

Abstract
Vascular cognitive impairment and dementia (VCID) is the second most common form of dementia behind Alzheimer's disease (AD). It is estimated that 40% of AD patients also have some form of VCID. One promising therapeutic for AD is anti-Aβ immunotherapy, which uses antibodies against Aβ to clear it from the brain. While successful in clearing Aβ and improving cognition in mice, anti-Aβ immunotherapy failed to reach primary cognitive outcomes in several different clinical trials. We hypothesized that one potential reason the anti-Aβ immunotherapy clinical trials were unsuccessful was due to this high percentage of VCID comorbidity in the AD population. We used our unique model of VCID-amyloid comorbidity to test this hypothesis. We placed 9-month-old wild-type and APP/PS1 mice on either a control diet or a diet that induces hyperhomocysteinemia (HHcy). After being placed on the diet for 3 months, the mice then received intraperotineal injections of either IgG2a control or 3D6 for another 3 months. While we found that treatment of our comorbidity model with 3D6 resulted in decreased total Aβ levels, there was no cognitive benefit of the anti-Aβ immunotherapy in our AD/VCID mice. Further, microhemorrhages were increased by 3D6 in the APP/PS1/control but further increased in an additive fashion when 3D6 was administered to the APP/PS1/HHcy mice. This suggests that the use of anti-Aβ immunotherapy in patients with both AD and VCID would be ineffective on cognitive outcomes.
SIGNIFICANCE STATEMENT Despite significant mouse model data demonstrating both pathological and cognitive efficacy of anti-Aβ immunotherapy for the treatment of Alzheimer's disease, clinical trial outcomes have been underwhelming, failing to meet any primary endpoints. We show here that vascular cognitive impairment and dementia (VCID) comorbidity eliminates cognitive efficacy of anti-Aβ immunotherapy, despite amyloid clearance. Further, cerebrovascular adverse events of the anti-Aβ immunotherapy are significantly exacerbated by the VCID comorbidity. These data suggest that VCID comorbidity with Alzheimer's disease may mute the response to anti-Aβ immunotherapy.
Keywords: amyloid, immunotherapy, microglia, microhemorrhage, mixed dementia, vascular cognitive impairment
Introduction
Alzheimer's disease (AD) is the most common form of dementia and is characterized pathologically by amyloid plaques, composed of aggregated amyloid β (Aβ) peptide, and neurofibrillary tangles, comprised of hyperphosphorylated tau (Braak and Braak, 1995; Alzheimer's Association, 2015). Vascular cognitive impairment and dementia (VCID) is the second most common form of dementia and results from disruptions in blood flow to the brain, such as stroke, hypoperfusion of the brain, microhemorrhages, and vasogenic edema (Gorelick et al., 2011). VCID and AD are not mutually exclusive though; it is estimated that 40% of AD patients also have some form of VCID (Bowler et al., 1998; Zekry et al., 2002; Langa et al., 2004; Van Iterson et al., 2015).
The most common hypothesis for the progression of AD is the amyloid cascade hypothesis, which states that Aβ aggregation leads to hyperphosphorylation of tau and tangle formation, which then leads to neurodegeneration (Hardy and Higgins, 1992; Hardy and Selkoe, 2002). As the leading hypothesis for the progression of AD, amyloid deposition and brain Aβ have become popular targets for many AD therapeutics. Immunotherapy targeting Aβ has been one of the most promising therapeutic interventions for AD since its initial report in 1999 (Schenk et al., 1999). Anti-Aβ immunotherapy uses antibodies targeting Aβ to reduce brain Aβ burden. Proposed mechanisms of action include microglial recruitment and Fcγ receptor-mediated phagocytosis (Bard et al., 2000; Wilcock et al., 2003), catalytic disaggregation and solublization (Solomon et al., 1997), and the peripheral sink mechanism (DeMattos et al., 2001). The relative contribution of each mechanism on the overall efficacy of the antibody likely relies on the isotype and epitope of the specific antibody (Bard et al., 2000; Bacskai et al., 2001; Wilcock et al., 2003; Wilcock and Colton, 2009; Zago et al., 2012).
While preclinical mouse model studies have repeatedly demonstrated significant efficacy of anti-Aβ immunotherapy, both in reducing amyloid burden and improving cognition (Schenk et al., 1999; Morgan et al., 2000; Wilcock et al., 2004a, b), the clinical trials have remained underwhelming, failing to meet primary endpoints for efficacy in a number of trials to date (Salloway et al., 2014; Siemers et al., 2016). While there has been discussion in the field that one reason for the lack of efficacy in clinical trials is that the disease is too advanced, we hypothesize that one reason for the lack of efficacy in clinical trials is the comorbidity of other dementia-causing pathologies, such as VCID, as well as AD pathology.
To test our hypothesis, we are using our recently developed mouse model of amyloid deposition comorbid with VCID (APP/PS1 + HHcy) (Sudduth et al., 2014), which develops additive cognitive deficits and redistribution of amyloid favoring the deposition of cerebral amyloid angiopathy (CAA). Using our comorbidity model, we administered 3D6, an anti-Aβ antibody, or IgG2a, a control antibody to test passive immunotherapy efficacy in a mixed dementia mouse model. We found that there was no cognitive benefit of immunotherapy in our comorbidity model despite comparable reductions in total amyloid burden. This was accompanied by a reduced immune response to the immunotherapy.
Materials and Methods
Animals.
Female and male wild-type (WT) and APP/PS1 mice (C57BL/6 mice carrying human APPSwe and PS1-dE9 mutations) (Jankowsky et al., 2004) were bred in house and aged 9 months. Thirty-five WT mice and 33 APP/PS1 mice were placed on either a diet with low levels of folate, vitamins B6 and B12 and enriched with methionine (HHcy study group) or a control diet with normal levels of folate, vitamins B6 and B12 and methionine (control study group). The HHcy diet was Harlan Teklad TD97345, and the control diet was Harlan Teklad 5001 C (both from Harlan Laboratories). Once mice had received the diet for 3 months, they began weekly intraperitoneal injections of either 3D6 or IgG2a at 10 mg/kg (Eli Lilly) for 3 months. The anti-Aβ antibody 3D6, which is an IgG2b isotype, binds to both soluble and insoluble Aβ at the extreme amino terminus (Aβ1–5) (Racke et al., 2005; Demattos et al., 2012). Although the control antibody and the anti-Aβ antibody are not similar isotypes, they both have similar immune functions, with IgG2a actually having a higher affinity for FcγR1 (Gessner et al., 1998). Control IgG2a antibodies have been shown to have no effect on amyloid burden (Demattos et al., 2012). This study was approved by the University of Kentucky Institutional Animal Care and Use Committee and conformed to the National Institutes of Health Guide for the care and use of animals in research. Behavioral testing and MRI were also performed, and an experimental timeline with the number of animals per group is shown in Figure 1A, B.
Figure 1.
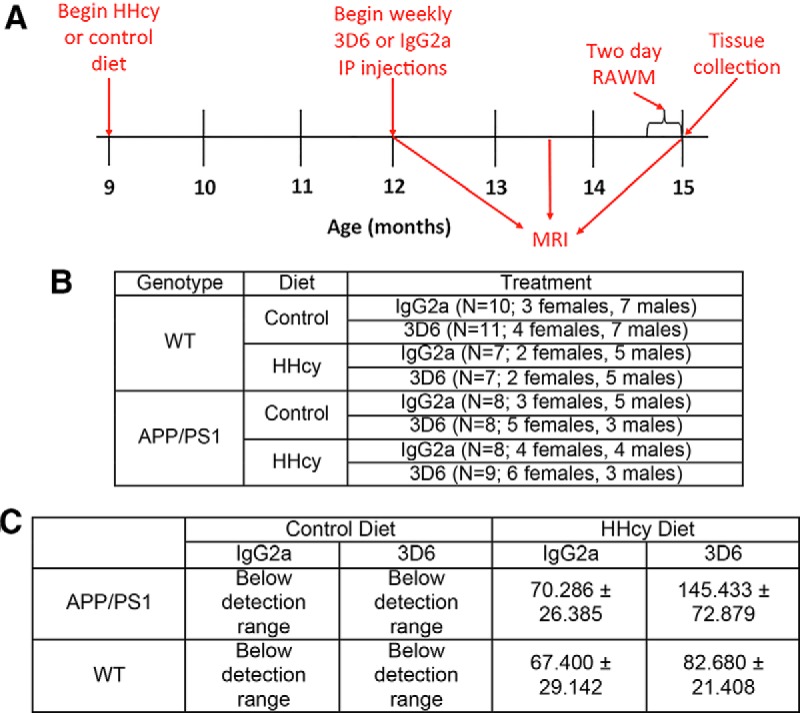
Experimental setup. A, Mice were 9 months old when they began either the control or HHcy diet. After being on the diet for 3 months, mice began a 3 month treatment of weekly intraperitoneal injections of 3D6 or IgG2a at 10 mg/kg. MRI was performed 3 months after starting the diet but before intraperitoneal injection treatment, halfway through treatment, and immediately before tissue collection. The 2 d radial arm water maze was also performed before tissue collection. B, Number of animals per experimental group. C, Quantification of plasma total homocysteine levels.
Behavior testing.
Radial arm water maze (RAWM) testing was performed at the University of Kentucky Rodent Behavior Core. The 2 d RAWM was performed as previously described (Alamed et al., 2006). Briefly, a six arm maze was submerged in a pool of water with a goal platform placed at the end of one arm. Each mouse was subjected to 15 trials each day, with the mouse starting in a different arm for each trial and the goal platform remaining in the same arm. The number of errors (the number of incorrect arm entries) was counted over a 60 s period. Errors were averaged for 3 trials resulting in 10 blocks over the 2 d. Blocks 1–5 comprise day 1 trials, whereas blocks 6–10 comprise day 2 trials.
MRI.
A subset of study mice were imaged by T2* MRI. Three or four mice from each group were imaged 3 months after starting diet but before beginning intraperitoneal antibody injections, again halfway through intraperitoneal antibody injections, and a third time immediately before tissue collection. Mice were imaged with a 7-T Bruker ClinScan MRI system (Bruker) with an MRI CryoProbe, delivering 2.5× the signal to noise of a standard room temperature radiofrequency coil, located at the Magnetic Resonance Imaging and Spectroscopy Center at the University of Kentucky. Fourteen coronal slices were acquired with a FLASH sequence with a TR 165 ms, TE 15.3 ms, 25 degree flip angle, 448 × 448 matrix, 0.4 mm thick, 20% gap, 0.033 mm × 0.033 mm resolution, 10 averages, and TA 24 min. Mice were anesthetized with 1.3% isoflorane using an MRI compatible vaporizer. They were then positioned prone and held in place on the scanning bed using tooth and ear bars. The animals were maintained at 37° with a water heated scanning bed. Body temperature, heart, and respiration rates were monitored. T2* MRI images were analyzed by one blinded investigator who identified abnormalities that resembled hemorrhagic infarcts. These infarcts were counted, and this number was normalized to the number of images counted to provide a per section count.
Tissue processing and histology.
After a lethal injection of Beuthanasia-D, blood was collected for plasma and mice were perfused intracardially with 25 ml normal saline. Brains were removed rapidly and bisected in the midsaggital plane with the left side being immersion fixed in 4% PFA for 24 h. The right side was dissected with the frontal cortex and hippocampus flash frozen in liquid nitrogen and then stored at −80°C. The left hemibrain was passed through a series of 10%, 20%, and 30% sucrose solutions for cryoprotection before sectioning. Using a sliding microtome, 25 μm frozen horizontal sections were collected and stored free floating in 1× DPBS-containing sodium azide at 4°C.
Eight sections spaced 600 μm apart were selected for free floating immunohistochemistry for CD11b (rat monoclonal, AbD Serotec, 1:1000 dilution) and total Aβ (rabbit polyclonal, Invitrogen, 1:3000 dilution). Immunohistochemistry was performed as previously described (Wilcock et al., 2008). Stained sections were mounted, air dried overnight, dehydrated, and coverslipped in DPX (Electron Microscopy Sciences). Analysis was performed by measuring the percent area occupied by positive immunostain using the Nikon Elements AR image analysis system as described previously (Sudduth et al., 2012).
Eight sections spaced 600 μm apart were mounted on slides and stained for Congo red and Prussian blue as previously described (Wilcock et al., 2004b, 2006). Neutral red was used as a counterstain for Prussian blue. Congo red analysis was performed using the Zeiss Axio Scan.Z1 Slide Scanner (Carl Zeiss Microscopy) and the Nikon Elements AR image analysis system.
Plasma was analyzed for total homocysteine by the Clinical Laboratory of the University of Kentucky Hospital Laboratories.
Aβ ELISA.
Biochemical analysis of Aβ levels was performed as previously described (Weekman et al., 2014). Briefly, soluble protein was extracted from the right frontal cortex using PBS with complete protease and phosphatase inhibitor (Pierce Biotechnology). After centrifugation, the supernatant was labeled the “soluble” extract, and the pellet was homogenized in 70% formic acid. After centrifugation and neutralization with 1 m Tris-HCl, the supernatant was labeled the “insoluble” extract. Protein concentrations were determined using the BCA protein assay kit (Pierce Biotechnology). The Meso-Scale Discovery multiplex ELISA system was used to measure Aβ1–38, Aβ1–40, and Aβ1–42 levels in the soluble and insoluble extracts (V-PLEX Aβ Peptide Panel 1 (6E10) Kit; MSD).
Quantitative RT-PCR.
RNA was extracted from the right hippocampus using the E.Z.N.A. Total RNA kit (Omega Bio-Tek) according to the manufacturer's instructions. RNA was quantified using the Biospec nano spectrophotometer. cDNA was produced using the cDNA High Capacity kit (ThermoFisher) according to the manufacturer's instructions. RT-PCR was performed using the Fast TaqMan Gene Expression assay (ThermoFisher). In each well of a 96 well plate, 0.5 μl cDNA (100 ng, based on the RNA concentrations) was diluted with 6.5 μl RNase-free water. One microliter of the appropriate gene probe was added along with 10 μl of Fast TaqMan to each well. Target amplification was performed using the ViiA7 (Applied Biosystems). All genes were normalized to 18S rRNA, and the fold change was determined using the −ΔΔCt method. Results from all APP/PS1 mice were compared with APP/PS1 mice on control diet with IgG2a treatment. Table 1 shows the genes tested along with their PMID and TaqMan ID.
Table 1.
Genes for real-time PCR
| Gene of interest | PMID | TaqMan ID |
|---|---|---|
| IL1β | NM_008361.3 | Mm0.222830 |
| TNFα | NM_013693.3 | Mm0.1293 |
| IL12a | NM_008351.2 | Mm0.103783 |
| IL12b | NM_008352.2 | Mm0.239707 |
| Marco | NM_010766.2 | Mm0.1856 |
| IL10 | NM_010548.2 | Mm0.874 |
| ARG1 | NM_007482.3 | Mm0.154144 |
| YM1 | NM_009892.2 | Mm0.387173 |
| Fizz | NM_020509.3 | Mm0.441868 |
| IL1Ra | NM_031167.5 | Mm0.882 |
| CD86 | NM_019388.3 | Mm0.1452 |
| FcγR1 | NM_010186.5 | Mm0.150 |
| FcγR3 | NM_010188.5 | Mm0.22119 |
| TGF1β | NM_011577.1 | Mm0.248380 |
| MRC1 | NM_008625.2 | Mm0.2019 |
| MMP14 | NM_008608.3 | Mm0.280175 |
| MMP2 | NM_008610.2 | Mm0.29564 |
| TIMP2 | NM_011594.3 | Mm0.206505 |
| MMP3 | NM_010809.1 | Mm0.4993 |
| MMP9 | NM_013599.3 | Mm0.4406 |
| TIMP1 | NM_001044384.1 | Mm0.8245 |
Analysis.
Data are presented as mean ± SEM. Statistical analysis was performed using the JMP statistical analysis software program (SAS Institute). RAWM data were analyzed by repeated-measures ANOVA. We also performed Student's t test on individual block data for the radial arm water maze analysis. For other data, one-way ANOVA and Student's t test were performed. Statistical significance was assigned when the p value was <0.05.
Results
Administration of a diet deficient in folate, vitamins B6 and B12 and enriched in methionine to 9-month-old WT and APP/PS1 mice for 6 months resulted in elevated plasma homocysteine levels as shown in Figure 1C. In C57BL/6 mice, a level of homocysteine between 5 and 12 μm is considered normal. Elevated levels of homocysteine can be categorized as mild with levels between 12 and 30 μm, moderate with levels between 30 and 100 μm, and severe with levels >100 μm. The mice on control diet had plasma homocysteine levels below the detection range of the clinical assay. Mice on our normal house chow also have levels below the detection range showing that mice on the control diet have a normal level of homocysteine. Plasma homocysteine levels in our mice on the HHcy diet reached moderate to severe levels ranging from 67 to 145 μm in all cases, thus inducing hyperhomocysteinemia. Statistically, the only comparison showing significant differences was between the APP/PS1 mice on control diet receiving 3D6 and the APP/PS1 mice on the HHcy diet receiving the 3D6 treatment (Fig. 1C).
We performed the 2 d RAWM to identify changes in spatial memory. Over the 2 d of testing, the WT mice on control diet with IgG2a treatment learned the task. They started the task making an average of three errors in the first block and ended with less than one error in the last block (Fig. 2A). WT mice on the HHcy diet had similar cognitive deficits as previously reported (Sudduth et al., 2013), with no effect of 3D6 or IgG2a (Fig. 2A). APP/PS1 mice on control diet with IgG2a treatment did not learn the task over the 2 d and were significantly impaired compared with WT mice on control diet with IgG2a treatment by block 10, as expected from APP/PS1 mice of this age (Fig. 2B). APP/PS1 mice on control diet with 3D6 treatment began the task with an average of ∼4 errors in block 1 and were making less than one error by the last block, indicating that they did learn the task and they were indistinguishable from the WT mice on control diet with IgG2a treatment. APP/PS1 mice on the HHcy diet with either IgG2a or 3D6 treatment did not learn the task, making a similar amount of errors in the first and last block. At the end of day 2, they made significantly more errors than the WT mice on control diet with IgG2a treatment or the APP/PS1 mice on control diet with 3D6 treatment as shown in the block 10 data in Figure 2D. The APP/PS1/HHcy mice did not show the additive effects we have previously reported in this mixed pathology APP/PS1/HHcy mouse (Sudduth et al., 2014); however, as can be seen from the learning curve of the APP/PS1 mice, there was essentially a “ceiling effect,” likely due to the older age of the mice in the current study. Therefore, we were unable to detect any potential worsening resulting from the HHcy component.
Figure 2.

The 3D6 treatment does not improve cognition in APP/PS1 mice on the HHcy diet. Two day radial arm water maze data are shown for WT mice (A) and APP/PS1 mice (B). The mean number of errors per trial was calculated for each block; each block is the average of three trials. *p < 0.05, compared with all groups on each graph (one-way ANOVA). **p < 0.01, compared with all groups on each graph (one-way ANOVA). Block 10 data are graphed for the WT mice (C) and the APP/PS1 mice (D). C, **p < 0.01, compared with WT, control, IgG2a and WT, control, and 3D6. D, **p < 0.01, compared with WT, control, IgG2a and APP/PS1, control, and 3D6.
Total Aβ immunohistochemistry was used to determine the efficacy of an anti-Aβ immunotherapy to reduce Aβ levels in our comorbidity model. In the frontal cortex, total Aβ was significantly reduced in the APP/PS1 mice on control diet with 3D6 treatment compared with APP/PS1 mice on control diet with IgG2a treatment (Fig. 3E). Levels were slightly reduced in the APP/PS1 mice on the HHcy diet with 3D6 treatment but did not reach significance. In the hippocampus, total Aβ was significantly reduced in both the APP/PS1 mice on control and HHcy diet with 3D6 treatment compared with APP/PS1 mice on control diet with IgG2a treatment (Fig. 3A–E). When APP/PS1 mice on HHcy diet with control antibody were compared with APP/PS1 mice on HHcy diet with 3D6 antibody, there was no significant difference. Biochemical levels of Aβ were also measured and are shown in Figure 3F. No significant differences were found among any of the groups, likely due to the high variation among the individual samples.
Figure 3.

Total Aβ is reduced by 3D6 treatment. Representative images of Aβ staining in the hippocampus of APP/PS1, control, and IgG2a (A), APP/PS1, control, and 3D6 (B), APP/PS1, HHcy, and IgG2a (C), and APP/PS1, HHcy, and 3D6 (D). A, The cornu ammonis (CA) 1, CA3, and dentate gyrus (DG) are labeled for orientation. Scale bar: A, 120 μm. E, Quantification of percentage positive stain in the frontal cortex and hippocampus. *p < 0.05, compared with APP/PS1, control, and IgG2a. F, Biochemical quantification of soluble and insoluble Aβ1–38, Aβ1–40, and Aβ1–42 ± SEM.
Congo red analysis showed that amyloid levels were reduced in the parenchyma following 3D6 treatment in APP/PS1 mice on both the control diet and the HHcy diet when they were compared with APP/PS1 mice on control diet with control antibody (Fig. 4E). This effect was more pronounced in the hippocampus than the frontal cortex. In contrast to the total amyloid reductions, the APP/PS1 mice on the HHcy diet showed significant increases in CAA relative to the APP/PS1 mice on control diet. This effect was not influenced by 3D6 administration, replicating our previous report that HHcy leads to redistribution of amyloid to the vasculature (Sudduth et al., 2014). The 3D6 administration in the APP/PS1 mice on control diet caused a modest increase in CAA relative to control antibody.
Figure 4.

HHcy redistributes amyloid to the vasculature. Representative images of Congo red staining in the hippocampus of APP/PS1, control, and IgG2a (A), APP/PS1, control, and 3D6 (B), APP/PS1, HHcy, and IgG2a (C), and APP/PS1, HHcy, and 3D6 (D). A, The CA1, CA3, and DG are labeled for orientation. Scale bar: A, 200 μm. Representative images of Congo red staining in the cortex of APP/PS1, control, and IgG2a (E), APP/PS1, control, and 3D6 (F), APP/PS1, HHcy, and IgG2a (G), and APP/PS1, HHcy, and 3D6 (H). Scale bar: E, 50 μm. Black arrows indicate Congo staining around a vessel (CAA). I, Quantification of percentage positive stain in the frontal cortex and hippocampus. *p < 0.05, compared with APP/PS1, control, and IgG2a. **p < 0.01, compared with APP/PS1, control, and IgG2a.
We used T2* MRI imaging to detect microhemorrhages as described previously (Sudduth et al., 2013). At the first imaging session, 3 months into the diet but immediately before the initiation of anti-Aβ immunotherapy, we found modest, nonsignificant increases in microhemorrhages resulting from the administration of the HHcy-inducing diet, consistent with our previous reports (Fig. 5B). Six weeks into immunotherapy treatment, we found that there were significant increases in microhemorrhages resulting from the HHcy inducing diet; however, the control IgG2a and 3D6 groups were still comparable. In contrast, 12 weeks into immunotherapy treatment, immediately before cognitive testing and tissue harvest, we found that microhemorrhage numbers were significantly greater in the APP/PS1 mice on the HHcy or control diet receiving 3D6 antibody than either the APP/PS1 mice on control diet or the HHcy diet receiving IgG2a treatment.
Figure 5.

The 3D6 increases MRI detected microhemorrhages. A, Representative T2* images of 3D6-treated mice. Arrows indicate a microhemorrhage. B, Quantification of the number of microhemorrhages per section over the 3 imaging sessions. *p < 0.05, compared with APP/PS1, control, and IgG2a. **p < 0.01, compared with APP/PS1, control, and IgG2a.
Prussian blue histological analysis of microhemorrhages showed a significant increase in the number of microhemorrhages in each of the groups compared with APP/PS1 mice on control diet with IgG2a treatment (Fig. 6B). The APP/PS1 mice on the HHcy diet with IgG2a treatment also had significantly more microhemorrhages compared with the 3D6-treated APP/PS1 mice on control diet. Also, the APP/PS1 mice on the HHcy diet with 3D6 treatment had a significant increase in microhemorrhages compared with all the other groups. The majority of microhemorrhages were located in the parietal and occipital cortex with no differences in location between any of the groups. Images of a positive Prussian microhemorrhage are shown in Figure 6A.
Figure 6.

The 3D6 and HHcy increase Prussain blue detected microhemorrhages. A, Representative images of Prussian blue-positive microhemorrhages in the frontal cortex. Scale bar, 120 μm. B, Quantification of the mean number of microhemorrhages per section. **p < 0.01, compared with APP/PS1, control, and IgG2a. Black bars represent significant differences between connecting groups.
One of the proposed mechanisms for anti-Aβ immunotherapy's reduction of Aβ involves activation of microglia leading to phagocytosis of Aβ. We performed immunohistochemistry for CD11b, which labels both activated and resting microglia, to determine whether there was an increase in total microglia. In both the frontal cortex and the hippocampus, CD11b was slightly increased in the APP/PS1 mice on control diet with 3D6 treatment compared with the APP/PS1 mice on control diet with IgG2a treatment (Fig. 7E), but this was not significant. CD11b staining was significantly reduced in the frontal cortex in the APP/PS1 mice on the HHcy diet with 3D6 treatment compared with the APP/PS1 mice on control diet with 3D6 treatment. In the hippocampus, the APP/PS1 mice on the HHcy diet with IgG2a or 3D6 treatment had significantly reduced levels of CD11b staining compared with the APP/PS1 mice on control diet with 3D6 treatment (Fig. 7A–E). The amount of CD11b staining was also slightly reduced in the APP/PS1 mice on the HHcy diet with IgG2a or 3D6 treatment compared with APP/PS1 mice on control diet with IgG2a treatment.
Figure 7.

CD11b staining is decreased in the HHcy groups. Representative images of CD11b staining in the hippocampus of APP/PS1, control, and IgG2a (A), APP/PS1, control, and 3D6 (B), APP/PS1, HHcy, and IgG2a (C), and APP/PS1, HHcy, and 3D6 (D). A, The CA1, CA3, and DG are labeled for orientation. Scale bar: A, 120 μm. Higher-magnification images of CD11b staining in the dentate gyrus of APP/PS1, control, and IgG2a (E), APP/PS1, control, and 3D6 (F), APP/PS1, HHcy, and IgG2a (G), and APP/PS1, HHcy, and 3D6 (H). Scale bar: E, 50 μm. I, Quantification of percentage positive stain in the frontal cortex and hippocampus.
Using several genetic markers specific to a proinflammatory, wound healing/repair or immune complex-mediated macrophage phenotype (Mantovani et al., 2005; Walker and Lue, 2015), we characterized the neuroinflammatory response. The data in Figure 8 are shown as a fold change from the APP/PS1 mice on control diet with IgG2a treatment. The APP/PS1 mice on control diet with 3D6 treatment showed a significant increase in several proinflammatory markers (specifically IL1β and TNFα) and increases in several immune complex-mediated genes (FcγR1 and FcγR3) (Fig. 8A,C). The APP/PS1 mice on the HHcy diet with IgG2a treatment showed elevations in several wound healing/repair genes (ARG1) (Fig. 8B). The APP/PS1 mice on the HHcy diet with 3D6 treatment only showed significant changes in some immune complex-mediated genes (FcγR1 and FcγR3) (Fig. 8C). There were also several significant increases in the expression of the MMP2 and MMP9 system markers as shown in Figure 8D. The APP/PS1 mice on control diet with 3D6 treatment showed a significant increase in MMP2, MMP3, and MMP9. The APP/PS1 mice on the HHcy diet showed an increase in MMP9 expression as well.
Figure 8.

HHcy reduced inflammatory markers and increased the MMP system markers in both the IgG2a and 3D6 groups. Data are shown as a fold change from APP/PS1, control, and IgG2a. Relative gene expression for proinflammatory markers (A), would healing/repair markers (B), immune complex-mediated markers (C), and MMP2 and MMP9 system markers (D). *p < 0.05, compared with APP/PS1, control, and IgG2a. **p < 0.01, compared with APP/PS1, control, and IgG2a.
Discussion
While AD is the most common form of dementia, VCID is the second most common, and it is estimated that 40% of AD patients also have some form of VCID (Bowler et al., 1998; Zekry et al., 2002; Langa et al., 2004; Van Iterson et al., 2015). One promising therapeutic for AD is anti-Aβ immunotherapy, which uses antibodies against Aβ to clear it from the brain. In mice, anti-Aβ immunotherapy significantly reduced Aβ levels and improved cognitive outcomes (Schenk et al., 1999; Morgan et al., 2000; Wilcock et al., 2004a, b); however, in clinical trials, anti-Aβ immunotherapy failed to reach primary cognitive outcomes in several different clinical trials (Salloway et al., 2014; Siemers et al., 2016). In addition, anti-Aβ immunotherapy has been associated with adverse cerebrovascular events. In mice, these manifested as microhemorrhages (Pfeifer et al., 2002; Wilcock et al., 2004b; Racke et al., 2005), whereas in clinical trials, it presented as vasogenic edema, detected using MRI imaging techniques. These adverse events were termed amyloid related imaging abnormalities (ARIA). Under this term, they were called ARIA-H (hemorrhagic) and ARIA-E (vasogenic edema) (Sperling et al., 2011). We hypothesized that one potential reason the anti-Aβ immunotherapy clinical trials were unsuccessful, both cognitively and with respect to the ARIA, was due to this high percentage of VCID comorbidity in the AD population. We used our unique model of VCID-amyloid comorbidity to test this hypothesis.
We placed 9-month-old WT and APP/PS1 mice on either a control diet or a diet that induces HHcy. Our laboratory has previously shown that a diet deficient in vitamins B6 and B12 and folate and enriched in methionine induces HHcy (Sudduth et al., 2014), which is a risk factor for VCID and cardiovascular disease (Bostom et al., 1999; Eikelboom et al., 1999). After being placed on the diet for 3 months, the mice then received intraperitoneal injections of either IgG2a or 3D6 for another 3 months. While we found that treatment of our comorbidity model with 3D6 resulted in decreased total Aβ levels, there was no cognitive benefit of the anti-Aβ immunotherapy in our AD/VCID mice. Further, microhemorrhages were increased by 3D6 in the APP/PS1 on control diet but further increased in an additive fashion when 3D6 was administered to the APP/PS1/HHcy mice.
Total Aβ deposition, which is mostly diffuse deposition, was reduced in the 3D6-treated animals, both APP/PS1 and APP/PS1/HHcy compared with APP/PS1 controls. Total Aβ deposition in the APP/PS1 mice on HHcy diet with 3D6 were not significantly different compared with APP/PS1 mice on HHcy diet with IgG2a, although they did show trends for reduction. We found that amyloid distribution was altered when we examined Congo red staining. When we looked at parenchymal versus vascular Congophilic amyloid deposition using our previously described method (Wilcock et al., 2006), we saw that the HHcy diet resulted in an apparent redistribution of the amyloid to the vasculature, as CAA, replicating our previous finding (Sudduth et al., 2014). We did not find that 3D6 altered this outcome. Interestingly, despite the comparable levels of CAA between the 3D6-treated and control antibody-treated APP/PS1/HHcy mice, microhemorrhages were significantly increased in the 3D6-treated mice, indicating that the microhemorrhages do not occur simply due to the presence of CAA, but that the anti-Aβ antibody causes a local reaction at the vessel, resulting in leakage.
We have previously shown that MMP activation, in particular MMP9, is associated with tight junction breakdown and microhemorrhages, both in response to immunotherapy (Wilcock et al., 2011) and in the HHcy model (Sudduth et al., 2013, 2014). MMP9 has been implicated in cerebrovascular injury, including hemorrhagic transformation after stroke and microhemorrhage occurrence in CAA (Lee et al., 2003; Jickling et al., 2014). In our study, both 3D6 and HHcy significantly increased gene expression of MMP9, similar to previous studies. MMP3, an activator of MMP9, was also increased in the 3D6-treated control APP/PS1 mice.
It is unclear what mechanism is responsible for the increased MMP9 and MMP3 in the current study. MMP9 and MMP3 can be activated by proinflammatory cytokines, such as IL1β and TNFα (Galis et al., 1994; Vecil et al., 2000). As a result of this literature and our previous data (Sudduth et al., 2013; Sudduth et al., 2014), we explored neuroinflammation in the current study. Our findings were intriguing and suggest that the immunotherapy did not result in an overt inflammatory response by the microglia. TNFα and IL1β were only significantly increased in the 3D6-treated control APP/PS1 mice and not in either of the HHcy-treated groups. Indeed, both the HHcy-treated groups had a slight reduction in CD11b staining and did not show any changes in the majority of markers for any of the neuroinflammatory states. In our previous study, we showed that the APP/PS1 mice on the HHcy diet had a neuroinflammatory shift from wound healing and repair to a proinflammatory state (Sudduth et al., 2014). Because of the older age of the mice in this study (15 months rather than 12 months), and the addition of a treatment arm, the mice could have progressed past this neuroinflammatory shift. Unfortunately, we are only able to assess the changes postmortem, and thus cannot know what changes preceded those reported here. Based on our data, we hypothesize that either there is a decrease in the numbers of microglial cells or the microglia could be senescent. The constant activation from high Aβ levels, long-term HHcy, and weekly administration of antibodies could force the microglia into senescence, resulting in the decreased CD11b staining and lack of immune activation. It is also possible that the decreased CD11b staining, which also labels other myeloid cells like monocytes, could also result from HHcy suppression of infiltration of peripheral cells, although this seems unlikely because the HHcy diet results in a microhemorrhage induction. Therefore, while MMP9 may be responsible for the tight junction breakdown leading to microhemorrhages, proinflammatory cytokines do not seem to be the stimulating factor in the current study.
Previous anti-Aβ immunotherapy studies have shown that the use of anti-Aβ antibodies significantly improves cognitive outcomes in mice (Schenk et al., 1999; Morgan et al., 2000; Wilcock et al., 2004a, b). While we saw that 3D6 treatment significantly reduced the number of errors APP/PS1 mice on control diet made making then indistinguishable from the WT controls, the presence of HHcy impeded those benefits. The APP/PS1 mice on the HHcy diet with 3D6 treatment did not benefit cognitively from the anti-Aβ immunotherapy. Indeed, they were not significantly different from the APP/PS1 mice on the HHcy diet with IgG2a treatment by the end of the second day. This lack of cognitive benefits could be due to the increase in the number of microhemorrhages seen in the 3D6-treated HHcy mice compared with the 3D6 or HHcy controls, or the increased levels of CAA. We cannot rule out that there may have been a benefit to the APP/PS1/HHcy that was undetected in our selected behavioral task due to wild-type HHcy mice also reaching a ceiling effect. Future studies will try to develop more sensitive behavioral tasks that may discriminate these effects. Despite the Aβ reductions in the APP/PS1/HHcy mice, the increase in vascular damage and leakage appears to negate any cognitive benefits of clearing the Aβ. This suggests that anti-Aβ immunotherapy in patients with VCID comorbid with AD may be ineffective with respect to functional outcomes. Prescreening subjects for cerebrovascular pathologies before enrollment in anti-Aβ immunotherapy clinical trials may reduce the adverse cerebrovascular events seen during treatment, and this could result in significantly improved cognitive outcomes. Although anti-Aβ immunotherapy trials are moving toward treatment of prodromal AD, VCID still remains a concern because the immunotherapy could hasten any vascular dysfunction that is present but asymptomatic.
Footnotes
This work was supported by Fellowship F31NS092202 to E.M.W. and National Institutes of Health Grant 1RO1NS079637 to D.M.W. These studies were assisted by the Magnetic Resonance Imaging and Spectroscopy Center at the University of Kentucky (National Institutes of Health shared instruments Grant 1S10RR029541). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Eli Lilly for providing the 3D6 and IgG2a antibodies for this study.
The authors declare no competing financial interests.
References
- Alamed J, Wilcock DM, Diamond DM, Gordon MN, Morgan D. Two-day radial-arm water maze learning and memory task: robust resolution of amyloid-related memory deficits in transgenic mice. Nat Protoc. 2006;1:1671–1679. doi: 10.1038/nprot.2006.275. [DOI] [PubMed] [Google Scholar]
- Alzheimer's Association. 2015 Alzheimer's disease facts and figures. Alzheimers Dement. 2015;11:332–384. doi: 10.1016/j.jalz.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–372. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Bostom AG, Rosenberg IH, Silbershatz H, Jacques PF, Selhub J, D'Agostino RB, Wilson PW, Wolf PA. Nonfasting plasma total homocysteine levels and stroke incidence in elderly persons: the Framingham Study. Ann Intern Med. 1999;131:352–355. doi: 10.7326/0003-4819-131-5-199909070-00006. [DOI] [PubMed] [Google Scholar]
- Bowler JV, Munoz DG, Merskey H, Hachinski V. Fallacies in the pathological confirmation of the diagnosis of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1998;64:18–24. doi: 10.1136/jnnp.64.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–278. doi: 10.1016/0197-4580(95)00021-6. discussion 278–284. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, Hole JT, Forster BM, McDonnell PC, Liu F, Kinley RD, Jordan WH, Hutton ML. A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer's disease mice. Neuron. 2012;76:908–920. doi: 10.1016/j.neuron.2012.10.029. [DOI] [PubMed] [Google Scholar]
- Eikelboom JW, Lonn E, Genest J, Jr, Hankey G, Yusuf S. Homocyst(e)ine and cardiovascular disease: a critical review of the epidemiologic evidence. Ann Intern Med. 1999;131:363–375. doi: 10.7326/0003-4819-131-5-199909070-00008. [DOI] [PubMed] [Google Scholar]
- Galis ZS, Muszynski M, Sukhova GK, Simon-Morrissey E, Unemori EN, Lark MW, Amento E, Libby P. Cytokine-stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circ Res. 1994;75:181–189. doi: 10.1161/01.RES.75.1.181. [DOI] [PubMed] [Google Scholar]
- Gessner JE, Heiken H, Tamm A, Schmidt RE. The IgG Fc receptor family. Ann Hematol. 1998;76:231–248. doi: 10.1007/s002770050396. [DOI] [PubMed] [Google Scholar]
- Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, Lu A, Sharp FR. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 2014;34:185–199. doi: 10.1038/jcbfm.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langa KM, Foster NL, Larson EB. Mixed dementia: emerging concepts and therapeutic implications. JAMA. 2004;292:2901–2908. doi: 10.1001/jama.292.23.2901. [DOI] [PubMed] [Google Scholar]
- Lee JM, Yin KJ, Hsin I, Chen S, Fryer JD, Holtzman DM, Hsu CY, Xu J. Matrix metalloproteinase-9 and spontaneous hemorrhage in an animal model of cerebral amyloid angiopathy. Ann Neurol. 2003;54:379–382. doi: 10.1002/ana.10671. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–346. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, Guenzler V, Miloslavsky M, Wang D, Lu Y, Lull J, Tudor IC, Liu E, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N Engl J Med. 2014;370:322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Siemers ER, Sundell KL, Carlson C, Case M, Sethuraman G, Liu-Seifert H, Dowsett SA, Pontecorvo MJ, Dean RA, Demattos R. Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer's disease patients. Alzheimers Dement. 2016;12:110–120. doi: 10.1016/j.jalz.2015.06.1893. [DOI] [PubMed] [Google Scholar]
- Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci U S A. 1997;94:4109–4112. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Jack CR, Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367–385. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudduth TL, Wilson JG, Everhart A, Colton CA, Wilcock DM. Lithium treatment of APPSwDI/NOS2−/− mice leads to reduced hyperphosphorylated tau, increased amyloid deposition and altered inflammatory phenotype. PLoS One. 2012;7:e31993. doi: 10.1371/journal.pone.0031993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudduth TL, Powell DK, Smith CD, Greenstein A, Wilcock DM. Induction of hyperhomocysteinemia models vascular dementia by induction of cerebral microhemorrhages and neuroinflammation. J Cereb Blood Flow Metab. 2013;33:708–715. doi: 10.1038/jcbfm.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudduth TL, Weekman EM, Brothers HM, Braun K, Wilcock DM. Beta-amyloid deposition is shifted to the vasculature and memory impairment is exacerbated when hyperhomocysteinemia is induced in APP/PS1 transgenic mice. Alzheimers Res Ther. 2014;6:32. doi: 10.1186/alzrt262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Iterson EH, Snyder EM, Joyner MJ, Johnson BD, Olson TP. Intrathecal fentanyl blockade of afferent neural feedback from skeletal muscle during exercise in heart failure patients: influence on circulatory power and pulmonary vascular capacitance. Int J Cardiol. 2015;201:384–393. doi: 10.1016/j.ijcard.2015.08.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecil GG, Larsen PH, Corley SM, Herx LM, Besson A, Goodyer CG, Yong VW. Interleukin-1 is a key regulator of matrix metalloproteinase-9 expression in human neurons in culture and following mouse brain trauma in vivo. J Neurosci Res. 2000;61:212–224. doi: 10.1002/1097-4547(20000715)61:2<212::AID-JNR12>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther. 2015;7:56. doi: 10.1186/s13195-015-0139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekman EM, Sudduth TL, Abner EL, Popa GJ, Mendenhall MD, Brothers HM, Braun K, Greenstein A, Wilcock DM. Transition from an M1 to a mixed neuroinflammatory phenotype increases amyloid deposition in APP/PS1 transgenic mice. J Neuroinflammation. 2014;11:127. doi: 10.1186/1742-2094-11-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Colton CA. Immunotherapy, vascular pathology, and microhemorrhages in transgenic mice. CNS Neurol Disord Drug Targets. 2009;8:50–64. doi: 10.2174/187152709787601858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004a;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004b;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Gordon MN, Morgan D. Quantification of cerebral amyloid angiopathy and parenchymal amyloid plaques with Congo red histochemical stain. Nat Protoc. 2006;1:1591–1595. doi: 10.1038/nprot.2006.277. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, Previti ML, Gharkholonarehe N, Vitek MP, Colton CA. Progression of amyloid pathology to Alzheimer's disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J Neurosci. 2008;28:1537–1545. doi: 10.1523/JNEUROSCI.5066-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Morgan D, Gordon MN, Taylor TL, Ridnour LA, Wink DA, Colton CA. Activation of matrix metalloproteinases following anti-Abeta immunotherapy: implications for microhemorrhage occurrence. J Neuroinflammation. 2011;8:115. doi: 10.1186/1742-2094-8-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zago W, Buttini M, Comery TA, Nishioka C, Gardai SJ, Seubert P, Games D, Bard F, Schenk D, Kinney GG. Neutralization of soluble, synaptotoxic amyloid beta species by antibodies is epitope specific. J Neurosci. 2012;32:2696–2702. doi: 10.1523/JNEUROSCI.1676-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zekry D, Hauw JJ, Gold G. Mixed dementia: epidemiology, diagnosis, and treatment. J Am Geriatr Soci. 2002;50:1431–1438. doi: 10.1046/j.1532-5415.2002.50367.x. [DOI] [PubMed] [Google Scholar]