

Abstract
Members of the tristetraprolin (TTP) family of RNA binding proteins are found in all major eukaryotic groups. Family members from plants through humans can bind AU-rich elements in target mRNAs with high affinity. In mammalian cells, these proteins then promote deadenylation and decay of target transcripts. Four such proteins are found in the mouse, of which the best studied is TTP. When its gene is disrupted in the mouse, the animals develop a severe syndrome of arthritis, autoimmunity, cachexia, dermatitis, and myeloid hyperplasia. Conversely, recent overexpression studies have demonstrated protection against several experimental models of immune inflammatory disease. This endogenous anti-inflammatory protein could serve as the basis for novel approaches to therapy of similar conditions in humans.
Keywords: inflammation, tumor necrosis factor, mRNA decay, deadenylation, autoimmunity
The need for improved therapies for autoimmune and inflammatory diseases
The term “autoimmune diseases” includes many common and debilitating diseases that are of major public health importance. Well known diseases of this type include rheumatoid arthritis, multiple sclerosis, Crohn’s disease, psoriasis and psoriatic arthritis, ankylosing spondylitis, dermatomyositis, type 1 diabetes, and many others. These conditions often respond to non-steroidal anti-inflammatory drugs, glucocorticoids, or antimetabolites, but the responses are often incomplete, and side effects can be severe.
Recently, there has been a revolution in therapy for some of these conditions, using “biologics”, often humanized antibodies or binding proteins directed at individual cytokines known to be involved in specific diseases [1]. For example, anti-tumor necrosis factor α (TNFα) antibody-based biologics are now a mainstay for the treatment of rheumatoid arthritis and Crohn’s disease [2, 3]. However, even in the conditions that respond to these biologics, there are drawbacks: Because they are made with recombinant DNA technology, they are expensive to produce; because they are proteins, they need to be injected; they risk raising anti-drug antibodies; and some increase the risk of serious infections or reactivated tuberculosis [4].
Another drawback to the use of modern biologics is that they typically target a single cytokine, as in the use of anti-TNFα therapies, when it might be beneficial to inhibit the pathways activated independently by other cytokines. For these and other reasons, novel therapeutic approaches are needed for these diseases.
In this review, we will describe recent work supporting the possibility of harnessing an endogenous anti-inflammatory pathway involving the protein tristetraprolin (TTP), an mRNA binding protein that inhibits the production of many pro-inflammatory cytokines. This possibility was first suggested by the profound systemic inflammatory syndrome that was observed in TTP knockout mice, and the discovery that this syndrome was due, at least in part, to the overexpression of the potent pro-inflammatory cytokine TNFα in the TTP deficient animals [5–7]. Conversely, recent TTP overexpression experiments have demonstrated protection against the development of several experimental models of inflammatory disease [8]. These studies in animal models have raised the possibility that increasing TTP levels in humans might be a desirable therapeutic goal for the eventual treatment of immune and inflammatory diseases.
The TTP family of RNA binding proteins
TTP (also known as NUP475, GOS24, and TIS11) is encoded by the ZFP36 gene in humans [9]. It is probably the best understood of a family of three proteins in humans, which includes the proteins encoded by ZFP36L1 and ZFP36L2. Orthologues of TTP are found in all vertebrates examined to date, with the exception of birds, which seem to have “lost” the gene expressing TTP at some point in their evolution [10]. By definition, members of this protein family contain a tandem CCCH zinc finger (TZF) domain, and all seem to act in the same way: The proteins, through their TZF domains, bind to adenine/uridine-rich elements (AREs) in the 3’-untranslated regions (3’UTRs) of specific mRNAs, and then promote their turnover [6]. The mechanism of the increased rates of mRNA decay has not yet been clearly elucidated, although the proteins are known to promote the removal of the polyA tail of their target mRNAs, a process known as deadenylation, the rate limiting step in mRNA decay in eukaryotes [11]. This step is thought to involve the recruitment of 3’–5’-exonucleases, or deadenylases, possibly through recruitment of the NOT1 scaffolding protein to the extreme C-terminus of the proteins [12, 13]. NOT1 in turn is bound to at least two deadenylases [14], and its binding to the C-terminus of TTP family proteins may at least partially explain the ability of the proteins to promote mRNA deadenylation and decay (Fig. 1).
Fig. 1. Critical domains of TTP.
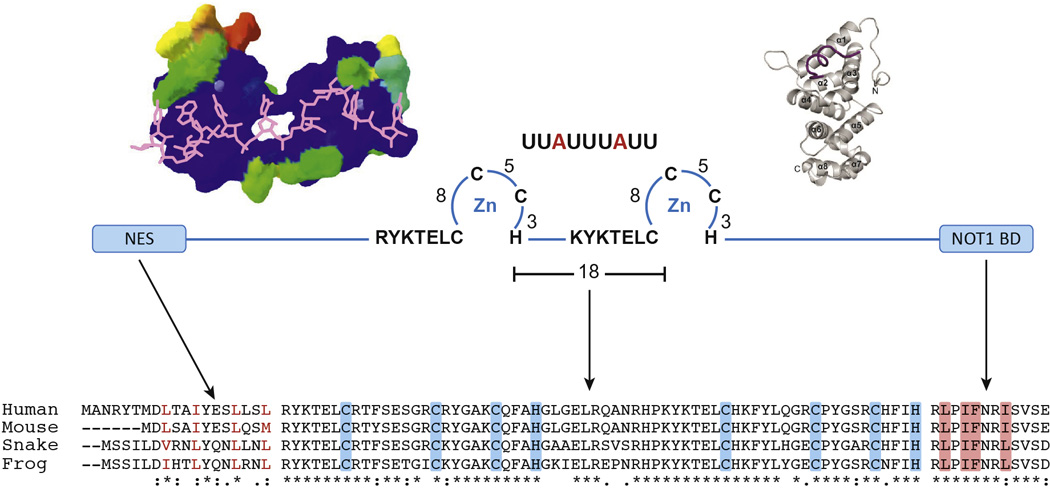
In the center of the figure is a schematic diagram of three critical domains of TTP: The N terminal nuclear export sequence (NES), the central tandem zinc finger domain (TZF domain), and the C terminal NOT1 binding domain (NOT1 BD). The key cysteines and histidines in each finger, as well as the conserved lead-in sequences, are indicated for the TZF domain, as is the ideal 9 base RNA binding site that is a component of many AU-rich regions in TTP target mRNAs. At the upper left is shown a structural model of the human TTP TZF domain bound to the same 9 base RNA sequence as shown at the top; RNA is in magenta in this figure. This structural model is taken from [46], with permission, and is based on the original structure of the ZFP36L2 (TIS11D) TZF domain [16]. At the upper right is a diagram of the crystal structure of the C-terminal NOT1 binding domain peptide of TTP (maroon), binding to the three internal helices from the human NOT1 protein (beige); this structure is taken from [12], with permission. At the bottom are shown parts of an amino acid sequence alignment done with Clustal Omega of human TTP with its orthologues from mouse, snake and frog. According to Clustal conventions, asterisks at the bottom indicate sequence identity at that site; a colon indicates a conserved residue; and a period indicates a less well conserved residue. Shown are the N termini of the proteins, with their conserved NES sequences; in this case, the branched chain amino acids are colored red. The TZF domain alignment is shown in the middle, with the critical cysteines and histidines shaded in blue. The NOT1 binding domains are shown to the right, with the sequences representing the extreme C termini of the proteins. Highlighted in orange are the amino acid residues that are inserted into the hydrophobic groove of the NOT1 protein central domain (upper right) [12]. The spaces in the sequence alignments represent gaps of various sizes. The sequences are from the following GenBank accession numbers: Human (Homo sapiens), NP_003398.2 ; mouse (Mus musculus), NP_035886.1 ; snake (Protobothrops mucrosquamatus (Taiwan habu)), XP_015676155.1; and frog (Xenopus tropicalis), NP_001106542.1.
The ideal TTP binding site sequence in mRNA is UUAUUUAUU; however, variations in this sequence can still mediate high affinity binding of TTP family proteins [15]. Recent nuclear magnetic resonance studies have shown that this mRNA sequence is unstructured when it binds to the TZF domain of TTP family members [16]. The TZF domains of proteins from very distant species, such as humans and plants, have high sequence identity and identical internal spacing; indeed, they can substitute for the endogenous domain in the yeast Schizosaccharomyces pombe [17]. Based on studies in yeast, fruit flies and other organisms, it appears that the basic functions of binding to AREs and promoting mRNA decay are conserved in the family members from most eukaryotes, with functional specificity conferred by patterns of expression and other factors.
Some of the important sequence domains within TTP are highlighted in Figure 2.
Fig. 2. Model of proposed TTP mechanism of action.
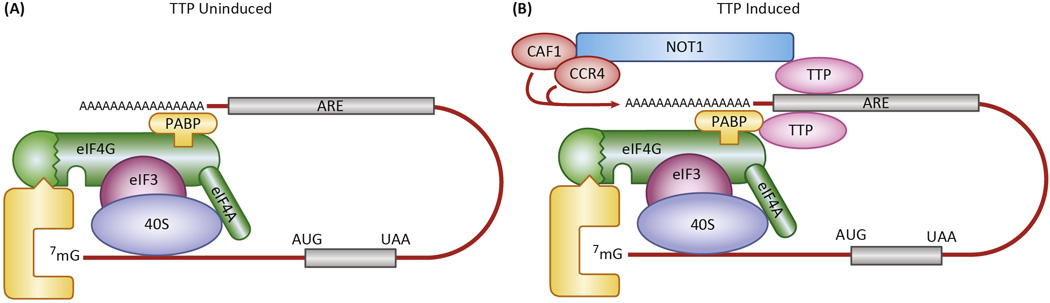
The diagram shows a schematic representation of the “closed-loop” model [47] of a typical mRNA being translated in the absence of TTP (left), as in serum deprived fibroblasts or macrophages, and after the induction of TTP (right), as in serum-stimulated fibroblasts or LPS-stimulated macrophages. TTP binds to the AREs of its target mRNAs, often on multiple sites, and interacts with both polyA binding protein (PABP) as a possible means of localizing near the polyA tail, and NOT1, which can bring its associated deadenylases CAF1 and CCR4 into proximity with the 3’ end of the polyA tail. The net result of the TTP binding is destabilization and decreased translation of the mRNA. See the text for further details.
TTP loss of function experiments indicate a role in controlling inflammation
The role of TTP to promote mRNA decay was first determined in cells and tissues derived from TTP knockout (KO) mice. TTP-deficient mice appeared healthy at birth, but soon developed a profound inflammatory syndrome that included cachexia, arthritis, conjunctivitis, dermatitis, myeloid hyperplasia, splenomegaly, and autoimmunity [7]. The involvement of tumor necrosis factor alpha (TNFα) was suspected because of the similarity of this phenotype to those seen in previous mouse models of TNFα overexpression. Injection of the mice with anti-TNFα antibodies shortly after birth prevented development of the syndrome; it rapidly returned when antibodies were withdrawn [7]. A similar protective effect was achieved by knocking out both types of TNFα receptor along with TTP [18]. Macrophages derived from the TTP KO mice over-secreted TNFα upon stimulation with lipopolysaccharide (LPS), and this was associated with increased levels of TNFα mRNA in the cells [5]. Finally, the mechanism of the increased TNFα mRNA, and the increased biosynthesis and secretion of the TNFα protein, was shown to be stabilization of the TNFα mRNA in the absence of TTP [6]. This effect was mediated by direct binding of TTP to the “AU-rich element” (ARE) present in the 3’UTR of the TNFα mRNA, a 70 nucleotide region that is highly conserved among mammals that is approximately 280 b 5’ of the polyA tail. The ARE of the TNFα mRNA 3’UTR from both human and mouse contains several copies of the ideal TTP binding site sequence described above.
An additional study evaluated the effect of TTP deficiency in bone marrow-derived stromal cells on a second cytokine whose mRNA contained a similar ARE, that encoding granulocyte-macrophage colony-stimulating factor (GM-CSF). That study established GM-CSF mRNA as a TTP target, and also demonstrated that deadenylation of the mRNA was involved in TTP-promoted decay [11].
TTP was originally identified in fibroblasts as the product of a gene induced by treatment with insulin, growth factors, phorbol esters and serum [19–22]. Fibroblasts derived from the TTP KO mice did not secrete TNFα, but instead exhibited stabilization of several other transcripts containing AREs, leading to increased levels of the mRNA and increased synthesis of the protein [23]. An example was IER3, the product of an inducible gene that is thought to be involved in regulation of blood pressure and Th17 cell differentiation, among other physiological roles [24]. More recently, fibroblasts were also shown to respond to TNFα by synthesizing and releasing a number of cytokines and chemokines involved in leukocyte chemotaxis, many of which are encoded by mRNAs that are direct TTP targets [25].
These studies established that TTP could control the stability of different target transcripts in three different cell types. However, because of the major contribution of TNFα to the TTP deficiency phenotype, the phenotype of mice with myeloid-specific TTP deficiency was evaluated [26]. Surprisingly, these mice appeared normal under laboratory conditions. However, when they were stimulated with low doses of LPS that had little effect on circulating TNFα or signs of endotoxemia in normal mice, the myeloid-deficient TTP KO mice developed severe endotoxin shock that was associated with blood levels of TNFα more than 100-fold higher than those seen in the control mice. Similar results were reported by another group [27]. These studies demonstrated that myeloid cell-specific TTP deficiency did not mimic the phenotype of total TTP deficiency, suggesting that other cell types could be involved in the pathogenesis of the TTP deficiency syndrome.
More recently, Goriely and colleagues showed that knocking out the p 19 subunit of interleukin 23 (IL23) could protect TTP-deficient mice from developing the TTP deficiency syndrome [28]. IL23 is largely secreted by dendritic cells and is a stimulus for the activation of the IL17 pathway in a subset of T cells, and these authors showed that the p19 mRNA was a target of TTP. IL17 itself is a major pro-inflammatory cytokine, and knocking its gene out in TTP-deficient mice also largely protected them from the TTP deficiency syndrome. As in macrophages, TTP is also a potent modulator of TNFα secretion from dendritic cells after LPS stimulation; a recent study found that TTP was the most potent negative regulator of LPS-stimulated TNFα production identified in a genome-wide screen of CRISPR-induced loss of function mutants in dendritic cells [29].
Thus, it appears that several cell types in addition to macrophages use TTP to control the stability of specific mRNAs in those cells. Indeed, a large number of mRNAs in various cell types have been identified or suggested as TTP targets [9]. Thus, the TTP deficiency syndrome should be viewed as not just a “macrophage problem”, but a problem affecting many cells in the body, which probably contribute in many yet-known ways to the eventual severe pathology of the syndrome.
TTP gain of function experiments demonstrate a protective role in experimental models of inflammatory disease
These experiments raised the possibility that overexpression of TTP might prevent or delay the onset of immune or inflammatory diseases in mice and possibly in man. To achieve overexpression in mice, we initially attempted to generate transgenic mice, in which strong general promoters were used to drive expression of a TTP cDNA. These attempts all ended in embryonic lethality. This result should not be surprising, since one of the key features of TTP expression, and the attribute that led to its cloning in the first place, is its extreme “inducibility” in response to external stimuli. In many cell types, TTP expression is almost absent in unstimulated, quiescent cells. Both mRNA and protein increase rapidly and dramatically after stimulation, with mRNA levels peaking at about 45 minutes and then falling back towards the baseline, while protein levels reached more of a plateau after 60 minutes or so. After stimulation, the mRNA in macrophages and fibroblasts is extremely unstable, with half lives of decay after treatment of cells with the transcription inhibitor actinomycin D of 15–20 minutes [23]. Recent studies in human diploid fibroblasts under normal growth conditions demonstrated similar extreme lability of the human TTP mRNA in these cells [30]. It seems likely that this extreme ability to be regulated is important in normal physiology, as well as in responses of the organism to, e.g., infectious stimuli. It therefore might be expected that high-level, constitutive, unregulated expression of the gene might have deleterious effects on the organism.
Another attempt to evaluate the possible beneficial effects of TTP overexpression was reported by Kirkwood and colleagues [31]. These investigators studied a rat model of periodontitis that involved intra-oral injection of LPS from the bacterium Actinobacillus actinomycetemcomitans, a prominent causative agent in human periodontitis. This treatment resulted in localized bone loss, infiltration of inflammatory cells, and overproduction of inflammatory cytokines. When adenovirus-expressed TTP was delivered to the same site, there were marked reductions in bone loss and inflammatory cell infiltration, as well as decreases in local cytokine levels (Table 1). Although TTP expression was apparently not regulated in this experiment, it demonstrated that increased localized TTP might have beneficial effects on an inflammatory condition.
Table 1.
Beneficial effects of TTP overexpression in immune disease models.
| Model | Species | Method | Result | Reference |
|---|---|---|---|---|
| Experimental periodontitis | Rat | Local adenovirus injection | Protection against bone loss; decreased cellular infiltrate. |
31 |
| Collagen antibody-induced arthritis |
Mouse | TTP ΔARE | Protection against joint destruction | 8 |
| Imiquimod-induced dermatitis | Mouse | TTP ΔARE | Decreased epidermal thickening; decreased inflammatory cell infiltration. |
8 |
| Experimental autoimmune encephalomyelitis |
Mouse | TTP ΔARE | Improved neurologic score; decreased weight loss. | 8 |
| LPS-induced cytokine levels | Mouse | TTP ΔARE | More rapid return to baseline of some serum cytokines | 8 |
More recently, we attempted to achieve systemic overexpression while leaving the regulation of the Zfp36 locus intact. To do this, we attempted to decrease TTP mRNA’s rapid rate of decay by removing lability-inducing sequences. Within the mouse mRNA 3’UTR there is an AU-rich region of approximately 130 b that is near to the 3’ end of the mRNA (Figure. 3). A similar sequence stretch can be found in the 3’UTR of the human TTP mRNA (Figure 3). Removing this region from the mouse TTP mRNA might be expected to increase its stability, resulting in still-regulated but overexpressed TTP protein in its normal sites. Similar reasoning has been used successfully before. For example, removal of the ARE from the TNFα mRNA in mice resulted in a severe TNFα overproduction syndrome and inflammatory bowel disease [32]. Smithies and colleagues [33, 34] achieved various levels of expression of their mRNAs and proteins of interest by inserting either labile or stable regions into the 3’UTRs of their target genes. Deletion of an AU-rich region of the interferon gamma mRNA 3’UTR stabilized this transcript, and resulted in increased secretion of interferon in the mouse [35].
Fig. 3. Sequences of mouse and human TTP mRNA 3’UTRs.

Shown is a Clustal Omega alignment of the mouse and human TTP 3’UTRs, starting from the stop codons (underlined). The AU-rich segment that was deleted in the TTPΔARE mice is in red type, and the polyadenylation signals in both mouse and human mRNAs are shaded yellow. The asterisks indicate nucleotide identity at that site.
Accordingly, we deleted the 130 base AU-rich region in the mouse Zfp36 locus using homologous recombination techniques [8]. This resulted in increased mRNA stability in serum-stimulated fibroblasts and LPS-stimulated macrophages derived from these mice (TTPΔARE mice) (Figure 4) and resulted in over-production of protein in the latter case. Most importantly, levels of immunoreactive protein were increased by several fold in various mouse tissues (Figure 4). Despite this level of overexpression, the mice gained weight normally and exhibited normal male and female fertility; to date, no abnormalities have been seen in these animals in terms of weight gain, general appearance, and tissue histology.
Fig. 4. Effects of the homozygous TTPΔARE deletion on TTP protein expression in macrophages and liver.
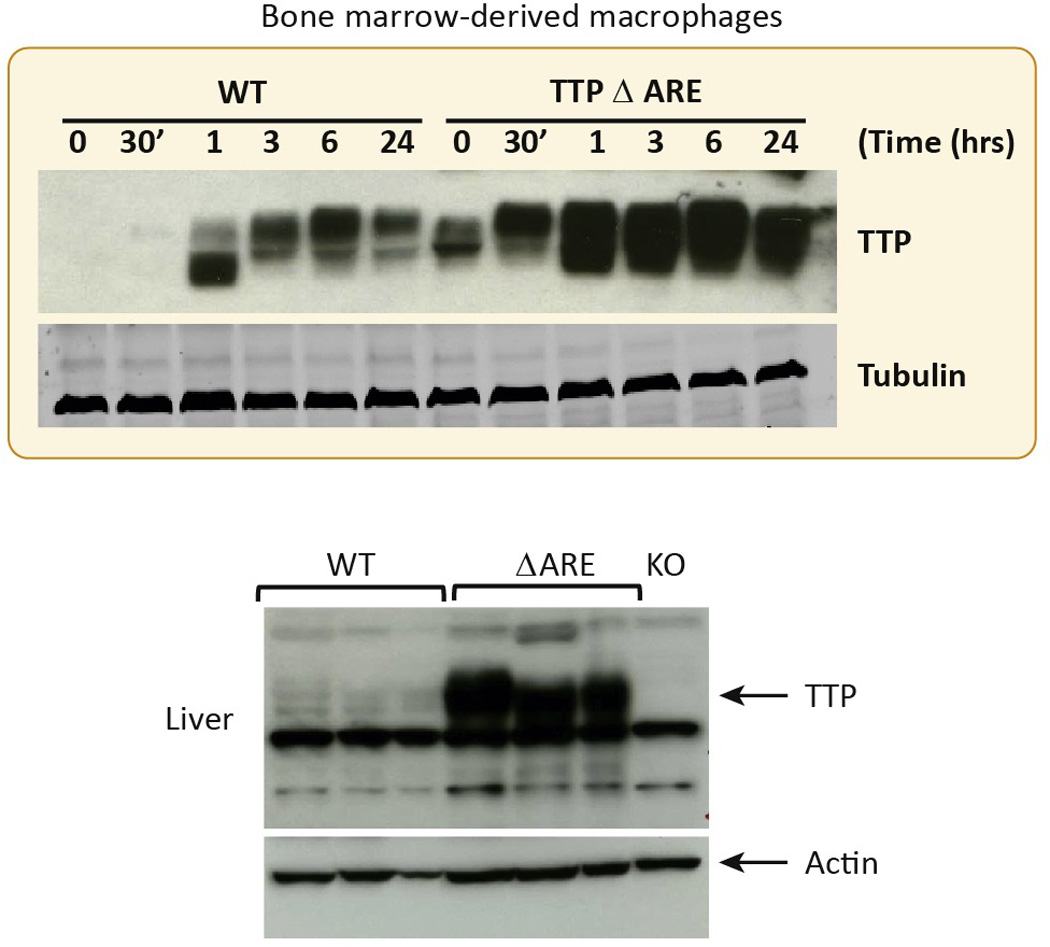
The top panel shows the responses of bone marrow derived macrophages from WT mice and their TTPΔARE counterparts to LPS stimulation. The time course of TTP protein expression is shown, as determined by western blotting. The different sizes of the protein bands are thought to represent variably phosphorylated forms of the protein. Immunoreactive tubulin is shown as a loading control. In the bottom panel is shown the expression of TTP protein in the livers of three TTPΔARE mice, along with livers from three WT mice and a single lane containing an equivalent amount of liver protein from a TTP KO mouse. Immunoreactive actin is shown as a loading control. Note the non-specific bands that appear in all samples. Taken from [8], with permission.
These mice were then subjected to four models of immune or inflammatory disease, designed to mimic a number of human conditions: Gram negative bacterial endotoxemia, rheumatoid arthritis, psoriasis, and multiple sclerosis. These studies are described in detail in [8], but will be summarized briefly here and in Table 1.
First, homozygous TTPΔARE mice were subjected to three different doses of LPS, and clinical responses and cytokine production were monitored for several hours. Although peak blood levels of IL-10 and IL-1B (both thought to be products of TTP target mRNAs) were decreased in the TTPΔARE mice, and returned to baseline more rapidly than in the control mice, the peak blood levels of TNFα were not significantly affected. Clinical responses to LPS were also no different in this acute disease model. One possible explanation for the lack of effect on TNFα production might be that TTP levels in cells producing TNFα were already so high in the normal mice after LPS stimulation that further increases would not promote faster TNFα mRNA decay.
The TTPΔARE mice were then subjected to three subacute disease models that involve more aspects of the immune system than the immediate innate response to LPS. One of these was collagen antibody-induced arthritis, a widely used model of human rheumatoid arthritis [36]. In this case, the TTPΔARE mice were almost completely protected against the development of the characteristic joint pathology, including infiltration of inflammatory cells, synovial hyperplasia, cartilage necrosis, bone erosion, and fibrosis, all of which were seen in the control mice.
The TTPΔARE mice were then challenged with imiquimod applied to the skin, widely used to induce a form of dermatitis that resembles psoriasis in humans [37]. The homozygous TTPΔARE mice exhibited strikingly reduced infiltration of inflammatory cells, particularly neutrophils, in both the dermis and the epidermis. These mice also exhibited decreased epidermal hyperplasia and hyperkeratosis, two characteristic histopathological features of this condition. Gene expression analysis showed reduced expression of various inflammatory mediators in the skin.
Finally, the mice were tested with a widely used model of experimental autoimmune encephalomyelitis (EAE), frequently used as a model of human multiple sclerosis [38]. The TTPΔARE mice overexpressing TTP were strikingly protected against the neurological dysfunction and weight loss associated with this model, as well as the characteristic pathological changes in brain and spinal cord.
Taken together with the results from the adenovirus injection experiments [31], the results of this study [8] provide proof of principle that increasing levels of TTP throughout the body by this means can prevent the occurrence or decrease the severity of certain models of immune inflammatory disease. The relative ineffectiveness of this genetic manipulation to protect against the “cytokine storm” of acute endotoxemia suggests the possibility that myeloid cell TTP levels under these LPS-stimulated conditions are already so high in the normal mice that increasing them modestly would have no additional effect on TNFα mRNA and protein levels. However, the data showed that constitutively increased TTP levels were much more effective against the three subacute disease models tested. Whether this positive result was dependent on the ability of the Zfp36 locus to remain in its normal regulatory chromosomal environment is a subject that will require further study.
Strategies to enhance TTP expression
The experiments with the TTPΔARE mice demonstrated that modest TTP overexpression from its endogenous genetic locus protected the animals against several models of inflammatory disease, while at the same time not obviously harming the mice. These data support the possibility that increasing TTP concentrations throughout the body might have beneficial effects in the treatment of human immune and inflammatory diseases. These diseases might include not only the classical autoimmune diseases listed above, but also other conditions in which inflammation is thought to play a pathogenetic role, including infectious diseases, inflammation related to cancer, immunotherapy of cancer, type 2 diabetes and obesity, atherosclerosis, and many others. How might one achieve increased TTP levels in humans? That will be the focus here, but other potential avenues for therapy with TTP exist: for example, co-activator proteins have been identified that can increase the affinity of TTP for its mRNA targets [39]; and altering certain phosphorylated residues within the TTP protein sequence seems to decrease its binding to sequestering 14-3-3 proteins, thus making TTP itself more potent [40, 41].
How could levels of TTP be increased in humans? Localized treatments, as described in the adenovirus experiments cited above ([31], might be possible for specific conditions, for example, cutaneous therapy for psoriasis. It also might be possible to use virus-based gene therapy approaches with more widespread expression in certain instances. It is possible that cell- or lineage-specific overexpression of TTP might be sufficient to provide therapeutic benefits. For example, bone marrow transplantation from TTP KO mice into immune-deficient mice resulted in recapitulation of the original TTP KO phenotype after a lag of several months, suggesting that cells derived from hematopoietic progenitors were sufficient to transmit the mutant phenotype to the recipients [5]. Therefore, it might be possible to achieve therapeutic benefit by altering TTP mRNA stability in hematopoietic stem cells alone, perhaps by using CRISPR-CAS technology, already in use to modify human CD34+ hematopoietic stem cells [42, 43]. It should be possible to test this idea in mice by limiting the expression of the TTPΔARE allele to hematopoietic stem cells and evaluating the mice for similar protection against inflammatory disease.
For many conditions, we suspect that successful treatments may need to be delivered systemically. One approach to increasing TTP expression in most if not all cells and tissues would be to use an anti-sense RNA approach, with the idea of protecting the labile TTP mRNA from decay. Although antisense oligonucleotides have normally been used to promote mRNA decay, certain types of antisense oligonucleotides, such as “steric blocking oligonucleotides” [44, 45], could potentially be used to block access of RNA binding and decay-promoting proteins to the TTP mRNA 3’UTR. One of the advantages of this approach is that sequence-specific complementary targeting oligonucleotides could be used, potentially minimizing off-target effects. Limitations of this approach include cell and tissue permeability, instability of the oligonucleotides themselves, and others.
A final method would involve traditional high-throughput screening for small molecules that would increase TTP protein expression in cells and tissues. A cell-based screening program could use protein readouts that are agnostic to the mechanism by which the molecules stimulate TTP expression. Compounds would need to be counter-screened to exclude those that also stimulated TNFα production, as seen with LPS and other pathogen-associated molecular patterns. Such screens could identify novel compounds that could serve as leads for further structure-activity modifications, or new uses for approved drugs.
Concluding Remarks
TTP loss of function in mice leads to a severe inflammatory syndrome that is partially the result of TNFα mRNA stabilization and hypersecretion, but also involves many different cell types and cytokines in its complex pathogenesis. Conversely, TTP gain of function studies using the knock-in TTPΔARE mutation to stabilize the TTP mRNA, leading to regulated but increased expression, showed that this genetic manipulation strikingly decreased the severity of several subacute models of immune inflammatory disease in mice, without having obvious deleterious effects on the mice. These two types of experiments provide support for the concept that increasing the levels or activity of TTP in humans might have similar effects on immune and inflammatory diseases (see “Outstanding Questions”). Beneficial effects in these conditions might occur because of TTP’s ability to target several pro-inflammatory cytokines and their pathways at one time, perhaps improving on existing biologic agents that target single cytokines. This could represent an attractive approach to the treatment of these conditions, by increasing the levels or mimicking the activity of a specific, endogenous anti-inflammatory protein. Our hope is that these ideas will stimulate investigators both in and outside the pharmaceutical industry to try to harness this pathway for the development of new types of therapy.
Outstanding Questions Box.
Can we identify human polymorphisms or sequence variants that result in constitutive TTP overexpression, and are these beneficial in terms of disease prevention?
Are there deleterious consequences of long-term TTP overexpression in mouse models?
Are there related disease models that could be affected beneficially by TTP overexpression, including other immune, inflammatory or infectious disease models?
Could enhanced TTP expression be beneficial in the treatment of other conditions in which inflammation is thought to play an important role, including obesity, type 2 diabetes, and various forms of cancer?
What methods, other than germline genetic manipulation, can be used to achieve localized or systemic overexpression of TTP?
Can we identify small molecules that could increase local or systemic levels of TTP without deleterious side effects?
Fig. 5. Schematic representation of steps in TTP accumulation that could be affected by proposed therapies.

Shown schematically are steps in the biosynthesis and decay of TTP mRNA and protein that might be influenced by novel therapies designed to increase its overall accumulation. Small molecules might be identified that increase transcription of ZFP36, the human gene encoding TTP, that would have minimal effects on other genes that are often co-induced with TTP, such as pro-inflammatory cytokines. Other small molecules might be able to enhance splicing of the single ZFP36 intron. Changes in TTP mRNA stability might be influenced by “steric-blocking” oligonucleotides, which would inhibit the normally very rapid decay of the TTP mRNA. Small molecules might be identified that decrease the rate of decay of TTP protein and promote its overall accumulation in cells. Cell-based screening programs could be conducted without targeting specific steps in this biosynthetic pathway.
Trends Box.
Autoimmune diseases remain major causes of death and disability. Traditional forms of therapy are sometimes ineffective, and can have significant side effects.
“Biological” therapies, that often involve the parenteral administration of recombinant antibodies or binding proteins that target specific pro-inflammatory cytokines, have revolutionized therapy for these diseases, but they have drawbacks of their own, including the expense of production, the need for injection, and others.
Novel therapies for these conditions are still needed.
The RNA binding protein tristetraprolin (TTP) is an important endogenous inhibitor of inflammation. It acts by destabilizing mRNAs encoding pro-inflammatory cytokines and inhibiting their biosynthesis.
Increasing local or systemic levels of TTP in mice can prevent or decrease the severity of several inflammatory disease models.
Acknowledgments
We are grateful to the members of our laboratory for many useful discussions, and to Drs. Mike Fessler and Don Cook for helpful comments on the manuscript. Supported by the Intramural Program of the National Institute of Environmental Health Sciences, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chastek B, et al. A Retrospective Cohort Study Comparing Utilization and Costs of Biologic Therapies and JAK Inhibitor Therapy Across Four Common Inflammatory Indications in Adult US Managed Care Patients. Advances in therapy. 2016;33:626–642. doi: 10.1007/s12325-016-0312-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. 2016;12:49–62. doi: 10.1038/nrrheum.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palladino MA, et al. Anti-TNF-alpha therapies: the next generation. Nature reviews. Drug discovery. 2003;2:736–746. doi: 10.1038/nrd1175. [DOI] [PubMed] [Google Scholar]
- 4.Ramiro S, et al. Safety of synthetic and biological DMARDs: a systematic literature review informing the 2013 update of the EULAR recommendations for management of rheumatoid arthritis. Annals of the rheumatic diseases. 2014;73:529–535. doi: 10.1136/annrheumdis-2013-204575. [DOI] [PubMed] [Google Scholar]
- 5.Carballo E, et al. Bone marrow transplantation reproduces the tristetraprolin-deficiency syndrome in recombination activating gene-2 (−/−) mice. Evidence that monocyte/macrophage progenitors may be responsible for TNFalpha overproduction. The Journal of clinical investigation. 1997;100:986–995. doi: 10.1172/JCI119649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carballo E, et al. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 7.Taylor GA, et al. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4:445–454. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 8.Patial S, et al. Enhanced stability of tristetraprolin mRNA protects mice against immune-mediated inflammatory pathologies. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:1865–1870. doi: 10.1073/pnas.1519906113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brooks SA, Blackshear PJ. Tristetraprolin (TTP): interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim Biophys Acta. 2013;1829:666–679. doi: 10.1016/j.bbagrm.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lai WS, et al. Life without TTP: apparent absence of an important anti-inflammatory protein in birds. American journal of physiology. Regulatory, integrative and comparative physiology. 2013;305:R689–R700. doi: 10.1152/ajpregu.00310.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carballo E, et al. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood. 2000;95:1891–1899. [PubMed] [Google Scholar]
- 12.Fabian MR, et al. Structural basis for the recruitment of the human CCR4-NOT deadenylase complex by tristetraprolin. Nature structural & molecular biology. 2013;20:735–739. doi: 10.1038/nsmb.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandler H, et al. Not1 mediates recruitment of the deadenylase Caf1 to mRNAs targeted for degradation by tristetraprolin. Nucleic acids research. 2011;39:4373–4386. doi: 10.1093/nar/gkr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu K, et al. Insights into the structure and architecture of the CCR4-NOT complex. Frontiers in genetics. 2014;5:137. doi: 10.3389/fgene.2014.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brewer BY, et al. RNA sequence elements required for high affinity binding by the zinc finger domain of tristetraprolin: conformational changes coupled to the bipartite nature of Au-rich MRNA-destabilizing motifs. The Journal of biological chemistry. 2004;279:27870–27877. doi: 10.1074/jbc.M402551200. [DOI] [PubMed] [Google Scholar]
- 16.Hudson BP, et al. Recognition of the mRNA AU-rich element by the zinc finger domain of TIS11d. Nature structural & molecular biology. 2004;11:257–264. doi: 10.1038/nsmb738. [DOI] [PubMed] [Google Scholar]
- 17.Wells ML, et al. Functional equivalence of an evolutionarily conserved RNA binding module. The Journal of biological chemistry. 2015;290:24413–24423. doi: 10.1074/jbc.M115.673012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carballo E, Blackshear PJ. Roles of tumor necrosis factor-alpha receptor subtypes in the pathogenesis of the tristetraprolin-deficiency syndrome. Blood. 2001;98:2389–2395. doi: 10.1182/blood.v98.8.2389. [DOI] [PubMed] [Google Scholar]
- 19.DuBois RN, et al. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. The Journal of biological chemistry. 1990;265:19185–19191. [PubMed] [Google Scholar]
- 20.Lai WS, et al. Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. The Journal of biological chemistry. 1990;265:16556–16563. [PubMed] [Google Scholar]
- 21.Varnum BC, et al. Nucleotide sequence of a cDNA encoding TIS11, a message induced in Swiss 3T3 cells by the tumor promoter tetradecanoyl phorbol acetate. Oncogene. 1989;4:119–120. [PubMed] [Google Scholar]
- 22.Ma Q, Herschman HR. A corrected sequence for the predicted protein from the mitogen-inducible TIS11 primary response gene. Oncogene. 1991;6:1277–1278. [PubMed] [Google Scholar]
- 23.Lai WS, et al. Novel mRNA targets for tristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Mol Cell Biol. 2006;26:9196–9208. doi: 10.1128/MCB.00945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arlt A, Schafer H. Role of the immediate early response 3 (IER3) gene in cellular stress response, inflammation and tumorigenesis. European journal of cell biology. 2011;90:545–552. doi: 10.1016/j.ejcb.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Qiu LQ, et al. Tristetraprolin (TTP) coordinately regulates primary and secondary cellular responses to proinflammatory stimuli. J Leukoc Biol. 2015;97:723–736. doi: 10.1189/jlb.3A0214-106R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiu LQ, et al. Myeloid-specific tristetraprolin deficiency in mice results in extreme lipopolysaccharide sensitivity in an otherwise minimal phenotype. Journal of immunology. 2012;188:5150–5159. doi: 10.4049/jimmunol.1103700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kratochvill F, et al. Tristetraprolin-driven regulatory circuit controls quality and timing of mRNA decay in inflammation. Molecular systems biology. 2011;7:560. doi: 10.1038/msb.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molle C, et al. Tristetraprolin regulation of interleukin 23 mRNA stability prevents a spontaneous inflammatory disease. J Exp Med. 2013;210:1675–1684. doi: 10.1084/jem.20120707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parnas O, et al. A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell. 2015;162:675–686. doi: 10.1016/j.cell.2015.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiu LQ, et al. Global analysis of posttranscriptional gene expression in response to sodium arsenite. Environmental health perspectives. 2015;123:324–330. doi: 10.1289/ehp.1408626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patil CS, et al. Targeting mRNA stability arrests inflammatory bone loss. Molecular therapy : the journal of the American Society of Gene Therapy. 2008;16:1657–1664. doi: 10.1038/mt.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kontoyiannis D, et al. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 33.Kakoki M, et al. Altering the expression in mice of genes by modifying their 3' regions. Developmental cell. 2004;6:597–606. doi: 10.1016/s1534-5807(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 34.Hathaway CK, et al. High Elmo1 expression aggravates and low Elmo1 expression prevents diabetic nephropathy. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:2218–2222. doi: 10.1073/pnas.1600511113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hodge DL, et al. IFN-gamma AU-rich element removal promotes chronic IFN-gamma expression and autoimmunity in mice. Journal of autoimmunity. 2014;53:33–45. doi: 10.1016/j.jaut.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nandakumar KS, Holmdahl R. Efficient promotion of collagen antibody induced arthritis (CAIA) using four monoclonal antibodies specific for the major epitopes recognized in both collagen induced arthritis and rheumatoid arthritis. Journal of immunological methods. 2005;304:126–136. doi: 10.1016/j.jim.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 37.Flutter B, Nestle FO. TLRs to cytokines: mechanistic insights from the imiquimod mouse model of psoriasis. European journal of immunology. 2013;43:3138–3146. doi: 10.1002/eji.201343801. [DOI] [PubMed] [Google Scholar]
- 38.Miller SD, Karpus WJ. Experimental autoimmune encephalomyelitis in the mouse. In: Coligan John E, et al., editors. Current protocols in immunology. Unit 15. Chapter 15. 2007. p. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kedar VP, et al. Direct binding of specific AUF1 isoforms to tandem zinc finger domains of tristetraprolin (TTP) family proteins. The Journal of biological chemistry. 2012;287:5459–5471. doi: 10.1074/jbc.M111.312652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ross EA, et al. Dominant Suppression of Inflammation via Targeted Mutation of the mRNA Destabilizing Protein Tristetraprolin. Journal of immunology. 2015;195:265–276. doi: 10.4049/jimmunol.1402826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tiedje C, et al. The RNA-binding protein TTP is a global post-transcriptional regulator of feedback control in inflammation. Nucleic acids research. 2016 doi: 10.1093/nar/gkw474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hendel A, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature biotechnology. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mandal PK, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell stem cell. 2014;15:643–652. doi: 10.1016/j.stem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kole R, et al. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nature reviews. Drug discovery. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ward AJ, et al. Nonsense-mediated decay as a terminating mechanism for antisense oligonucleotides. Nucleic acids research. 2014;42:5871–5879. doi: 10.1093/nar/gku184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carrick DM, et al. The tandem CCCH zinc finger protein tristetraprolin and its relevance to cytokine mRNA turnover and arthritis. Arthritis research & therapy. 2004;6:248–264. doi: 10.1186/ar1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kahvejian A, et al. The mRNA closed-loop model: the function of PABP and PABP-interacting proteins in mRNA translation. Cold Spring Harbor symposia on quantitative biology. 2001;66:293–300. doi: 10.1101/sqb.2001.66.293. [DOI] [PubMed] [Google Scholar]