were of around 0.7 in a 1 cm path-length cuvette and allowed a fine determination of protein concentration using the corresponding extinction coefficients. These concentrations were finally used for the MRE calculation leading to Fig 2a. Fluorescence emission spectra of 20 μM holoprotein or apoprotein, in 150 mM NaCl, 100 mM MES, pH 6.5, were recorded at 10°C from 275 to 750 nm or from 290 to 500 nm, respectively, with excitation at 270 nm, using a Cary Eclipse spectrofluorometer (Varian).
Thermal denaturation followed by spectroscopy
Holo and apo SpFld thermal denaturation was followed, from 8 to 90°C, by CD. Unfolding curves for the holoprotein were acquired at 220 nm (far-UV CD) and at 272 nm (near-UV CD) with heating rates of 1°C min-1. For the apoprotein, the wavelengths used were 221 and 285 nm, respectively. For near-UV, a 1-cm path-length quartz cuvette was used and the samples were prepared with the same buffer and protein concentration used for recording the CD spectra, while for far-UV CD a 0.1-cm path-length cuvette and 80 μM protein solutions were used. The thermal denaturation of the holoprotein was followed, from 8 to 80°C, by fluorescence in the visible region (excitation and emission of 464 and 525 nm, respectively) associated to FMN release coupled to protein unfolding, using a heating rate of 1°C min-1. Protein concentration and buffer conditions were those used for acquiring the fluorescence spectrum (20 μM for both, holo- and apoprotein). Each individual curve was fitted to a two-state model, as previously described [53, 54], using the following equations:
| (1) |
| (2) |
where “S” is the observed spectroscopic signal at a given temperature, and “SN(U) + mN(U) × T” represents the temperature-dependent intrinsic signal of either the native (N subscript) or the unfolded (U subscript) protein, R is the ideal gas constant, T is the absolute temperature, and ΔG is the unfolding Gibbs energy change, which can be expressed in terms of the mid-transition temperature Tm, the unfolding enthalpy change ΔHm, and the unfolding heat capacity change ΔCP,m. Because all the individual thermal unfolding curves of either the apo- or the holoprotein acquired following different spectroscopic signals could be superimposed, each group of curves (from either holo or apoflavodoxin) was also globally fitted to the two-state unfolding model, as described [53, 54].
Thermal denaturation followed by differential scanning calorimetry
Differential scanning calorimetry (DSC) assays were performed on 40 μM apo SpFld in the absence or presence of FMN (40 and 1000 μM). Measurements (from 10 to 100°C at a scanning rate of 1°C min-1) were performed in a VP-DSC microcalorimeter (MicroCal LLC, Northampton, MA), and rescans were recorded after cooling to 10°C. The reference and sample solutions were degassed and carefully loaded into the cells to avoid bubble formation. Baselines were recorded before each assay with the reference and sample cells filled with buffer (150 mM NaCl, 100 mM MES, pH 6.5). Data analysis, using the software package ORIGIN (OriginLab), was based on the model-free van´t Hoff analysis. Briefly, from the thermal unfolding assay the mid-transition temperature Tm and the calorimetric enthalpy ΔHcal could be determined and, from those values, the van’t Hoff enthalpy was estimated as:
| (3) |
where <ΔCP,tr>max is the maximal value of the excess molar heat capacity of the protein [55]. The van’t Hoff enthalpy is equivalent to the unfolding enthalpy obtained from analysis of spectroscopic techniques, and: a) it is equal to the calorimetric enthalpy if the thermal unfolding follows a two-state mechanism; b) it is smaller than the calorimetric enthalpy if the thermal unfolding follows a non-two-state mechanism with partially folded intermediates; and c) it is higher than the calorimetric enthalpy if the thermal unfolding follows a two-state mechanism where the native protein self-associates in oligomers [56]. Once the appropriate unfolding model has been determined, the unfolding curve can be analyzed accordingly.
FMN binding by isothermal titration calorimetry
Isothermal titration calorimetry (ITC) measurements were carried out in a MicroCal Auto-iTC200 (MicroCal/GE Healthcare) using carefully degassed cofactor and protein solutions. A 300 μM solution of FMN (Sigma-Aldrich, >95% pure) dissolved in 150 mM NaCl, 100 mM MES, pH 6.5 was titrated into 20 μM apo SpFld, in the same buffer. Intervals of 700 s between injections were allowed to guarantee recovery of the base line. Data were analyzed with a one-site binding model implemented in the Origin (OriginLab) software package. Binding titrations were performed at 15, 20 and 25°C to obtain the binding heat capacity as the temperature derivative of the observed binding enthalpy. Additional assays required for assessing proton exchange processes coupled to ligand binding were performed in 150 mM NaCl 50 mM MOPS pH 6.5 at 25°C. The buffer-independent binding enthalpy ΔH0, and the net number of protons exchanged nH were estimated by performing assays using buffers with different ionization enthalpies and linear regression using eq 4:
| (4) |
where ΔHobs is the enthalpy obtained in a given assay and ΔHion is the ionization enthalpy of the buffer.
Dithionite titration of SpFld holoflavodoxin
To observe the spectra of the different redox states of SpFld, a sodium dithionite reduction was performed at 10°C using 20 μM protein in 150 mM NaCl, 100mM MES buffer, pH 6.5 under pseudo-anaerobic conditions. A concentrated anaerobic sodium dithionite solution (50 mM) was prepared in the same buffer by sequential evacuation and re-equilibration with oxygen-free argon. Pseudo-anaerobic conditions were achieved in the spectrophotometer cell by argon flux. Stepwise SpFld reduction was achieved by addition of 2.5–10 ml aliquots of sodium dithionite to the protein sample. After each addition, the UV-Vis spectrum was recorded in a Cary 100 spectrophotometer (Varian).
Crystallization and data collection
Crystallization trials were carried out using the sitting-drop vapor diffusion method at 18°C. The drops were prepared by mixing 0.5 μL of protein solution containing either holo (62 mg ml-1) or apoprotein (127 mg ml-1) in 100 mM MES, 150 NaCl, pH 6,5 buffer with 0.5 μL of the mother liquor. Crystals of the holoprotein (though these crystals contain selenomethionine, they are referred here as our holo form) were grown in 45% 2-Methyl-2,4-pentanediol (MPD), 100 mM Bis-Tris pH 5.5 and 200 mM calcium chloride, apoprotein form crystals were obtained in 10% glycerol and 2 M ammonium sulphate. Crystals were formed after 3–8 days. Best apoprotein and holoprotein SpFld crystals were immersed, respectively, in the same reservoir solution or in the same solution with 20% of glycerol for cryoprotection. All diffraction datasets were collected at the Diamond synchrotron light source (DLS).
Structure determination, phase calculation, model building and refinement
Initial molecular replacement trials were attempted using MOLREP [57] with the data from a holo crystal processed as H3 (note that the holo form contains SeMet residues) and the structure of Desulfovibrio desulfuricans flavodoxin (PDB code: 3F9O) as search model; however, no improvement in Rfactor and Rfree was observed during refinement. The program SOLVE [58] was used for initial phasing by single anomalous dispersion (SAD). Despite the data collection had been performed using a suboptimal wavelength (λ = 0.976 Å), the positions of the 3 Se atoms were determined. Subsequent phasing and solvent flattening were carried out using SOLVE and RESOLVE [59] as implemented in PHENIX [60]. A solvent flattened Se-SAD derived electron density map was used for automatic construction of the holo SpFld structure with RESOLVE. The automatic building process resulted in a model formed by 76 residues in 11 chains. Remarkably, one of the chains was an alpha helix that appeared at the same position in the previous MR attempt. Then, we built a more complete model with the program PHENIXAutoBuild and using the initial MR solution as initial model in combination with the Se-SAD density map obtained previously. The resulting model contained 143 residues positioned in 2 chains, with Rfact and Rfree of 0.29 and 0.36 respectively. We completed the model manually with Coot [61] and refined with REFMAC [62], obtaining Rfact and Rfree values of 0.25 and 0.33 respectively. Finally, the combination of adding FMN, water molecules and new rounds of model building in Coot and TLS restrained refinement in REFMAC, rendered Rfact and Rfree values of 0.187 and 0.228, respectively.
To solve the structure of the SpFld apoprotein, molecular replacement was performed using MOLREP[58] with the holoprotein SpFld structure as starting model and the data in the space group P212121. Iterative rounds of manual building in Coot and TLS restrained refinement in REFMAC were also performed, and the final model was validated with PROCHEK [63]. All figures were generated with PyMoL (http://wwwrcsb.org). Coordinates and structure factors have been deposited in the protein data bank under accession code 5LJI and 5LJL for the holo and apo form, respectively.
Supporting Information
(PNG)
{kind=link}
(TIF)
Note that FMN induces the formation of a larger pocket in the holo form (in which FMN locates) in comparison with a smaller pocket present in the apo structure.
(TIF)
(TIF)
Acknowledgments
We acknowledge financial support from BFU2010-16297 and BFU2010-19504 [Ministerio de Ciencia e Innovación Spain], BFU2013-47064-P, BIO2014-57314-REDT and CTQ2013-44367-C2-2-P [Ministerio de Economía y Competitividad, Spain], and DGA (Protein Targets B89). We also thank synchrotron radiation sources DLS (Oxford), and in particular beamline I04-1 (experiment number MX8035-3 and MX8035-11). The research leading to these results has also received funding from the FP7 (2007–2013) under BIOSTRUCTX-7687. A.R.C. was funded by a Banco Santander Central Hispano/Universidad de Zaragoza predoctoral fellowship. M. C-G was recipient of a predoctoral fellowship from the Government of Aragón. We thank Dr. Ana Sánchez for help with the dithionite titration.
Data Availability
Data are available at https://www.ebi.ac.uk/pdbe/entry/pdb/5lji, https://www.ebi.ac.uk/pdbe/entry/pdb/5ljl.
Funding Statement
We acknowledge financial support from BFU2010-16297 and BFU2010-19504 [Ministerio de Ciencia e Innovación Spain], BFU2013-47064-P, BIO2014-57314-REDT and CTQ2013-44367-C2-2-P [Ministerio de Economía y Competitividad, Spain], and DGA (Protein Targets B89). We also thank synchrotron radiation sources DLS (Oxford), and in particular beamline I04-1 (experiment number MX8035-3 and MX8035-11). The research leading to these results has also received funding from the FP7 (2007-2013) under BIOSTRUCTX-7687. A.R.C. was funded by a Banco Santander Central Hispano/Universidad de Zaragoza predoctoral fellowship. M. C-G was recipient of a predoctoral fellowship from the Government of Aragón. We thank Dr. Ana Sánchez for help with the dithionite titration.
References
- 1.AlonsoDeVelasco E, Verheul AF, Verhoef J, Snippe H. Streptococcus pneumoniae: virulence factors, pathogenesis, and vaccines. Microbiological Reviews, 1995. 59(4): p. 591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawson RJ, von Wachenfeldt C, Haq I, Perkins J, Munro AW. Expression and characterization of the two flavodoxin proteins of Bacillus subtilis, YkuN and YkuP: biophysical properties and interactions with cytochrome P450 BioI. Biochemistry, 2004. 43(39): p. 12390–12409. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell AM, Mitchell TJ. Streptococcus pneumoniae: virulence factors and variation. Clinical Microbiology and Infection, 2010. 16: p. 411–418. 10.1111/j.1469-0691.2010.03183.x [DOI] [PubMed] [Google Scholar]
- 4.Martin M, Turco JH, Zegans ME, Facklam RR, Sodha S, Elliott JA, et al. An Outbreak of Conjunctivitis Due to Atypical Streptococcus pneumoniae. New England Journal of Medicine, 2003. 348(12): p. 1112–1121. [DOI] [PubMed] [Google Scholar]
- 5.WHO P. Pneumococcal vaccines WHO position paper -2012- Recommendations. Vaccine. 30(32): p. 4717–4718. 10.1016/j.vaccine.2012.04.093 [DOI] [PubMed] [Google Scholar]
- 6.Pérez-Dorado I, Bortolotti A, Cortez N, Hermoso J. Structural and phylogenetic analysis of Rhodobacter capsulatus NifF: Uncovering general features of nitrogen-fixation (nif)-flavodoxins. International Journal of Molecular Sciences, 2013. 14(1): p. 1152–1163. 10.3390/ijms14011152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liñares J, Ardanuy C, Pallares R, Fenoll A. Changes in antimicrobial resistance, serotypes and genotypes in Streptococcus pneumoniae over a 30-year period. Clinical Microbiology and Infection, 2010. 16: p. 402–410. 10.1111/j.1469-0691.2010.03182.x [DOI] [PubMed] [Google Scholar]
- 8.Croucher NJ, Finkelstein JA, Pelton SI, Mitchell PK, Lee GM, Parkhill J, et al. Population genomics of post-vaccine changes inpneumococcal epidemiology. Nature genetics, 2013. 45(6): p. 656–663. 10.1038/ng.2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molzen TE, Burghout P, Bootsma HJ, Brandt CT, van der Gaast-de J, Christa E, et al. Genome-wide identification of Streptococcus pneumoniae genes essential for bacterial replication during experimental meningitis. Infection and Immunity, 2011. 79(1): p. 288–297. 10.1128/IAI.00631-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, et al. Complete Genome Sequence of a Virulent Isolate of Streptococcus pneumoniae. Science, 2001. 293(5529): p. 498–506. [DOI] [PubMed] [Google Scholar]
- 11.Zhang R, Lin Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Research, 2009. 37(Database issue): p. D455–D458. 10.1093/nar/gkn858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerdes SY, Scholle MD, Campbell JW, Bala G, Ravasz E, Daugherty MD, et al. Experimental Determination and System Level Analysis of Essential Genes in Escherichia coli MG1655. Journal of Bacteriology, 2003. 185(19): p. 5673–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular Systems Biology, 2006. 2: p. 2006.0008–2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langridge GC, Gemma C, Phan MD, Turner DJ, Perkins TT, Parts L, et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Research, 2009. 19(12): p. 2308–2316. 10.1101/gr.097097.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akerley BJ, Rubin EJ, Novick VL, Amaya K, Judson N, Mekalanos JJ, et al. A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proceedings of the National Academy of Sciences of the United States of America, 2002. 99(2): p. 966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cameron DE, Urbach JM, Mekalanos JJ. A defined transposon mutant library and its use in identifying motility genes in Vibrio cholerae. Proceedings of the National Academy of Sciences of the United States of America, 2008. 105(25): p. 8736–8741. 10.1073/pnas.0803281105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freigang J, Diederichs K, Schäfer KP, Welte W, Paul R. Crystal structure of oxidized flavodoxin, an essential protein in Helicobacter pylori. Protein Science: A Publication of the Protein Society, 2002. 11(2): p. 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sancho J. Flavodoxins: sequence, folding, binding, function and beyond. Cellular and Molecular Life Sciences CMLS, 2006. 63(7): p. 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hill S, Kavanagh EP. Roles of nifF and nifJ gene products in electron transport to nitrogenase in Klebsiella pneumoniae. Journal of Bacteriology, 1980. 141(2): p. 470–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bianchi V, Eliasson R, Fontecave M, Mulliez E, Hoover DM, Matthews RG, et al. Flavodoxin Is Required for the Activation of the Anaerobic Ribonucleotide Reductase. Biochemical and Biophysical Research Communications, 1993. 197(2): p. 792–797. [DOI] [PubMed] [Google Scholar]
- 21.Fujii K, Huennekens FM. Activation of methionine synthetase by a reduced triphosphopyridine nucleotide-dependent flavoprotein system. Journal of Biological Chemistry, 1974. 249(21): p. 6745–6753. [PubMed] [Google Scholar]
- 22.Ifuku O, Ifuku O, Koga N, Haze SI, Kishimoto J, Wachi Y. Flavodoxin is Required for Conversion of Dethiobiotin to Biotin in Escherichia coli. European Journal of Biochemistry, 1994. 224(1): p. 173–178. [DOI] [PubMed] [Google Scholar]
- 23.Wong KK, Murray BW, Lewisch SA, Baxter MK, Ridky TW, Ulissi-DeMario L, et al. Molecular properties of pyruvate formate-lyase activating enzyme. Biochemistry, 1993. 32(51): p. 14102–14110. [DOI] [PubMed] [Google Scholar]
- 24.Cremades N, Bueno M, Toja M, Sancho J, et al. Towards a newtherapeutic target: Helicobacter pylori flavodoxin. Biophysical Chemistry, 2005. 115(2–3): p. 267–276. [DOI] [PubMed] [Google Scholar]
- 25.St. Maurice M, Croxen MA, Sisson G, Sancho J, Hoffman PS. Flavodoxin:quinone reductase (FqrB): a redox partner of pyruvate:ferredoxin oxidoreductase that reversibly couples pyruvate oxidation to NADPH production in Helicobacter pylori and Campylobacter jejuni. Journal of Bacteriology, 2007. 189(13): p. 4764–4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martínez-Júlvez M, Cremades N, Bueno M, Pérez-Dorado I, Maya C, Cuesta-López S, et al. Common conformational changes in flavodoxins induced by FMN and anion binding: The structure of Helicobacter pylori apoflavodoxin. Proteins: Structure, Function, and Bioinformatics, 2007. 69(3): p. 581–594. [DOI] [PubMed] [Google Scholar]
- 27.Cremades N, Bueno M, Neira JL, Velázquez-Campoy A, Sancho J. Conformational Stability of Helicobacter pylori Flavodoxin: Fit to Function at pH 5. Journal of Biological Chemistry, 2008. 283(5): p. 2883–2895. [DOI] [PubMed] [Google Scholar]
- 28.Cremades N, Velazquez-Campoy A, Freire E, Sancho J. The flavodoxin from Helicobacter pylori: structural determinants of thermostability and FMN cofactor binding. Biochemistry, 2008. 47(2): p. 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cremades N, Sancho J. Molten Globule and Native State Ensemble of Helicobacter pylori Flavodoxin: Can Crowding, Osmolytes or Cofactors Stabilize the Native Conformation Relative to the Molten Globule? Biophysical Journal, 2008. 95(4): p. 1913–1927. 10.1529/biophysj.108.130153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ayuso-Tejedor S, Abián O, Velázquez-Campoy A, Sancho J. Mechanism of FMN Binding to the Apoflavodoxin from Helicobacter pylori. Biochemistry, 2011. 50(40): p. 8703–8711. 10.1021/bi201025y [DOI] [PubMed] [Google Scholar]
- 31.Cremades N, Velázquez-Campoy A, Martínez-Júlvez M, Neira JL, Pérez-Dorado I, Hermoso J, et al. Discovery of specific flavodoxin inhibitors as potential therapeutic agents against Helicobacter pylori infection. ACS Chemical Biology, 2009. 4(11): p. 928–938. 10.1021/cb900166q [DOI] [PubMed] [Google Scholar]
- 32.Galano JJ, Alías M, Pérez R, Velázquez-Campoy A, Hoffman PS, Sancho J. Improved flavodoxin inhibitors with potential therapeutic effects against Helicobacter pylori infection. Journal of Medicinal Chemistry, 2013. 56(15): p. 6248–6258. 10.1021/jm400786q [DOI] [PubMed] [Google Scholar]
- 33.Ayuso-Tejedor S, Garcia-Fandiño R, Orozco M, Sancho J, Bernadó P, Structural Analysis of an Equilibrium Folding Intermediate in the Apoflavodoxin Native Ensemble by Small-Angle X-ray Scattering. Journal of Molecular Biology, 2011. 406(4): p. 604–619. 10.1016/j.jmb.2010.12.027 [DOI] [PubMed] [Google Scholar]
- 34.Burkhart BM, Ramakrishnan B, Yan H, Reedstrom RJ, Markley JL, Straus NA, et al. Structure of the trigonal form of recombinant oxidized flavodoxin from Anabaena 7120 at 1.40 A resolution. Acta Crystallographica. Section D, Biological Crystallography, 1995. 51: p. 318–330. [DOI] [PubMed] [Google Scholar]
- 35.Rao ST, Shaffie F, Yu C, Satyshur KA, Stockman BJ, Markley JL, et al. Structure of the oxidized long-chain flavodoxin from Anabaena 7120 at 2 A resolution, in Protein Science: A Publication of the Protein Society. 1992, Cold Spring Harbor Laboratory Press; p. 1413–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alagaratnam S, Pouderoyen GV, Pijning T, Dijkstra BW, Cavazzini D, Rossi GL, et al. A crystallographic study of Cys69Ala flavodoxin II from Azotobacter vinelandii: Structural determinants of redox potential. Protein Science: A Publication of the Protein Society, 2005. 14(9): p. 2284–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Analytical Biochemistry, 1989. 182: p. 319–326. [DOI] [PubMed] [Google Scholar]
- 38.Mayhew SG, Massey V. Purification and Characterization of Flavodoxin from Peptostreptococcus elsdenii. Journal of Biological Chemistry, 1969. 244(4): p. 794–802. [PubMed] [Google Scholar]
- 39.Khan PC. The interpretation of near-ultraviolet circular dichroism. Methods in Enzymology, 1979. 61: p. 339–378. [DOI] [PubMed] [Google Scholar]
- 40.D'Anna JA, Tollin G. Studies of flavine-protein interaction in flavoproteins using protein fluorescence and circular dichroism. Biochemistry, 1972. 11(6): p. 1073–1080. [DOI] [PubMed] [Google Scholar]
- 41.Pace CN, Shirley B, Thomson J. Measuring the conformational stability of a protein, in Protein structure: A practical Apporoach, Creighton T.E., Editor. 1989. p. 311–330. [Google Scholar]
- 42.Murray TA, Swenson RP. Mechanism of flavin mononucleotide cofactor binding to the Desulfovibrio vulgaris flavodoxin. 1. Kinetic evidence for cooperative effects associated with the binding of inorganic phosphate and the 5´-phosphate moiety of the cofactor. Biochemistry, 2003. 42(8): p. 2307–2316. [DOI] [PubMed] [Google Scholar]
- 43.Ayuso-Tejedor S, Angarica VE, Bueno M, Campos LA, Abián O, Bernadó P, et al. Design and Structure of an Equilibrium Protein Folding Intermediate: A Hint into Dynamical Regions of Proteins. Journal of Molecular Biology, 2010. 400(4): p. 922–934. 10.1016/j.jmb.2010.05.050 [DOI] [PubMed] [Google Scholar]
- 44.López-Llano J, Maldonado S, Jain S, Lostao A, Godoy-Ruiz R, Sanchez-Ruiz JM, et al. The Long and Short Flavodoxins: II. The role of the differentiating loop in apoflavodoxin stability and folding mechanism. Journal of Biological Chemistry, 2004. 279(45): p. 47184–47191. [DOI] [PubMed] [Google Scholar]
- 45.Campos LA, Garcia-Mira MM, Godoy-Ruiz R, Sanchez-Ruiz JM, Sancho J. Do Proteins Always Benefit from a Stability Increase? Relevant and Residual Stabilisation in a Three-state Protein by Charge Optimisation. Journal of Molecular Biology, 2004. 344(1): p. 223–237. [DOI] [PubMed] [Google Scholar]
- 46.Lamazares E, Clemente I, Bueno M, Velázquez-Campoy A, Sancho J. Rational stabilization of complex proteins: a divide and combine approach. Scientific Reports, 2015. 5: p. 9129 10.1038/srep09129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu Y, Li Y, Zhang X, Guo X, Xia B, Jin C, et al. Solution Structures and Backbone Dynamics of a Flavodoxin MioC from Escherichia coli in both Apo- and Holo-forms: IMPLICATIONS FOR COFACTOR BINDING AND ELECTRON TRANSFER. Journal of Biological Chemistry, 2006. 281(46): p. 35454–35466. [DOI] [PubMed] [Google Scholar]
- 48.Holm L, Park J. DaliLite workbench for protein structure comparison. Bioinformatics, 2000. 16(6): p. 566–567. [DOI] [PubMed] [Google Scholar]
- 49.Genzor CG, Perales-Alcón A, Sancho J, Romero A. Closure of a tyrosine/tryptophan aromatic gate leads to a compact fold in apo flavodoxin. Nature Structural Biology, 1996. 3(4): p. 329–332. [DOI] [PubMed] [Google Scholar]
- 50.Di Guilmi AM, Dessen AA. New approaches towards the identification of antibiotic and vaccine targets in Streptococcus pneumoniae. EMBO Reports, 2002. 3(8): p. 728–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reynolds CA, Finkelstein JA, Ray GT, Moore MR, Huang SS, et al. Attributable healthcare utilization and cost of pneumonia due to drug-resistant streptococcus pneumonia: a cost analysis. Antimicrobial Resistance and Infection Control, 2014. 3: p. 16 10.1186/2047-2994-3-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edmondson DE, Tollin G. Chemical and physical characterization of the Shethna flavoprotein and apoprotein and kinetics and thermodynamics of flavin analog binding to the apoprotein. Biochemistry, 1971. 10(1): p. 124–32. [DOI] [PubMed] [Google Scholar]
- 53.Irún MP, Maldonado S, Sancho J. Stabilization of apoflavodoxin by replacing hydrogen-bonded charged Asp or Glu residues by the neutral isosteric Asn or Gln. Protein Engineering, 2001. 14(3): p. 173–181. [DOI] [PubMed] [Google Scholar]
- 54.Sancho J. The stability of 2-state, 3-state and more-state proteins from simple spectroscopic techniques… plus the structure of the equilibrium intermediates at the same time. Archives of Biochemistry and Biophysics, 2013. 531(1–2): p. 4–13. 10.1016/j.abb.2012.10.014 [DOI] [PubMed] [Google Scholar]
- 55.Privalov PL, Khechinashvili NN. A thermodynamic approach to the problem of stabilization of globular protein structure: A calorimetric study. Journal of Molecular Biology, 1974. 86(3): p. 665–684. [DOI] [PubMed] [Google Scholar]
- 56.Cooper A, ed. Differential scanning microcalorimetry Protein-Ligand Interactions: hydrodynamics and calorimetry ed. Harding S.E. and Chowdhry B.Z.. 2000, Oxford New York: Oxford University Press; 287–318. [Google Scholar]
- 57.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 66(Pt 1): p. 22–5. 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 58.Terwilliger TC. Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr, 1999. 55(Pt 4): p. 849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr, 2000. 56(Pt 8): p. 965–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy Airlie J, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr, 2002. 58(Pt 11): p. 1948–54. [DOI] [PubMed] [Google Scholar]
- 61.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr, 2004. 60(Pt 12 Pt 1): p. 2126–32. [DOI] [PubMed] [Google Scholar]
- 62.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr, 1997. 53(Pt 3): p. 240–55. [DOI] [PubMed] [Google Scholar]
- 63.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, 1993. 26(2): p. 283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PNG)
(TIF)
Note that FMN induces the formation of a larger pocket in the holo form (in which FMN locates) in comparison with a smaller pocket present in the apo structure.
(TIF)
(TIF)
Data Availability Statement
Data are available at https://www.ebi.ac.uk/pdbe/entry/pdb/5lji, https://www.ebi.ac.uk/pdbe/entry/pdb/5ljl.