

Abstract
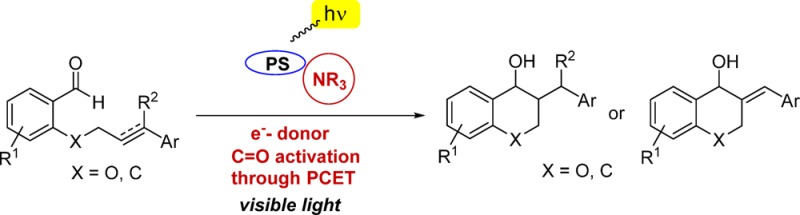
A visible light photoredox-catalyzed aldehyde olefin cyclization is reported. The method represents a formal hydroacylation of alkenes and alkynes and provides chromanol derivatives in good yields. The protocol takes advantage of the double role played by trialkylamines (NR3) which act as (i) electron donors for reducing the catalyst and (ii) proton donors to activate the substrate via a proton-coupled electron transfer.
Carbon–carbon and carbon–heteroatom bond-forming reactions represent powerful transformations useful for the assembly of simple as well as sophisticated molecular architectures. Among the variety of established methods reported so far, the generation of ketyl radical species by the reductive umpolung reaction of carbonyl derivatives and their application in carbon–carbon bond-forming reactions represents an appealing route for the preparation of complex molecules. The ketyl–olefin coupling, as first described by Molander and Kenny,1 was promoted by samarium diiodide as a stoichiometric single-electron reducing agent and enhanced by its large reduction potential (up to −2.05 V in the presence of HMPA).2 Alternatively, organotin compounds as reducing agents have been used in either stoichiometric3,4 or catalytic amounts,5 in this case requiring an additional metal hydride. Other relevant procedures involve the use of zinc/trimethylchlorosilane as reagent,6 or titanocene complexes,7 well-known as pinacol-coupling promoters. Among the ketyl–olefin couplings reported, the major protocols imply the use of stoichiometric amounts of (transition) metals.8
Recently, the possibility of generating ketyl radicals by visible light activation9−13 was shown to be a feasible alternative to the common methods, avoiding the use of toxic and/or environmentally unfriendly reducing reagents and yet enabling access to a wide range of complex alcohols. However, the reduction of aldehydes (E1/2red = −2.17 V vs Ag/AgNO3 for benzaldehydes),14 possessing an exceptionally negative redox potential, represents an obstacle for the most commonly employed visible light photocatalysts. In this context, Pandey reported the visible-light-promoted one-electron reductive activation of aldehydes and ketones and their intramolecular addition to activated olefins (five examples) with a system consisting of DCA (9,10-dicyanoanthracene) and PPh3 or DCA, DMN (1,5-dimethoxynaphthalene) and ascorbic acid.11a
Knowles and co-workers reported a catalytic protocol in which ketones are successfully reduced to ketyl radicals by the concerted transfer of both a proton and an electron.11b This phenomenon is named proton-coupled electron transfer (PCET).15
In an attempt to demonstrate the active role of proton donors in the reduction step, Knowles and co-workers also reported an asymmetric aza-pinacol cyclization of ketones and hydrazones where the binding effect of the chiral phosphoric acids led to the formation of enantioenriched vicinal amino alcohols.11c
Recently, our group described the photoredox-catalyzed pinacol coupling of aldehydes, ketones, and aldimines through hydrogen-bonding activation.12a With this work, we disclose the possibility of generating a long-lived ketyl radical using tertiary amines as the electron and proton source, replacing the combination of Hantzsch ester and diphenyl phosphate (DPP).
Based on our previous work,12a we envisioned the possibility of performing the ketyl radical addition on unsaturated bonds triggered by visible light, which would initiate the single-electron reduction from a photoexcited polypyridyl iridium complex to a carbonyl group (aldehyde or ketone). We postulated that the so-generated radical intermediate would be able to undergo subsequent intramolecular addition to a pendant alkene or alkyne moiety (Scheme 1).
Scheme 1. Photoredox-Catalyzed Reduction of Aldehydes through PCET.

Herein, we report the realization of this activation mode in the context of a catalytic intramolecular reductive coupling of aldehydes with alkenes and alkynes. This transformation allows the synthesis of substituted 3-benzylchroman-4-ols (5), containing a chroman unit, that exhibit anti picornavirus activity, commonly responsible for the upper respiratory tract infections in humans.16
Remarkably, we observed that tertiary amines not only provided the desired products in higher yields but also proved to be crucial for the reaction to proceed.
We began our studies by evaluating conditions for the cyclization of 2-(cinnamyloxy)benzaldehyde (1a) to chromanol 2a in the presence of Ir(ppy)2(dtb-bpy)PF6 (PC 3, 1 mol %), trialkylamines 4a–c as reductive quencher (RQ), and irradiation with a blue LED light source (11W, λmax = 450 nm). As shown in Table S1 (Supporting Information), the reaction proceeded with good yield in the presence of 2.5 equiv of tributylamine (4b, entry 5). Solvent evaluation confirmed acetonitrile to be the best solvent, whereas aprotic (entries 1 and 2) and protic (entry 3) polar solvents were shown to be unsuccessful as reaction media. Further screening of electron donors in acetonitrile confirmed Hünig base (4c) to be the best among the trialkylamines tested, providing the desired product in 80% yield (entry 6). As previously mentioned, the use of Hantzsch esters 5a and 5b alone or in combination with 10 mol % of DPP did not lead to the expected product (entries 7–10). A series of control experiments demonstrated that the reaction did not take place in the absence of light, catalyst, or 3. With the optimized conditions in hand (acetonitrile, rt, 1 mol % PC 3), we began to evaluate a variety of substrates (Table 1).
Table 1. Scope of the Reactiona–c.
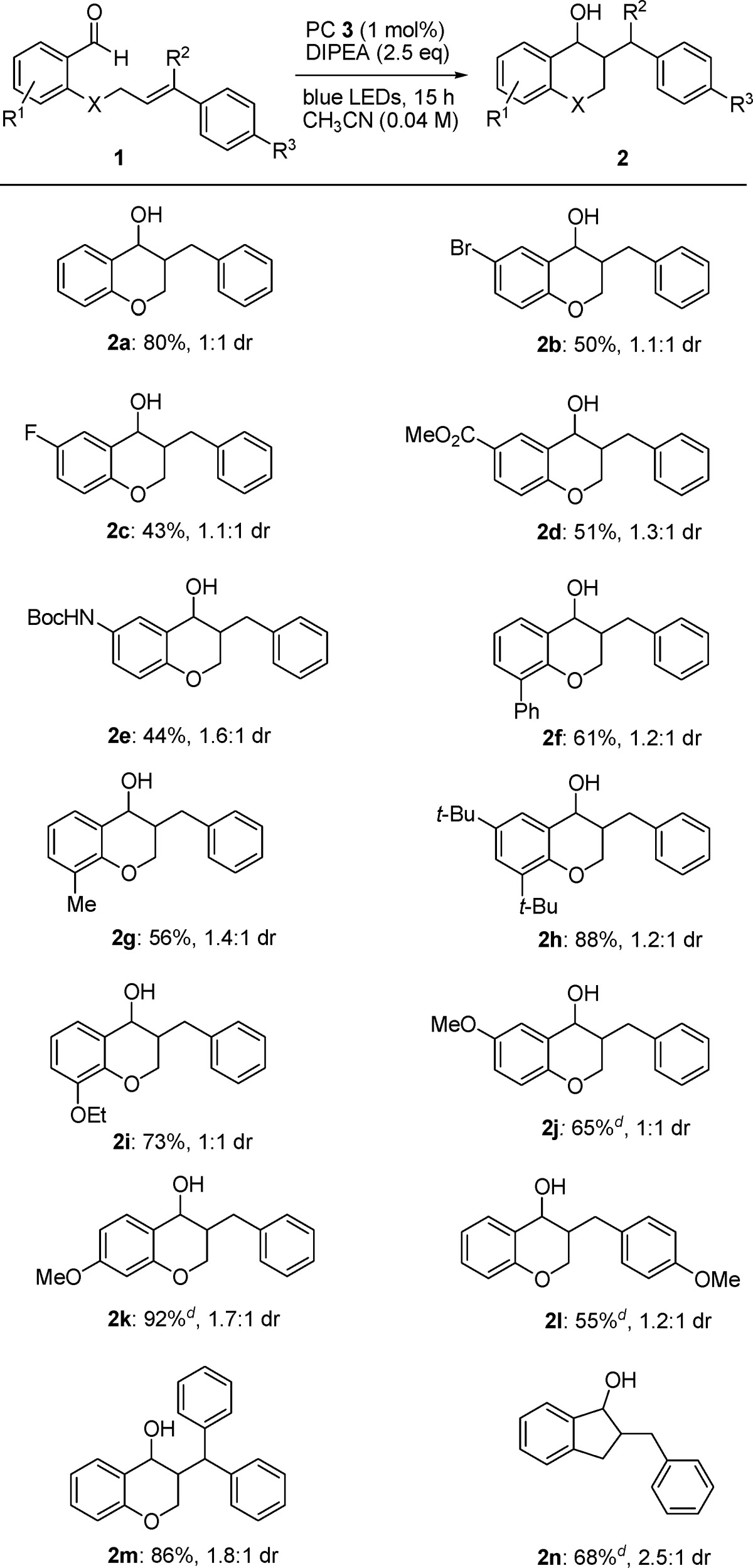
All reactions of 1 (0.12 mmol) with DIPEA (0.3 mmol) were carried out in the presence of PC 3 (1 mol %) in acetonitrile (3.0 mL) under irradiation with blue LEDs (11 W, 450 nm) for 15 h at 25 °C.
Isolated yields are reported.
The dr is given for anti/syn ratio.
Reaction performed using 2.5 mol % of photocatalyst.
As shown in Table 1, the reaction tolerates a wide variety of substituents, confirming the ability of the ketyl radical to couple to the olefin in the presence of both electron-withdrawing and -donating groups on the aromatic aldehyde moiety.
Following the development of a successful formal hydroacylation we investigated the reaction in which the olefin was replaced by an alkyne group (Table 2). Also in this case, the reaction gave the expected products (2o and 2p) in good yields.
Table 2. Scope of the Intramolecular Ketyl–Alkyne Cyclizationa.
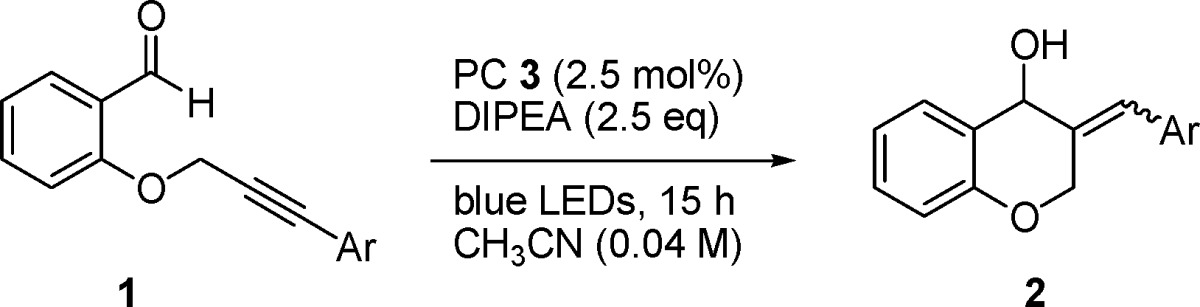
| entry | product | yieldb (%) | E/Z |
|---|---|---|---|
| 1 | Ar = m-MePh (2o) | 57 | 1:1 |
| 2 | Ar = p-OMePh (2p) | 59 | 1:1 |
All reactions of 1 (0.12 mmol) with DIPEA (0.3 mmol) were carried out in the presence of PC 3 (2.5 mol %) in acetonitrile (3.0 mL) under irradiation with blue LEDs (11 W, 450 nm) for 18 h at 25 °C.
Isolated yields are reported.
Regarding the reaction mechanism, we propose the following: Irradiation with visible light results in the formation of the reductive species Ir2+. Hünig base (i-Pr2NEt) acts as a sacrificial electron donor; therefore, the single-electron oxidation weakens the adjacent C–H bond17 and the 1,2-H-shift becomes energetically favored. The following deprotonation through the tertiary amine provides the ammonium derivative, which is now able to engage in a proton-coupled electron transfer with the substrate, lowering its potential for Ir2+ reduction and generation of a neutral ketyl intermediate. Alternatively, the ammonium radical could also activate the carbonyl functionality. The ketyl radical is able to add to the unsaturated bond, forming the chroman ring and a benzyl radical which upon hydrogen atom transfer leads to the final product (Scheme 2).
Scheme 2. Plausible Reaction Mechanism.

Conclusions
In conclusion, we report a ketyl–olefin coupling for the preparation of substituted 3-benzylchroman-4-ols, promoted by visible light photoredox catalysis. Importantly, we uncovered trialkylamines as a cheap and readily available alternative to the previously reported electron–proton donor system consisting of Hantzsch ester/Brønsted acid. By employing the trialkylamine/photocatalyst system, we have been able to develop an efficient intramolecular ketyl–olefin and ketyl–alkyne coupling employing readily prepared substrates. The formal hydroacylation protocol provides the chromanol derivatives in good yield under mild reaction conditions and with low catalyst loadings. Efforts are currently underway to expand this concept to further transformations.
Experimental Section
General Information
All reactions were performed with oven-dried glassware and under an inert atmosphere (argon) unless otherwise stated. Acetonitrile was distilled from calcium hydride and stored over 4 Å molecular sieves under nitrogen/argon atmosphere. Tetrahydrofuran was distilled from Solvona/benzophenone. Pentane was distilled in a standard distillation apparatus. Other solvents were used as purchased unless otherwise stated. Commercial reagents were used as purchased without further purification. Organic solutions were concentrated under reduced pressure on a rotary evaporator. Chromatographic purification of products was carried out using silica gel (230–400 mesh). Thin-layer chromatography was carried out using aluminum plates coated with silica (230–400 mesh) which were visualized under UV light (254 nm) or by staining with aqueous potassium permanganate solutions or vanillin alcoholic solution. 1H NMR spectra were recorded in deuterated solvents on spectrometers at 300, 400, or 600 MHz with residual protic solvent as the internal standard. 13C NMR spectra were recorded in deuterated solvents on spectrometers at 75, 101, or 151 MHz with the central peak of the deuterated solvent as the internal standard. Chemical shifts (δ) are given in parts per million (ppm), and coupling constants (J) are given in hertz (Hz) rounded to the nearest 0.1 Hz. The 1H NMR spectra are reported as δ downfield from tetramethylsilane (multiplicity, coupling constant J, number of protons). The 13C NMR spectra are reported as δ. For the F-containing compound (2c), a simultaneously proton and fluorine decoupled 13C NMR spectrum was recorded. Assignments are aided by the use of DEPT-135, COSY and HMQC spectra where necessary. IR spectra were recorded on a FTIR spectrometer. Low resolution mass spectra (EI/CI) were recorded on a mass spectrometer with (EI) or (CI) detectors. High-resolution mass spectrometry (HRMS) was performed by EI using a double-focusing mass spectrometer and by ESI using a hybrid ion-trap mass spectrometer. Melting points were recorded at ambient pressure and are uncorrected.
General Procedure for the Synthesis of Chromanols
A Schlenk tube was charged with aldehyde 1 (1 equiv), catalyst PC 3 (1 or 2.5 mol %), and degassed DIPEA (2.5 equiv). It was capped, evacuated, and backfilled with argon. Subsequently, degassed acetonitrile (3 mL) was added via syringe. The vial was placed in a 100 mL beaker containing a blue LED strip glued on the inner wall, and the reaction mixture was stirred for 15 h. After this time, the solvent was evaporated, and the reaction mixture was purified on silica gel (gradient pentane/EtOAc 95:5).
3-Benzylchroman-4-ol (2a)18
The title compound was synthesized according to the general procedure employing 1a (0.12 mmol, 28.6 mg), 1 mol % (1.1 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 80% (23 mg); anti/syn 1:1; 1H NMR (600 MHz, CDCl3) δ 7.34–7.18 (m, 14H), 6.96 (td, J = 7.5, 0.9 Hz, 1H), 6.90 (dd, J = 10.6, 4.2 Hz, 2H), 6.85 (d, J = 8.5 Hz, 1H), 4.51 (dd, J = 9.4, 3.1 Hz, 2H), 4.23 (dd, J = 11.1, 2.6 Hz, 1H), 4.14–4.05 (m, 2H), 3.98 (dd, J = 11.1, 4.2 Hz, 1H), 2.89 (dd, J = 13.8, 8.4 Hz, 1H), 2.75–2.64 (m, 2H), 2.54 (dd, J = 13.8, 9.3 Hz, 1H), 2.40–2.29 (m, 1H), 2.28–2.17 (m, 1H), 1.92 (bs, 1H), 1.74 (bs, 1H); 13C NMR (151 MHz, CDCl3) δ 154.3, 154.2, 139.2, 139.1, 130.2, 130.1, 129.9, 129.8, 129.1 (2C), 128.6, 128.5, 126.3 (2C), 124.1, 123.2, 120.9, 120.5, 116.9 (2C), 67.6, 64.9 (2C), 64.6, 41.5, 40.0, 34.6, 32.8; FT-IR νmax(ATR) cm–1 3372, 2922, 1582, 1485, 1450, 1302, 1264, 1222, 1119, 1074, 1039, 909, 748, 698; m/z (EI) 240 ([M]+, 100), 91 ([PhCH2]+, 28); HRMS (ESI) calcd for [C16H16O2 + Na]+ 263.10425, found 263.10419.
3-Benzyl-6-bromochroman-4-ol (2b)
The title compound was synthesized according to the general procedure employing 1b (0.12 mmol, 38.1 mg), 2.5 mol % (2.74 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 50% (19.1 mg); anti/syn 1.1:1; 1H NMR (400 MHz, CD3OD) δ 7.45 (d, J = 2.4 Hz, 1H, syn), 7.34 (d, J = 3.1 Hz, 1.1 × 1H, anti), 7.31–7.13 (m, 12.6H), 6.73–6.65 (m, 2.1H), 4.47 (d, J = 3.4 Hz, 1H, syn), 4.36 (d, J = 5.0 Hz, 1.1 × 1H, anti), 4.13 (dd, J = 11.2, 2.8 Hz, 1.1 × 1H, anti), 4.03–3.94 (m, 2H, syn), 3.89 (dd, J = 11.1, 5.5 Hz, 1.1 × 1H, anti), 2.87 (dd, J = 13.6, 7.1 Hz, 1H, syn), 2.76 (dd, J = 13.8, 6.1 Hz, 1H, syn), 2.55 (dd, J = 13.6, 8.3 Hz, 1.1 × 1H, anti), 2.45 (dd, J = 13.7, 9.3 Hz, 1.1 × 1H, anti), 2.25–2.17 (m, 1H, syn), 2.16–2.08 (m, 1.1 × 1H, anti); 13C NMR (101 MHz, CD3OD) δ 153.5 (2C), 139.4, 139.2, 132.4 (2C), 131.60 (2C), 128.8, 128.7, 128.1 (2C), 127.0, 126.1, 126.0, 125.8, 118.1 (2C), 111.9, 111.5, 66.4, 65.0, 64.8, 64.2, 41.5, 40.2, 34.2, 32.1; FT-IR νmax(ATR) cm–1 2926, 1479, 1407, 1260, 1224, 1191, 1085, 1055, 1021, 819, 748, 699; m/z (EI) 319 ([M]+, 9), 91 ([PhCH2]+, 100); HRMS (EI) calcd for [C16H15O279Br]+ 318.02499, found 318.02492.
3-Benzyl-6-fluorochroman-4-ol (2c)
The title compound was synthesized according to the general procedure employing 1c (0.12 mmol, 30.8 mg), 1 mol % (1.1 mg) of PC 3 and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 43% (13.2 mg); mp =102–104 °C; anti/syn 1.1:1; 1H NMR (600 MHz, CD3OD) δ 7.31–7.23 (m, 6.5 H), 7.21–7.15 (m, 4.3 H), 7.06 (dd, J = 9.0, 3.1 Hz, 1.1 × 1H, anti), 6.96 (dd, J = 8.9, 3.1 Hz, 1H, syn), 6.89 (dtd, J = 11.6, 8.5, 3.1 Hz, 2H), 6.78–6.71 (m, 2H), 4.48 (d, J = 3.5 Hz, 1H, syn), 4.37 (d, J = 5.1 Hz, 1.1 × 1H, anti), 4.12 (dd, J = 11.1, 2.8 Hz, 1.1 × 1H, anti), 4.01–3.94 (m, 2H, syn), 3.87 (dd, J = 11.1, 5.6 Hz, 1.1 × 1H, anti), 2.88 (dd, J = 13.6, 7.2 Hz, 1H, syn), 2.79 (dd, J = 13.8, 6.0 Hz, 1.1 × 1H, anti), 2.56 (dd, J = 13.6, 8.3 Hz, 1H, syn), 2.46 (dd, J = 13.8, 9.4 Hz, 1.1 × 1H, anti), 2.24–2.17 (m, 1H, syn), 2.16–2.11 (m, 1.1 × 1H, anti); 19F NMR (564 MHz, CD3OD) δ −125.8, −126.4; 13C NMR (101 MHz, CD3OD) δ 156.9, 156.6, 150.4, 150.3, 139.5, 139.2, 128.8, 128.7, 128.1, 128.0, 125.9 (2C), 125.8, 125.0, 117.2, 117.1, 115.5, 115.4, 115.3 (2C), 66.8, 65.0, 64.7, 64.4, 41.6, 40.3, 34.2, 32.1; FT-IR νmax(ATR) cm–1 3370, 2926, 2493, 1488, 1436, 1251, 1201, 1027, 736, 698; m/z (EI) 258 ([M]+, 48), 91 ([PhCH2]+, 100); HRMS (EI) calcd for [C16H15O2F]+ 258.10506, found 258.10537.
Methyl 3-Benzyl-4-hydroxychroman-6-carboxylate (2d)
The title compound was synthesized according to the general procedure employing 1d (0.12 mmol, 35.6 mg), 1 mol % (1.1 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 51% (18.2 mg); anti/syn 1.3:1; 1H NMR (400 MHz, CD3OD) δ 8.05 (d, J = 2.1 Hz, 1.3 × 1H, anti), 7.92 (d, J = 2.2 Hz, 1H, syn), 7.80 (ddd, J = 14.4, 8.6, 2.2 Hz, 2.3H), 7.32–7.11 (m, 11.5H), 6.81 (dd, J = 14.4, 8.6 Hz, 2.3H), 4.52 (d, J = 3.2 Hz, 1H, syn), 4.42 (d, J = 4.9 Hz, 1.3 × 1H, anti), 4.21 (dd, J = 11.2, 2.9 Hz, 1.3 × 1H, anti), 4.11–3.99 (m, 2H, syn), 3.96 (dd, J = 11.1, 5.3 Hz, 1.3 × 1H, anti), 3.84 (s, 1.3 × 3H, anti), 3.81 (s, 3H, syn), 2.88 (dd, J = 13.6, 7.1 Hz, 1H, syn), 2.75 (dd, J = 13.7, 6.1 Hz, 1.3 × 1H, anti), 2.56 (dd, J = 13.6, 8.3 Hz, 1H, syn), 2.43 (dd, J = 13.7, 9.3 Hz, 1.3 × 1H, anti), 2.26–2.13 (m, 2.3H); 13C NMR (101 MHz, CD3OD) δ 167.0 (2C), 158.6 (2C), 139.4, 139.1, 132.3, 132.2, 130.4, 130.3, 128.8, 128.7, 128.1 (2C), 126.0, 125.9, 124.8, 123.9, 122.1, 121.7, 116.3 (2C), 66.4, 65.4, 65.2, 64.2, 51.0, 50.9, 41.4, 40.2, 34.2, 32.2; FT-IR νmax(ATR) cm–1 2949, 1711, 1611, 1496, 1436, 1253, 1192, 1125, 1012, 767, 700; m/z (EI) 298 ([M]+, 12), 91 ([PhCH2]+, 40); HRMS (EI) calcd for [C18H18O4]+ 298.11996, found 298.12009.
tert-Butyl (3-Benzyl-4-hydroxychroman-6-yl)carbamate (2e)
The title compound was synthesized according to the general procedure employing 1e (0.12 mmol, 42.4 mg), 1 mol % (1.1 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 44% (18.7 mg); anti/syn 1.6:1; 1H NMR (600 MHz, CDCl3) δ 7.49–7.15 (m, 15.6H), 7.08 (dd, J = 8.8, 2.6 Hz, 1.6 × 1H, anti), 6.99 (dd, J = 8.8, 2.6 Hz, 1H, syn), 6.80 (d, J = 8.8 Hz, 1.6 × 1H, anti), 6.76 (d, J = 8.8 Hz, 1H, syn), 6.45–6.24 (m, 2.6H), 4.52–4.41 (m, 2.6H), 4.19 (dd, J = 11.0, 1.9 Hz, 1.6 × 1H, anti), 4.12–4.01 (m, 2H, syn), 3.92 (dd, J = 11.0, 3.9 Hz, 1.6 × 1H, anti), 2.86 (dd, J = 13.5, 8.6 Hz, 1H, syn), 2.73–2.61 (m, 2.6H), 2.51 (dd, J = 13.7, 9.5 Hz, 1.6 × 1H, anti), 2.31–2.26 (m, 1H, syn), 2.21–2.17 (m, 1.6 × 1H, anti), 1.91–1.62 (overlapping of two bs, 2.6H) 1.51 (s, 1.6 × 9H, anti), 1.48 (s, 9H, syn); 13C NMR (151 MHz, CDCl3) δ 153.2, 153.1, 150.3, 150.2, 139.2, 139.1, 131.4, 131.3, 131.0 (2C), 129.1 (2C), 128.5 (2C), 126.3 (2C), 124.4, 123.5, 121.2, 120.7, 117.2, 117.1, 80.4 (2C), 67.6, 65.1, 64.8, 64.6, 41.3, 40.0, 34.6, 32.7, 28.4, 28.3; FT-IR νmax(ATR) cm–1: 3350, 2981, 2932, 1696, 1499, 1454, 1367, 1309, 1241, 1218, 1163, 1023, 822, 733, 699; m/z (EI) 298 ([M – (CH3)3C]+, 100), 91 ([PhCH2]+, 67); HRMS (EI) calcd for [C16H16O2N1]+ ([M – Boc]+) 254.11756, found 254.11706.
3-Benzyl-8-phenylchroman-4-ol (2f)
The title compound was synthesized according to the general procedure employing 1f (0.12 mmol, 37.7 mg), 1 mol % (1.1 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 61% (23 mg); anti/syn 1.2:1; 1H NMR (600 MHz, CD3OD) δ7.48–7.44 (m, 1.2 × 1H, anti), 7.44–7.40 (m, 1H, syn), 7.37–7.05 (m, 24.2H), 7.00–6.94 (m, 1.2 × 1H, anti), 6.92–6.86 (m, 1H, syn), 4.49 (d, J = 2.7 Hz, 1H, syn), 4.44 (d, J = 4.1 Hz, 1.2 × 1H, anti), 4.15 (dd, J = 11.0, 2.6 Hz, 1.2 × 1H, anti), 4.00 (t, J = 10.9 Hz, 1H, syn), 3.92 (dd, J = 10.6, 3.5 Hz, 1H, syn), 3.86 (dd, J = 11.0, 4.5 Hz, 1.2 × 1H, anti), 2.85 (dd, J = 13.5, 7.5 Hz, 1H, syn), 2.70 (dd, J = 13.7, 6.4 Hz, 1.2 × 1H, anti), 2.55 (dt, J = 18.0, 9.0 Hz, 1H, syn), 2.51–2.44 (m, 1.2 × 1H, anti), 2.25–2.18 (m, 1H, syn), 2.17–2.12 (m, 1.2 × 1H, anti); 13C NMR (151 MHz, CD3OD) δ 151.1 (2C), 139.6, 139.4, 138.5, 138.4, 130.2, 130.1, 129.8 (2C), 129.7 (2C), 129.2 (2C), 128.8, 128.7, 128.1 (2C), 127.5 (2C), 126.4 (2C), 125.9, 125.8, 125.1, 123.9, 120.2, 119.9, 67.1, 64.8, 64.6, 64.5, 41.5, 40.3, 34.3, 32.5; FT-IR νmax(ATR) cm–1 1594, 1496, 1469, 1430, 1219, 1017, 754, 697; m/z (EI) 316 ([M]+, 63%), 91 ([PhCH2]+, 27%); HRMS (ESI) calcd for [C22H20O2 + Na]+ 339.13555, found 339.13525.
3-Benzyl-8-methylchroman-4-ol (2g)
The title compound was synthesized according to the general procedure employing 1g (0.12 mmol, 30.3 mg), 1 mol % (1.1 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 56% (17.1 mg); anti/syn 1.4:1; 1H NMR (600 MHz, CD3OD) δ 7.30–7.21 (m, 7.5H), 7.20–7.12 (m, 5.5H), 7.05–7.02 (m, 2.4H), 6.99 (d, J = 7.3 Hz, 1.4 × 1H, anti), 6.80 (t, J = 7.5 Hz, 1H, syn), 6.73 (t, J = 7.5 Hz, 1.4 × 1H, anti), 4.45 (d, J = 3.2 Hz, 1.4 × 1H, anti), 4.38 (d, J = 4.2 Hz, 1H, syn), 4.19 (dd, J = 11.0, 2.6 Hz, 1H, syn), 4.06–4.02 (m, 1.4 × 2H, anti), 3.94 (dd, J = 11.0, 4.6 Hz, 1H, syn), 2.87 (dd, J = 13.7, 7.4 Hz, 1.4 × 1H, anti), 2.68 (dd, J = 13.7, 6.5 Hz, 1H, syn), 2.58 (dd, J = 13.6, 8.1 Hz, 1.4 × 1H, anti), 2.45 (dd, J = 13.7, 9.1 Hz, 1H, syn), 2.16 (s, 3H, syn), 2.12 (s, 1.4 × 3H, anti) OH signals not observed; 13C NMR (151 MHz, CD3OD) δ 152.3, 152.2, 139.7, 139.5, 129.9, 129.8, 128.8, 128.7, 128.1 (2C), 127.9, 127.8, 125.8 (2C), 125.2 (2C), 123.9, 122.7, 119.6, 119.3, 66.9, 64.7, 64.6, 64.4, 41.8, 40.6, 34.3, 32.6, 14.8 (2C); FT-IR νmax(ATR) cm–1 3352, 2919, 1598, 1472, 1258, 1210, 1092, 1023, 747, 698; m/z (EI) 254 ([M]+, 59), 253 ([M – 1], 100), 91 ([PhCH2]+, 39); HRMS (EI) calcd for [C17H18O2]+ 254.13013, found 254.13046.
3-Benzyl-6,8-di-tert-butylchroman-4-ol (2h)
The title compound was synthesized according to the general procedure employing 1h (0.12 mmol, 42.1 mg), 1 mol % (1.1 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 88% (37.5 mg); anti/syn 1.2:1; 1H NMR (400 MHz, CD3OD) δ 7.33–7.07 (m, 15.4H), 4.48 (d, J = 3.5 Hz, 1H, syn), 4.39 (d, J = 3.3 Hz, 1.2 × 1H, anti), 4.13 (dd, J = 10.9, 2.5 Hz, 1.2 × 1H, anti), 4.03–3.94 (m, 2H, syn), 3.90 (ddd, J = 10.9, 4.0, 1.1 Hz, 1.2 × 1H, anti), 2.88 (dd, J = 13.5, 7.2 Hz, 1H, syn), 2.67 (dd, J = 13.6, 6.0 Hz, 1.2 × 1H, anti), 2.57 (dd, J = 13.5, 8.3 Hz, 1H, syn), 2.42 (dd, J = 13.6, 9.8 Hz, 1.2 × 1H, anti), 2.23–2.15 (m, 1H, syn), 2.14–2.08 (m, 1.2 × 1H, anti), 1.37 (s, 1.2 × 9H, anti), 1.33 (s, 9H, syn), 1.29 (s, 1.2 × 9H, anti), 1.25 (s, 9H, syn); 13C NMR (101 MHz, CD3OD) δ 150.6 (2C), 141.8, 141.5, 139.8, 139.6, 136.3, 136.1, 128.8 (2C), 128.0 (2C), 125.8, 125.7, 125.2, 124.7, 124.0, 123.1, 123.0, 122.4, 67.7, 65.5, 63.9, 63.0, 41.7, 40.6, 34.5, 34.5, 34.3, 33.7, 33.7, 32.5, 30.7 (2C), 28.9 (2C); FT-IR νmax(ATR) cm–1 2956, 2870, 1479, 1450, 1361, 1229, 1171, 1130, 1028, 882, 749, 701, 532; m/z (EI) 352 ([M]+, 65), 91 ([PhCH2]+, 15); HRMS (EI) calcd for [C24H32O2]+ 352.23968, found 352.23972.
3-Benzyl-8-ethoxychroman-4-ol (2i)
The title compound was synthesized according to the general procedure employing 1i (0.12 mmol, 33.8 mg), 1 mol % (1.1 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 73% (25 mg); anti/syn 1:1; 1H NMR (600 MHz, CDCl3) δ 7.36–7.14 (m, 10H), 6.95–6.77 (m, 6H), 4.48–4.47 (m, 2H), 4.29 (dd, J = 11.1, 2.7 Hz, 1H), 4.22 (ddd, J = 10.7, 3.7, 1.2 Hz, 1H), 4.15–4.04 (m, 6H), 2.88 (dd, J = 13.7, 8.4 Hz, 1H), 2.70 (dd, J = 14.0, 7.0 Hz, 1H), 2.68 (dd, J = 13.7, 7.2 Hz, 1H), 2.59 (dd, J = 13.9, 8.5 Hz, 1H), 2.35–2.29 (m, 1H), 2.22 (dqd, J = 12.9, 4.3, 2.8 Hz, 1H), 1.97 (bs, 2H), 1.47–1.45 (m, 6H); 13C NMR (151 MHz, CDCl3) δ 147.6, 147.5, 144.1, 144.0, 139.2, 139.1, 129.1 (2C), 128.5 (2C), 126.3 (2C), 125.0, 124.0, 121.8, 121.6, 120.4, 120.1, 112.6 (2C), 67.4, 65.4, 65.2, 64.7, 64.4, 64.3, 41.3, 39.8, 34.6, 32.9, 14.8 (2C); FT-IR νmax(ATR) cm–1 3506, 2921, 1585, 1478, 1391, 1315, 1255, 1214, 1080, 1030, 964, 900, 737; m/z (EI) 284 ([M]+, 23), 91 ([PhCH2]+, 100); HRMS (EI) calcd for [C18H20O3]+ 284.14070, found 284.14077.
3-Benzyl-6-methoxychroman-4-ol (2j)
The title compound was synthesized according to the general procedure employing 1j (0.12 mmol, 32.2 mg), 1 mol % (1.1 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 65% (21 mg); mp = 112–113 °C; anti/syn 1:1; 1H NMR (400 MHz, CD3OD) δ 7.30–7.13 (m, 10H), 6.88 (d, J = 3.0 Hz, 1H), 6.80–6.63 (m, 5H), 4.46 (d, J = 3.5 Hz, 1H), 4.36 (d, J = 4.6 Hz, 1H), 4.09 (dd, J = 11.0, 2.7 Hz, 1H), 3.99–3.89 (m, 2H), 3.84 (ddd, J = 11.0, 5.1, 0.7 Hz, 1H), 3.73 (s, 3H), 3.70 (s, 3H), 2.87 (dd, J = 13.6, 7.2 Hz, 1H), 2.74 (dd, J = 13.7, 6.2 Hz, 1H), 2.56 (dd, J = 13.6, 8.3 Hz, 1H), 2.47 (dd, J = 13.7, 9.3 Hz, 1H), 2.23–2.15 (m, 1H), 2.14–2.07 (m, 1H); 13C NMR (101 MHz, CD3OD) δ 153.7, 153.4, 148.2, 148.1, 139.7, 139.5, 128.8, 128.7, 128.0 (2C), 125.8, 125.7, 124.9, 123.9, 116.7 (2C), 115.5 (2C), 113.9, 113.8, 67.1, 64.8, 64.5 (2C), 54.7 (2C), 42.0, 40.7, 34.3, 32.3; FT-IR νmax(ATR) cm–1 2923, 2471, 1490, 1430, 1265, 1205, 1161, 1037, 807, 703; m/z (EI) 270 ([M]+, 71), 91 ([PhCH2]+, 33); HRMS (EI) calcd for [C17H18O3]+ 270.12505, found 270.12551.
3-Benzyl-7-methoxychroman-4-ol (2k)19
The title compound was synthesized according to the general procedure employing 1k (0.12 mmol, 32.2 mg), 2.5 mol % (2.74 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 92% (29.8 mg); mp = 100–103 °C; anti/syn 1.7:1; 1H NMR (400 MHz, CD3OD) δ 7.42–7.21 (m, 7.2H), 7.20–7.12 (m, 8H), 7.09 (d, J = 8.5 Hz, 1H), 6.51 (dd, J = 8.5, 2.5 Hz, 1.7 × 1H, anti), 6.44 (dd, J = 8.5, 2.5 Hz, 1H, syn), 6.35 (d, J = 2.5 Hz, 1.7 × 1H, anti), 6.30 (d, J = 2.5 Hz, 1H, syn), 4.41 (d, J = 3.1 Hz, 1H, syn), 4.34 (d, J = 3.9 Hz, 1.7 × 1H, anti), 4.15 (dd, J = 11.0, 2.6 Hz, 1.7 × 1H, anti), 4.11–3.93 (m, 2H, syn), 3.89 (ddd, J = 11.0, 4.2, 0.9 Hz, 1.7 × 1H, anti), 3.72 (s, 3H, anti), 3.69 (s, 3H, syn), 2.85 (dd, J = 13.6, 7.4 Hz, 1H, syn), 2.66 (dd, J = 13.7, 6.6 Hz, 1.7 × 1H, anti), 2.57 (dd, J = 13.6, 8.0 Hz, 1H, syn), 2.46 (dd, J = 13.7, 9.1 Hz, 1.7 × 1H, anti), 2.23–2.14 (m, 1H, syn), 2.14–2.03 (m, 1.7 × 1H, anti); 13C NMR (101 MHz, CD3OD) δ 160.7 (2C), 155.2 (2C), 139.6, 139.5, 131.1, 130.9, 128.8, 128.7, 128.0 (2C), 125.8, 125.7, 117.1, 115.6, 107.3, 106.8, 100.5 (2C), 66.3, 64.5, 64.2, 64.1, 54.3, 54.2, 42.0, 40.7, 34.2, 32.6; FT-IR νmax(ATR) cm–1: 3215, 2960, 1737, 1667, 1494, 1366, 1221, 1092, 1032, 734; m/z (EI) 270 ([M]+, 15), 91 ([PhCH2]+, 89); HRMS (EI) calcd for [C17H18O3]+ 270.12505, found 270.12534.
3-(4-methoxybenzyl)chroman-4-ol (2l)20
The title compound was synthesized according to the general procedure employing 1l (0.12 mmol, 32.2 mg), 2.5 mol % (2.74 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 55% (17.7 mg); mp =121–126 °C; anti/syn 1.2:1; 1H NMR (600 MHz, CDCl3) δ 7.32 (dd, J = 7.6, 1.4 Hz, 1.2 × 1H, anti), 7.25–7.17 (m, 5.4H), 7.11–7.08 (m, 1.2 × 1H, anti), 6.95 (td, J = 7.5, 0.9 Hz, 1.2 × 1H, anti), 6.91–6.82 (m, 7.4H), 4.50 (t, J = 3.5 Hz, 1H, syn), 4.48 (t, J = 4.1 Hz, 1.2 × 1H, anti), 4.21 (dd, J = 9.2, 4.6 Hz, 1.2 × 1H, anti) 4.11–4.06 (m, 2H, syn), 3.97 (dd, J = 11.0, 4.0 Hz, 1.2 × 1H, anti), 3.80 (s, 3H, syn), 3.79 (s, 1.2 × 3H, anti), 2.82 (dd, J = 13.8, 8.5 Hz, 1H, syn), 2.68–2.58 (m, 2.2H), 2.48 (dd, J = 14.0, 9.3 Hz, 1.2 × 1H, anti), 2.32–2.24 (m, 1H, syn), 2.21–2.14 (m, 1.2 × 1H, anti), 1.91 (bs, 1.2 × 1H, anti), 1.72 (bs, 1H, syn); 13C NMR (151 MHz, CDCl3) δ 158.1 (2C), 154.3, 154.2, 131.1, 131.0, 130.1 (2C), 130.0 (2C), 129.9, 129.7, 124.2, 123.2, 120.9, 120.5, 116.9 (2C), 114.0, 113.9, 67.6, 65.0, 64.9, 64.6, 55.3 (2C), 41.6, 40.1, 33.7, 31.9; FT-IR νmax(ATR) cm–1: 3392, 2928, 1609, 1582, 1510, 1486, 1450, 1298, 1246, 1176, 1031, 850, 813, 753; m/z (EI) 270 ([M]+, 27), 121 ([4-MeOPhCH2]+, 100); HRMS (EI) calcd for [C17H18O3]+ 270.12505, found 270.12548.
3-Benzhydrylchroman-4-ol (2m)
The title compound was synthesized according to the general procedure employing 1m (0.12 mmol, 37.7 mg), 1 mol % (1.1 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 86% (32.7 mg); anti/syn 1.8:1; 1H NMR (600 MHz, CDCl3) δ 7.43 (d, J = 7.2 Hz, 2H), 7.39 (d, J = 7.2 Hz, 2H), 7.35–7.18 (m, 26.7H), 7.12–7.09 (m, 3H), 6.97–6.85 (m, 5.5H), 4.44 (bs, 1H, syn), 4.41 (bs, 1.8 × 1H, anti), 4.27 (dd, J = 11.2, 2.3 Hz, 1.8 × 1H, anti), 4.14–4.06 (m, 2H, syn), 4.00 (ddd, J = 10.8, 3.5, 1.5 Hz, 1H, syn), 3.96–3.93 (m, 1.8 × 1H, anti), 3.70 (d, J = 12.4 Hz, 1.8 × 1H, anti), 2.91 (ddd, J = 11.9, 7.5, 3.2 Hz, 1H, syn), 2.79 (dq, J = 12.4, 2.3 Hz, 1.8 × 1H, anti), 2.00 (bs, 1.8 × 1H, anti), 1.80 (bs, 1H, syn); 13C NMR (151 MHz, CDCl3) δ 154.4 (2C), 142.6, 142.4, 142.3 (2C), 130.9, 130.5, 130.0, 129.9, 128.9 (2C), 128.8 (2C), 128.1 (2C), 127.9, 127.8, 126.8, 126.7, 126.6, 126.6, 124.0, 122.5, 121.0, 120.5, 117.1, 116.9, 65.8, 64.1, 64.0, 63.1, 49.6, 49.2, 43.0, 41.9; FT-IR νmax(ATR) cm–1: 3036, 2927, 1702, 1619, 1516, 1491, 1446, 1318, 1226, 1117, 1014, 972, 755; m/z (EI) 167 ([CHPh2]+, 59), 151 ([M – CPh2]+, 28); HRMS (EI) calcd for [C22H20O2]+ 316.14578, found 316.14636.
2-Benzyl-2,3-dihydro-1H-inden-1-ol (2n)
The title compound was synthesized according to the general procedure employing 1n (0.12 mmol, 26.7 mg), 2.5 mol % (2.74 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 68% (14.6 mg); anti/syn 2.5:1; 1H NMR (600 MHz, CDCl3) δ 7.41–7.35 (m, 4H), 7.35–7.29 (m, 10H), 7.28–7.21 (m, 14.5H), 7.19–7.15 (m, 3H), 5.00 (d, J = 5.4 Hz, 1H, syn), 4.95 (d, J = 6.6 Hz, 2.5 × 1H, anti), 3.14–3.07 (m, 4.5H), 3.01 (dd, J = 15.3, 7.3 Hz, 3H), 2.87–2.83 (m, 2.5H), 2.81–2.75 (m, 4H), 2.68 (dq, J = 14.0, 7.9 Hz, 1H, syn), 2.62–2.48 (m, 6H), 1.62 (bs, 3.5H); 13C NMR (151 MHz, CDCl3) δ 144.7, 144.5, 143.5, 141.5, 141.2, 140.6, 128.9 (2C), 128.7, 128.5, 128.4, 128.2, 126.8 (2C), 126.2, 125.9, 125.0, 124.8, 124.7, 123.9, 80.8, 76.3, 52.3, 46.9, 39.3, 35.9, 35.8, 35.0; FT-IR νmax(ATR) cm–1 3363, 2932, 1487, 1454, 1257, 1102, 1026, 804, 745, 697; m/z (EI) 223 ([M – 1]+, 21), 132 (M – [PhCH2]+, 100) 91 ([PhCH2]+, 80); HRMS (EI) calcd for [C16H16O]+ 224.11957, found 224.11989.
3-(3-Methylbenzylidene)chroman-4-ol (2o)
The title compound was synthesized according to the general procedure employing 1o (0.12 mmol, 30 mg), 2.5 mol % (2.74 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 59% (17.7 mg); E/Z 1:1; 1H NMR (400 MHz, CDCl3) δ 7.41 (dd, J = 7.7, 1.6 Hz, 1H), 7.31–7.18 (m, 6H), 7.16–7.07 (m, 2H), 7.06–7.00 (m, 2H), 7.03–6.8 (m, 6H), 6.77 (s, 1H), 5.32 (s, 1H), 5.19 (s, 1H), 4.97–4.86 (m, 3H), 4.57 (dd, J = 12.0, 0.9 Hz, 1H), 2.37 (s, 3H), 2.35 (s, 3H), 2.14 (bs, 1H), 2.05 (bs, 1H); 13C NMR (101 MHz, CDCl3) δ 154.4, 154.3, 138.1 (2C), 135.5, 135.4, 134.4, 133.4, 131.6, 130.3, 130.0, 129.8, 129.5 (2C), 129.2, 129.0, 128.7, 128.4, 128.3 (2C), 125.9, 125.8, 124.9, 124.0, 121.1 (2C), 117.2, 116.9, 69.3, 68.1, 63.0, 62.8, 21.4 (2C); FT-IR νmax(ATR) cm–1 3391, 2922, 1737, 1590, 1479, 1370, 1224, 993, 921, 756, 702; m/z (EI) 252 ([M]+, 17), 104 ([3-MePh]+, 25), 91 ([3-MePhCH]+, 32); HRMS calcd for [C17H16O2]+ 252.11448, found 252.11402.
3-(4-Methoxybenzylidene)chroman-4-ol (2p)21
The title compound was synthesized according to the general procedure employing 1p (0.12 mmol, 32 mg), 2.5 mol % (2.74 mg) of PC 3, and 2.5 equiv of DIPEA (38.8 mg). The product was purified by flash column chromatography: yield 57% (18.3 mg), E/Z 1:1; 1H NMR (600 MHz, CDCl3) δ 7.46–7.43 (m, 2H), 7.42–7.39 (m, 1H), 7.30–7.28 (m, 1H), 7.24–7.20 (m, 2H), 7.20–7.16 (m, 2H), 6.99–6.84 (m, 9H), 6.76 (s, 1H), 5.33 (s, 1H), 5.18 (s, 1H), 4.99–4.89 (m, 2H), 4.87 (dd, J = 12.0, 1.2 Hz, 1H), 4.59–4.55 (m, 1H), 3.83 (s, 3H), 3.82 (s, 3H), 2.20 (bs, 1H), 2.07 (bs, 1H); 13C NMR (151 MHz, CDCl3) δ 159.3, 159.1, 154.4, 154.3, 133.3, 133.2, 131.9, 131.3, 130.3, 130.2, 130.0, 129.8, 129.1, 128.8, 128.1, 128.0, 125.0, 124.1, 121.1 (2C), 117.3, 116.9, 113.9 (2C), 69.4, 68.2, 63.0, 62.8, 55.3 (2C); FT-IR νmax(ATR) cm–1 3393, 2958, 2929, 1606, 1509, 1485, 1458, 1246, 1177, 1031, 995, 828, 755; m/z (EI) 268 ([M]+, 25), 120 ([3-MeOPhCH]+, 100).
Acknowledgments
The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement No. 617044 (SunCatChem).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.6b01006.
1H and 13C NMR spectra of products (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Molander G. A.; Kenny C. Tetrahedron Lett. 1987, 28, 4367. 10.1016/S0040-4039(00)96511-0. [DOI] [Google Scholar]; b Molander G. A.; Kenny C. J. Am. Chem. Soc. 1989, 111, 8236. 10.1021/ja00203a027. [DOI] [Google Scholar]
- Values calculated vs Ag/AgNO3 as reference electrode in THF:; a Shabangi M.; Flowers R. A. II Tetrahedron Lett. 1997, 38, 1137. 10.1016/S0040-4039(97)00008-7. [DOI] [Google Scholar]; b Flowers R. A. II. Synlett 2008, 2008, 1427. 10.1055/s-2008-1078414. [DOI] [Google Scholar]
- Selected examples using stoichiometric amount of tributyltin:; a Beckwith A. L. J.; Roberts D. H. J. Am. Chem. Soc. 1986, 108, 5893. 10.1021/ja00279a039. [DOI] [PubMed] [Google Scholar]; b Ardisson J.; Ferezou J. P.; Julia M.; Pancrazi A. Tetrahedron Lett. 1987, 28, 2001. 10.1016/S0040-4039(00)96030-1. [DOI] [Google Scholar]; c Sugawara T.; Otter B. A.; Ueda T. Tetrahedron Lett. 1988, 29, 75. 10.1016/0040-4039(88)80020-0. [DOI] [Google Scholar]
- Enholm was the first to systematically explore the scope of this reaction:; a Enholm E. J.; Prasad G. Tetrahedron Lett. 1989, 30, 4939. 10.1016/S0040-4039(01)80548-7. [DOI] [Google Scholar]; b Enholm E. J.; Burroff J. A. Tetrahedron Lett. 1992, 33, 1835. 10.1016/S0040-4039(00)74155-4. [DOI] [Google Scholar]
- Hays D. S.; Fu G. C. J. Org. Chem. 1996, 61, 4. 10.1021/jo951827s. [DOI] [PubMed] [Google Scholar]
- Corey E. J.; Pyne S. G. Tetrahedron Lett. 1983, 24, 2821. 10.1016/S0040-4039(00)88033-8. [DOI] [Google Scholar]
- Estévez R. E.; Oller-López J. L.; Robles R.; Melgarejo C. R.; Gansäuer A.; Cuerva J. M.; Oltra J. E. Org. Lett. 2006, 8, 5433. 10.1021/ol0620390. [DOI] [PubMed] [Google Scholar]
- a Molander G. A.; Harris C. R. J. Org. Chem. 1998, 63, 812. 10.1021/jo971889d. [DOI] [PubMed] [Google Scholar]; b Corey E. J.; Pyne S. G. Tetrahedron Lett. 1983, 24, 2821. 10.1016/S0040-4039(00)88033-8. [DOI] [Google Scholar]; c Streuff J. Synthesis 2013, 45, 281. 10.1055/s-0032-1316840. [DOI] [Google Scholar]
- For selected reviews on photocatalysis, see:; a Hoffmann N. Chem. Rev. 2008, 108, 1052. 10.1021/cr0680336. [DOI] [PubMed] [Google Scholar]; b Balzani V.; Credi A.; Venturi M. ChemSusChem 2008, 1, 26. 10.1002/cssc.200700087. [DOI] [PubMed] [Google Scholar]; c Ravelli D.; Dondi D.; Fagnoni M.; Albini A. Chem. Soc. Rev. 2009, 38, 1999. 10.1039/b714786b. [DOI] [PubMed] [Google Scholar]; d Ravelli D.; Fagnoni M.; Albini A. Chem. Soc. Rev. 2013, 42, 97. 10.1039/C2CS35250H. [DOI] [PubMed] [Google Scholar]
- For selected reviews on photoredox catalysis, see:; a Zeitler K. Angew. Chem., Int. Ed. 2009, 48, 9785. 10.1002/anie.200904056. [DOI] [PubMed] [Google Scholar]; b Yoon T. P.; Ischay M. A.; Du J. Nat. Chem. 2010, 2, 527. 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]; c Narayanam J. M. R.; Stephenson C. R. J. Chem. Soc. Rev. 2011, 40, 102. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]; d Ravelli D.; Fagnoni M. ChemCatChem 2012, 4, 169. 10.1002/cctc.201100363. [DOI] [Google Scholar]; e Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pandey G.; Hajra S.; Ghorai M. K.; Kumar K. R. J. Org. Chem. 1997, 62, 5966. 10.1021/jo9702812. [DOI] [Google Scholar]; b Tarantino K. T.; Liu P.; Knowles R. R. J. Am. Chem. Soc. 2013, 135, 10022. 10.1021/ja404342j. [DOI] [PubMed] [Google Scholar]; c Rono L. J.; Yayla H. G.; Wang D. Y.; Armstrong M. F.; Knowles R. R. J. Am. Chem. Soc. 2013, 135, 17735. 10.1021/ja4100595. [DOI] [PubMed] [Google Scholar]; d Petronijević F. R.; Nappi M.; MacMillan D. W. C. J. Am. Chem. Soc. 2013, 135, 18323. 10.1021/ja410478a. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Griesbeck A. G.; Reckenthäler M. Beilstein J. Org. Chem. 2014, 10, 1143. 10.3762/bjoc.10.114. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Wang C.; Qin J.; Shen X.; Riedel R.; Harms K.; Meggers E. Angew. Chem., Int. Ed. 2016, 55, 685. 10.1002/anie.201509524. [DOI] [PubMed] [Google Scholar]
- a Nakajima M.; Fava E.; Loescher S.; Jiang Z.; Rueping M. Angew. Chem., Int. Ed. 2015, 54, 8828. 10.1002/anie.201501556. [DOI] [PubMed] [Google Scholar]; b Zhu Z.; Das A.; Bui L.; Zhou H. Z.; Curran D. P.; Rueping M. J. Am. Chem. Soc. 2013, 135, 1823. 10.1021/ja309580a. [DOI] [PubMed] [Google Scholar]; c Nakajima M.; Lefebvre Q.; Rueping M. Chem. Commun. 2014, 50, 3619. 10.1039/c4cc00753k. [DOI] [PubMed] [Google Scholar]; d Lefebvre Q.; Hoffmann N.; Rueping M. Chem. Commun. 2016, 52, 2493. 10.1039/C5CC09881E. [DOI] [PubMed] [Google Scholar]
- For early examples of photochemical generation of ketyl radicals and their intramolecular addition on carbon-carbon double bonds, see:; a Belotti D.; Cossy J.; Pete J.-P.; Portella C. Tetrahedron Lett. 1985, 26, 4591. 10.1016/S0040-4039(00)98759-8. [DOI] [Google Scholar]; b Belotti D.; Cossy J.; Pete J.-P.; Portella C. J. Org. Chem. 1986, 51, 4196. 10.1021/jo00372a018. [DOI] [Google Scholar]; For an overview for the photochemical generation of ketyl radicals and their applications, see:; c Cossy J.; Belotti D. Tetrahedron 2006, 62, 6459. 10.1016/j.tet.2006.03.062. [DOI] [Google Scholar]
- Ishitani O.; Yanagida S.; Takamuku S.; Pac C. J. Org. Chem. 1987, 52, 2790. 10.1021/jo00389a027. [DOI] [Google Scholar]
- For a review on PCET, see:Huynh M. H. V.; Meyer T. J. Chem. Rev. 2007, 107, 5004. 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti C.; Desideri N. Bioorg. Med. Chem. 2009, 17, 3720. 10.1016/j.bmc.2009.03.051. [DOI] [PubMed] [Google Scholar]
- Dinnocenzo J. P.; Banach T. E. J. Am. Chem. Soc. 1989, 111, 8646. 10.1021/ja00205a014. [DOI] [Google Scholar]
- Bentley J.; Nilsson P. A.; Parsons A. F. J. Chem. Soc. Perkin Trans. 1 2002, 12, 1461. 10.1039/b202077g. [DOI] [Google Scholar]
- Sangwan N. K.; Rastogi S. N. Indian J. Chem. B 1980, 19, 500. [Google Scholar]
- Gomis M.; Kirkiacharian B. S.; Likforman J.; Mahuteau J. Bull. Soc. Chim. Fr. 1988, 3, 585. [Google Scholar]
- Pivnenko N. S.; Orlov V. D.; Nodel’man O. A.; Mikhed’kina E. I.; Lavrushin V. F. J. Org. Chem. USSR (Engl. Trans.) 1980, 16, 1657. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.