

Abstract
Over the last decade, the field of imaging genomics has combined high-throughput genotype data with quantitative magnetic resonance imaging (QMRI) measures to identify genes associated with brain structure, cognition, and several brain-related disorders. Despite its successful application in different psychiatric and neurological disorders, the field has yet to be advanced in epilepsy. In this article we examine the relevance of imaging genomics for future genetic studies in epilepsy from three perspectives. First, we discuss prior genome-wide genetic mapping efforts in epilepsy, considering the possibility that some studies may have been constrained by inherent theoretical and methodological limitations of the genome-wide association study (GWAS) method. Second, we offer a brief overview of the imaging genomics paradigm, from its original inception, to its role in the discovery of important risk genes in a number of brain-related disorders, and its successful application in large-scale multinational research networks. Third, we provide a comprehensive review of past studies that have explored the eligibility of brain QMRI traits as endophenotypes for epilepsy. While the breadth of studies exploring QMRI-derived endophenotypes in epilepsy remains narrow, robust syndrome-specific neuroanatomical QMRI traits have the potential to serve as accessible and relevant intermediate phenotypes for future genetic mapping efforts in epilepsy.
Keywords: Endophenotypes, Epilepsy, Imaging genomics, Magnetic resonance imaging
Highlights
-
•
QMRI traits have the potential to serve as robust intermediate phenotypes for brain-related disorders.
-
•
Hippocampal volume is the most promising neuroimaging endophenotype for MTLE + HS.
-
•
Imaging genomics holds great promise in advancing epilepsy genetic research.
-
•
Studies are encouraged to explore the validity of QMRI traits as endophenotypes for epilepsy.
1. Introduction
Epilepsy is the most common serious neurological disorder. It comprises a heterogeneous group of epilepsy syndromes characterized by a shared predisposition to develop unprovoked epileptic seizures (Fisher et al., 2014). Based on the origin of seizure activity, epileptic seizures and syndromes can be focal or generalized (Berg et al., 2010). The underlying cause varies greatly across different epilepsy syndromes. Estimates from twin studies indicate a significant genetic role in most epilepsy syndromes (Miller et al., 1998, Kjeldsen et al., 2001). Newly developed techniques to estimate heritability from dense genotype data have indicated that at least 26% of the phenotypic variation in epilepsy is collectively explained by genetic variants occurring commonly in the general population (Speed et al., 2014). Despite the evidence of heritability, most epilepsy-causing genetic mutations have been identified by linkage analyses in families with Mendelian patterns of inheritance, which account for < 1% of all epilepsy syndromes. Common epilepsies often exhibit a complex or obscure inheritance pattern: causation may be attributable to multiple genetic variants interacting with each other and with environmental factors. Although steady progress is being made, the genetic determinants underlying most common epilepsy syndromes remain largely unknown.
2. Genetic studies of complex epilepsies - current constraints and limitations
Several single-gene mutations have been linked to familial forms of epilepsy (Hardies et al., 2016). For example, familial forms of genetic generalized epilepsies (GGEs) have been linked to mutations in the SLC2A1 gene that encodes GLUT1, a cell membrane glucose transporter (Striano et al., 2012). Mutations in LGI1 have been associated with familial focal epilepsies, including autosomal-dominant lateral temporal lobe epilepsy (ADLTLE) (Kalachikov et al., 2002). Interestingly, pleiotropy is not uncommon: a number of genes which when mutated can cause epilepsy are associated with diverse epilepsy phenotypes. For example, mutations in the genes SCN1A, SCN2A, SCN1B, KCNQ2, DEPDC5 and TBC1D24 can all cause a variety of epilepsies (Gambardella and Marini, 2009, Dibbens et al., 2013, Allen et al., 2014, Balestrini et al., 2016).
The genetic determinants of common epilepsy syndromes, where family history is lacking or suggests a complex inheritance pattern, are much less clearly defined. Their genetic architecture has been explored in many candidate gene studies, deep sequencing analyses and genome-wide association studies (GWASs; Helbig et al., 2008, Leu et al., 2016). Rare sequence and copy number variants of large effect have been identified in childhood absence epilepsies (Chen et al., 2003), GGEs (Helbig et al., 2009) and other epilepsy phenotypes (Heinzen et al., 2010, Klassen et al., 2011). GWAS has shown that common polymorphisms have a role in complex focal epilepsies with known or suspected brain lesions (Guo et al., 2012), mesial temporal lobe epilepsy (MTLE) with hippocampal sclerosis (Kasperaviciute et al., 2013) and complex generalized epilepsies (EPICURE Consortium et al., 2012). A recent meta-analysis of genome-wide data from 8696 epilepsy cases and 26,157 controls revealed two susceptibility genes, SCN1A and PCDH7, as risk loci across all forms of epilepsy, and a third risk locus (at VRK2 or FANCL) for generalized forms of the disorder (ILAE Consortium on Complex Epilepsies, 2014). Despite these promising findings, the replication rate and functional validation of susceptibility loci for complex epilepsies is poor (Leu et al., 2016).
Sample sizes need to be large for successful GWAS. Recent multisite collaborations, such as those led by the International League Against Epilepsy (ILAE) Consortium on Complex Epilepsies (http://www.ilae.org/Commission/class/consortium.cfm), address this by assembling larger cohorts of epilepsy cases and healthy controls, boosting statistical power. Even so, these genome-wide meta-analyses can aggregate data from diverse ethnic groups and samples can be clinically heterogeneous. Heterogeneity can make GWAS results harder to interpret, as the detected genome-wide signals may be driven by differences in linkage disequilibrium (LD) structure between sub-cohorts, uncontrolled effects of cryptic population stratification (McClellan and King, 2010), synthetic associations created by rare variants (Dickson et al., 2010, Wray et al., 2011), or overrepresented effects from specific epilepsy subtypes or sub-cohorts. Similarly, non-significant findings may be attributed, in part, to phenotype misclassifications (Manchia et al., 2013). GWAS will also miss any consistent effects of rare genetic variants (Gershon et al., 2011, Gibson, 2012).
GWAS was initiated based on a simple common disease-common variant (CDCV) hypothesis, which posited that complex disorders, such as epilepsy, are largely explained by a moderate number of variants occurring commonly (> 1%) in the general population (Botstein and Risch, 2003, Pritchard and Cox, 2002, Reich and Lander, 2001). This perspective has since evolved into several diverging hypotheses. Some argue that the genetic component of a disease is mainly due to a large number of common variants of small effect (the infinitesimal model). Others insist that it is due to a large number of rare variants of large effect (the rare allele model), and others still suggest a combination of genomic, environmental and epigenetic interactions (the broad sense heritability model; see Gibson, 2012, for a review). Recent genome-wide meta-analyses have integrated the broad-sense heritability hypothesis into their statistical designs using linear mixed models (LMM), which restrict heritability estimates to a certain class of single nucleotide polymorphisms (SNPs) in high LD with those on genotype platforms (Yang et al., 2010, Zaitlen and Kraft, 2012). Despite these advances, the number of novel genome-wide significant findings remains sparse, and the liability variance explained by each detected risk locus is low (often < 1%).
Typically, common susceptibility loci captured via case: control association studies exert subtle molecular and cellular alterations with remote effects on gene transcription, protein structure, or protein function (McCarthy and Hirschhorn, 2008). A complex clinical phenotype, such as seizure type or epilepsy syndrome, therefore tends to be indirectly predicted by any associated genotype. Although large-scale multisite meta-analyses may boost power to detect subtle biological effects, several creative alternatives may help to sidestep the complicated phenotype integrity of common epilepsy syndromes (Pal and Strug, 2014). One widely investigated alternative is the use of intermediate quantitative traits (QTs) that may be closer to the underlying biology of the disease than the clinical phenotype. These intermediate QTs are commonly referred to as endophenotypes.
3. Endophenotypes - definition and criteria
Endophenotypes are subclinical QTs, occupying the domain between disease phenotype and the genetic variants that influence disease risk (Gottesman and Gould, 2003). Unlike binary diagnostic outcomes (e.g. ‘patient’ versus ‘healthy control’), endophenotypes are quantifiable measures that are arguably closer to the genetic architecture of a disease than its clinical manifestation (e.g., seizure semiology) (Gould and Gottesman, 2006). In theory, endophenotypes allow us to deconstruct phenotypically heterogeneous syndromes, including epilepsy syndromes, into phenomena more reflective of the underlying genetic architecture, improving power to detect risk genes and their functional consequences (Walters and Owen, 2007).
Several criteria have been proposed to identify valid endophenotypes, since Gottesman & Shields' original (1973) conceptualization. Some corollaries and extensions continue to be discussed (see Miller and Rockstroh, 2013, for a review), but the following five criteria are generally accepted by the genetics community (Gould and Gottesman, 2006, Hasler et al., 2006). An endophenotype should be:
-
1.
Associated with illness in the relevant population
-
2.
State independent (i.e., it manifests in an individual regardless of whether an illness is active or not) and may require elicitation by a challenge (e.g., glucose tolerance test in relatives of patients with diabetes)
-
3.
Heritable
-
4.
Co-segregating with illness within families
-
5.
Present in non-affected family members at a higher rate than in the general population
In principle, endophenotypes may be biochemical, endocrinological, electrophysiological, cognitive, or anatomical in nature (Gould and Gottesman, 2006). A vast number of research tools have been applied to characterize relevant endophenotypes in brain-related disorders; ranging from eye movement recording (Mazhari et al., 2011), electroencephalography (EEG) and magnetic resonance imaging (MRI; Iacono and Malone, 2011), to behavioral measures and neuropsychological batteries (Glahn et al., 2004, Mann et al., 2009). MRI is arguably one of the most easily accessible and widely employed of these techniques. Indeed, in epilepsy, a large percentage of people with epilepsy undergo brain MRI as part of their clinical evaluation to look for potential underlying structural abnormality (e.g. malformation of cortical development or tumor). Quantitative MRI (QMRI) studies of people with epilepsy have characterized a wide range of subtle structural and functional brain alterations that are associated with particular epilepsy syndromes (Mueller et al., 2009). Some proportion of these subtle brain alterations may be due to early brain insults (e.g., central nervous system (CNS) infection or head trauma), disease-related factors (e.g. recurrent seizure activity or chronic medication use), or other environmental factors (Lewis et al., 2014). However, genetic mechanisms may also play a role, based on growing evidence from studies of asymptomatic first-degree relatives of epilepsy sufferers (Tsai et al., 2013).
4. QMRI traits as endophenotypes in genetic mapping studies: a promising future?
For a number of psychiatric illnesses, there is an ongoing quest to identify heritable, state-independent, neuroimaging-derived endophenotypes (Meyer-Lindenberg, 2010). For example, schizophrenia patients and their asymptomatic first-degree relatives appear to exhibit a similar pattern of brain structural alterations, including reduced prefrontal and temporal lobe grey matter (GM) volume (Cannon et al., 2002), vertical displacement of the corpus callosum (Narr et al., 2002), and inward deformity at the head of the hippocampus (Tepest et al., 2003). Similarly, in bipolar disorder, significant white matter (WM) alterations in the corpus callosum, posterior thalamic radiations and left superior longitudinal fasciculus have been reported in patients and their asymptomatic siblings (Sprooten et al., 2013). Also, in major depressive disorder, GM alterations within the ventral diencephalon have been associated, in part, with genetic risk factors (Glahn et al., 2012). Despite limitations attributed to variable sensitivity of different image analysis techniques and modest statistical power, several of these findings have been validated using larger sample sizes and standardized image processing algorithms (de Zwarte et al., 2016, Sprooten et al., 2016).
Many of the proposed neuroanatomical endophenotypes show moderate-to-high heritability in healthy and affected populations (Peper et al., 2007, Glahn et al., 2007, Goldman et al., 2008, den Braber et al., 2013). This has led some groups to pioneer a new research paradigm - known as imaging genomics - whereby neuroimaging-derived endophenotypes are used as quantitative phenotypes for genome-wide genetic mapping studies (Thompson et al., 2014). Under this imaging genomics model, a brain measure (such as the volume of a region of interest) is specified as a QT across a moderate-to-large sample of genome-wide genotyped participants. A regression model is then applied to test for an association between the additive allelic dosage of each SNP (independent variable) and the imaging phenotype of interest (dependent variable). Imaging genomics has successfully mapped the effects on the brain of a number of important genetic variants that are also associated with disease risk, sometimes using smaller samples than those required by traditional case-control association studies. In one of the first GWAS to be conducted directly on neuroimaging data, Potkin et al. (2009) analyzed genotypes and QMRI phenotype data from 381 Alzheimer's disease (AD) patients, collected as part of the Alzheimer's Disease Neuroimaging Initiative (ADNI). Although such a sample would rate as very small for a GWAS in general, the group still detected a significant association between variants in the TOMM40 and APOE genes and hippocampal volume (HV) in AD (Potkin et al., 2009). Shortly after the ADNI dataset was released to the public, over 100 discoveries were reported, relating differences in brain images to common variants in the BIN1, CLU, CR1 and PICALM genes - all of which were corroborated by imaging, cerebrospinal fluid (CSF) markers and cognitive phenotypes (see Shen et al., 2014, for a review).
In recent years, the field of imaging genomics has expanded into a number of multinational research networks. Some notable collaborations include the Early Growth Genetics (EGG) project, which identified a significant association between variants in the HMGA2 gene and head circumference at birth in a cohort of over 69,000 individuals (Horikoshi et al., 2013); the Enhancing Neuro Imaging through Meta-Analysis (ENIGMA) consortium (http://enigma.ini.usc.edu), which replicated the association between HMGA2 and intracranial volume, in addition to identifying a novel intergenic variant associated with HV in a sample of almost 8000 adult participants (Stein et al., 2012); and the Cohorts of Heart and Aging Research in Genomic Epidemiology (CHARGE) group, which reported variants associated with HV reduction (including a replication of the variant identified by ENIGMA) in a sample of over 9000 elderly participants (Bis et al., 2012).
There has been heated debate as to whether the penetrance of genetic variants is higher for these neuroimaging-derived endophenotypes than for complex, binary diagnostic outcomes (see Fig. 1; Franke et al., 2016). If effect sizes are greater for common variants that affect some key imaging traits, or if less of the genome explains more of the variance, imaging genomics may offer a more efficient method to discover genes involved in different neurological or psychiatric illnesses than traditional case–control association studies (Mier and Meyer-Lindenberg, 2009). Indeed, as imaging genomics collaborations, such as the ENIGMA consortium, continue to grow their sample sizes to over 30,000 participants, the discovery rate for novel genetic loci associated with important brain regions continues to increase (Hibar et al., 2015). The effects of loci associated with brain measures are actively being explored in many psychiatric and neurologic illnesses (Meyer-Lindenberg, 2010, Roussotte et al., 2014), and in animal models (Ashbrook et al., 2014). Imaging genomics in future may offer a new and powerful method to improve our understanding of epilepsy and its elusive genetic underpinnings. So far, several QMRI-based studies have attempted to validate an epilepsy-related neuroimaging endophenotype by exploring potential co-segregation of neuroanatomical and functional alterations in people with epilepsy and their asymptomatic first-degree relatives. The next section offers a comprehensive review of these investigations.
Fig. 1.
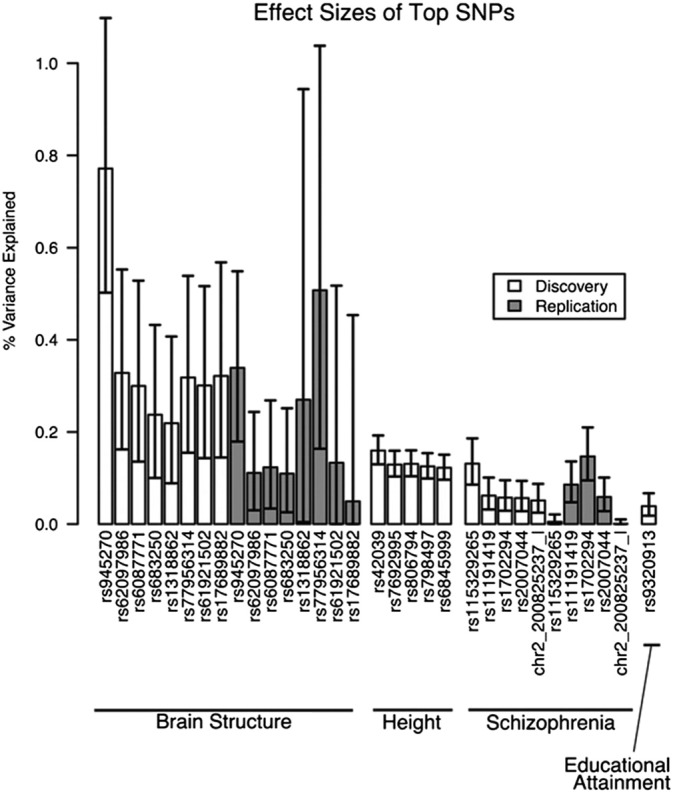
The maximum effect sizes for the top individual common genetic variants (both for discovery and replication samples) from genome-wide association studies of brain structure (for example, hippocampal volume), schizophrenia (using traditional case:control design), and other anthropomorphic traits including height and educational attainment are shown (Franke et al., 2016). For quantitative traits, effect sizes were measured in percent variance explained. For disease categories, effect sizes were measured in percent variance explained on the liability scale. [Image is reproduced with permission from nature publishing group (Nature Neuroscience, 2016)].
5. QMRI traits as endophenotypes for epilepsy
Over the last two decades, epilepsy-related EEG and brain QMRI traits have been evaluated as possible endophenotypes. Studies that examined the endophenotypic potential of QMRI traits in epilepsy are summarized in Table 1.
Table 1.
A summary of QMRI studies that investigated first-degree relatives of people with epilepsy (1998–2016).
| First author | Year | Sample | Method | Findings | Comment |
|---|---|---|---|---|---|
| Focal epilepsies | |||||
| Alhusaini | 2016 |
|
Surface-based morphometry (FreeSurfer) was applied to characterize temporal cortex morphology features | Altered temporal cortex morphology was identified in patients ipsilaterally within the anterio-medial temporal regions, including the entorhinal cortex, parahippocampal gyrus, and temporal pole. Subtle but similar pattern of morphology change was also noted in the asymptomatic siblings. This localized morphology alteration was related to volume loss that appeared driven by shared contraction in cerebral cortex surface area. | MTLE + HS patients and their asymptomatic siblings share a common pattern of temporal cortex morphologic alteration. |
| Whelan | 2015a |
|
DTI whole-brain voxel-wise statistics and deterministic tractography | Significant FA reductions in the corpus callosum (CC), bilateral superior longitudinal fasciculi (SLF), bilateral inferior longitudinal fasciculi (ILF), and ipsilateral corticospinal tract (CST) were noted in patients only. MD increases were observed in MTLE patients and their asymptomatic siblings in the ipsilateral SLF and CST. | SLF and CST microstructural alterations in patients with MRI-negative MTLE may partly be influenced by genetic factors. |
| Suemitsu | 2014 |
|
The degree of hippocampal T2 relaxometry changes was examined | In a cross-sectional design, elevated T2 relaxometry was identified in patients and intermediate values were noted in the asymptomatic relatives compared to controls. A longitudinal follow up over a mean of 4.4 year revealed significant increase in T2 relaxometry of the ipsilateral hippocampus exclusively in patients. With recurrent seizures |
Genetic factors may be involved in the development of some mild hippocampal abnormalities in FMTLE. |
| Tsai | 2013 |
|
Manual hippocampal volumetry | Mean hippocampal volume was smaller in the asymptomatic relatives compared to controls. Additionally, the asymptomatic relatives had more asymmetric hippocampi. This effect was greater in relatives of probands with a positive family history of epilepsy. | Small asymmetric hippocampi in healthy relatives of patients could represent a familial developmental variant that may predispose to the formation of MTLE + HS. |
| Alhusaini | 2013 |
|
FreeSurfer (an automated segmentation tool) | Volume deficits across the ipsilateral hippocampus, amygdala and thalamus were noted in MTLE + HS patients but not MRI-negative MTLE. These volume deficits were not present in the asymptomatic siblings of MTLE + HS patients. | Volume deficits for many subcortical structures in ‘sporadic’ MTLE + HS are largely driven by acquired factors. |
| Scanlon | 2013 |
|
FreeSurfer | A significant volume deficit in cerebral WM was common to patient and their siblings. | Cerebral WM volume deficit is common to MTLE + HS patients and their siblings. |
| Kobayashi | 2002 |
|
Manual hippocampal volumetry | Hippocampal atrophy was identified in 18 of 52 first-degree relatives of patients. Visual analysis of T1- and T2-weighted images showed additional classic MRI signs of HS in 14 of these 18 individuals | HS in FMTLE is influenced by a significant genetic predisposition. |
| Jackson | 1998 |
|
Manual hippocampal volumetry | HS was not identified in the unaffected twin using visual, volumetric, and T2 relaxometry criteria | The absence of HS in the unaffected twin is arguing against a strictly genetic hypothesis for HS. |
| Fernández | 1998 |
|
Manual hippocampal volumetry: the right/left ratios of hippocampal volumes (RHV) | All subjects with febrile convulsions who did not develop epilepsy and six clinically asymptomatic relatives showed asymmetric HV but normal hippocampal signal intensity. | A subtle, pre-existing hippocampal alteration may predispose to febrile convulsions and contribute to the development of subsequent HS. |
| Genetic generalized epilepsies (GGEs) | |||||
| Wandschneider | 2014 |
|
fMRI was applied to record brain activation during a working memory paradigm | The asymptomatic siblings of patients showed abnormal primary motor cortex and supplementary motor area co-activation with increasing cognitive load, as well as increased task-related functional connectivity between motor and prefrontal cognitive networks | Altered motor system activation and functional connectivity may represents a potential familial trait for JME |
5.1. QMRI traits as endophenotypes for focal epilepsies
Most studies exploring potential neuroimaging endophenotypes for epilepsy focus on focal syndromes, primarily MTLE. MTLE is the most prevalent form of medically intractable focal epilepsy. The most common pathology underlying MTLE is hippocampal sclerosis (HS), which is characterized histologically by cellular loss and synaptic reorganization within hippocampal subregions (Wieser and ILAE Commission on Neurosurgery of epilepsy, 2004). HS can be detected using MRI in up to 70% of MTLE patients through the identification of hippocampal atrophy and signal abnormalities (Blümcke et al., 2013). The underlying etiology of HS remains a subject of debate, although the latest evidence indicates an interaction of several polygenic and environmental factors (Lewis et al., 2014).
The heritability of HS has been examined by a number of investigators. The majority of these investigations were conducted on relatively small sample sizes. Fernández et al. (1998) investigated 23 family members of patients with MTLE associated with HS (MTLE + HS) and reported a significant hippocampal asymmetry in the asymptomatic family members who had experienced febrile seizures (FS) during childhood (Fernández et al., 1998). Hippocampal asymmetry was also identified in up to 50% of those family members who had never experienced FS. A follow up study by Kobayashi et al. (2002) examined 52 asymptomatic members of 11 families of patients with MTLE + HS. Significant hippocampal atrophy was identified in up to 34% of the asymptomatic relatives (Kobayashi et al., 2002). In addition, visual inspection of the T1- and T2-weighted images revealed classic MRI features of HS, including abnormal T2 signal abnormalities and altered internal structure of the hippocampus, in most of these relatives (Kobayashi et al., 2002). These two studies were complemented by a more recent investigation that explored HV in 32 asymptomatic first-degree relatives of MTLE + HS patients (Tsai et al., 2013). Here, the authors reported significantly reduced HV in the asymptomatic relatives of patients when compared to healthy controls (Tsai et al., 2013). This trend appeared stronger in the relatives of patients with a family history of epilepsy (see Fig. 2). Contrastingly, an independent investigation of MTLE + HS patients who reported no family history of seizures (i.e., sporadic cases) and their asymptomatic siblings (n = 50 discordant sibling pairs) reported no significant HV deficits in the asymptomatic siblings (Alhusaini et al., 2013). A trend of volume reduction was, however, noted in the hippocampus ipsilateral to the side of HS (Alhusaini et al., 2013). The authors attributed these findings to the possibly limited sensitivity of their automated segmentation technique relative to standardized manual tracing methods and to the small sample size (Alhusaini et al., 2016). Further evidence for the role of genetic contributions towards hippocampal structural abnormalities was recently illustrated by a study that applied T2-relaxometry to indirectly quantify the degree of hippocampal structural alteration in patients with MTLE + HS and their asymptomatic relatives (Suemitsu et al., 2014). The investigators reported increased hippocampal T2-relaxometry signal in patients relative to controls, and an intermediate relaxometry value for the asymptomatic relatives of patients (Suemitsu et al., 2014). Despite some inconsistencies, the findings of the above studies suggest a familial component to hippocampal structural alterations in MTLE + HS, particularly in patients with a strong family history for seizures.
Fig. 2.

The distribution of hippocampal volume in the asymptomatic relatives of MTLE + HS patients and healthy controls is displayed (Tsai et al., 2013). (A): The symptomatic relatives (red) had smaller hippocampal volume relative to the healthy controls (blue). The same data is displayed in (B), with 90% confidence ellipse of the controls displayed in the blue oval, and 90% confidence ellipse of the relatives of probands with a positive family history (FH) displayed in the red oval. [Image is reproduced with permission from Wolters Kluwer Health, Inc. (Neurology, 2013)]. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Over the last two decades, collective neuroimaging evidence has pointed to widespread structural alteration in MTLE + HS, extending beyond the hippocampus and mesial temporal lobe. Regional atrophy has been described in several subcortical GM structures (Bernasconi et al., 2003, McDonald et al., 2008), the ipsilateral temporal cortex (Moran et al., 2001, Alhusaini et al., 2012b), and several fronto-limbic cortical regions (Keller and Roberts, 2008, Mueller et al., 2009, Voets et al., 2011), suggesting an alteration in interconnected brain regions and localized brain networks (Bernhardt et al., 2013). The endophenotypic quality of these extra-hippocampal neuroanatomical alterations in MTLE has been explored by two separate studies (Alhusaini et al., 2013, Alhusaini et al., 2016). Using an automated segmentation method, the volume of five subcortical structures (amygdala, thalamus, caudate, putamen and globus pallidus) was compared between asymptomatic siblings of MTLE + HS patients and healthy controls (Alhusaini et al., 2013). No significant group differences were noted in the volume of any of the five subcortical structures, although a trend of reduced amygdalar volume was reported in the siblings of patients (Alhusaini et al., 2013). In a separate investigation, temporal cortex morphology was examined in MTLE + HS patients and their siblings (Alhusaini et al., 2016). A contraction in surface area was noted within the anterio-medial regions of the temporal cortex in patients and their asymptomatic siblings (Alhusaini et al., 2016). Interestingly, this altered surface area pattern was observed ipsilateral to the side of HS of the affected siblings, suggesting the possibility of a common driving process. WM tract integrity was also investigated in patients with MRI-negative MTLE and their siblings using diffusion-tensor imaging (DTI; Whelan et al., 2015a). Subtle microstructural alterations of the superior longitudinal fasciculus were reported in the asymptomatic siblings in a pattern similar to that displayed by patients (see Fig. 3), highlighting the endophenotypic potential of WM microstructural alterations in MTLE (Whelan et al., 2015a). Although replication studies to validate the above findings are yet to be conducted, these results provide preliminary evidence for the eligibility of QMRI neuroanatomical traits as endophenotypes for MTLE.
Fig. 3.

Whole-brain, voxel-wise comparisons of brain diffusion tensor imaging (DTI) measures, including fractional anisotropy (FA) and mean diffusivity (MD), in a group of patients with MRI-negative mesial temporal lobe epilepsy (MTLE), their asymptomatic siblings and healthy controls are shown (Whelan et al., 2015a). Patients with MTLE demonstrated patterns of reduced FA (illustrated in red) in voxel clusters encompassing the corpus callosum, ipsilateral anterior thalamic radiation, ipsilateral corticospinal tract (CST), bilateral superior longitudinal fasciculi (SLF), and bilateral inferior longitudinal fasciculi (ILF) compared to healthy controls. The asymptomatic siblings did not show significant FA compromise when compared to the same controls. Compared to controls, MTLE patients exhibited patterns of increased MD across the SLF bilaterally and the CST ipsilaterally (illustrated in blue). Similar patterns of increased MD along the ipsilateral SLF and ipsilateral CST (illustrated in yellow) were noted in the asymptomatic siblings. [Image is reproduced with permission from John Wiley and Sons (Epilepsia, 2015)]. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
5.2. QMRI traits as endophenotypes for genetic generalized epilepsies
GGEs (previously known as primary generalized epilepsies or idiopathic generalized epilepsies) refer to a group of characteristic epilepsy syndromes with a known or presumed genetic defect in the absence of either structural brain abnormality or an acquired pathology (Berg et al., 2010, Gallentine and Mikati, 2012). Although the biological basis of the majority GGE syndromes remains unknown, evidence from EEG and neuroimaging studies suggests a link to disruptions in thalamo-frontal brain networks (Duncan, 2005). Juvenile myoclonic epilepsy (JME) is the most common of the GGEs; accounting for 5% to 10% of all epilepsies. Although visual inspection of routine brain MRI in JME patients reveals no visible abnormality, QMRI studies have detected several subtle neuroanatomical abnormalities, such as thalamic volume loss and increased mesio-frontal and frontobasal GM concentration (Woermann et al., 1999, Betting et al., 2006, Alhusaini et al., 2013). These observations have been supported by positron emission tomography (PET) and proton magnetic resonance spectroscopy studies (Lin et al., 2009), indicating that the underlying mechanisms in JME may relate to aberrant frontothalamic structure (Duncan, 2005). Nonetheless, a clear set of GGE-specific neuroanatomical markers has not yet been fully characterized, and the endophenotypic property of frontothalamic alterations remains to be determined.
Recently, Wandschneider et al. (2014) applied functional MRI to explore the endophenotypic theory in JME (Wandschneider et al., 2014). In this study, brain activation during a working memory task was compared between 15 asymptomatic siblings of JME patients and 20 healthy controls. Abnormal primary motor cortex and supplementary motor area co-activation was noted in the asymptomatic siblings when compared to controls. This was consistent with a report of similar abnormal pattern of activation in JME patients using the same memory task (Vollmar et al., 2011), suggesting that abnormal motor cortex connectivity may represent a familial trait in JME. Although the specificity of this trait to JME was questioned (Berkovic and Jackson, 2014), the study was the first to explore the applicability of QMRI to identify endophenotypes in GGEs.
6. Discussion
The field of imaging genomics has expanded significantly over the last decade. Its application in epilepsy holds much promise. Characterizing epilepsy-relevant neuroimaging endophenotypes represents the first step towards expediting this research field. Unraveling the genetic determinants of relevant neuroimaging traits will undoubtedly provide valuable insight into the underlying mechanisms of the common epilepsy syndromes.
In Section 5, we discussed a number of neuroimaging traits that may fulfill the criteria of an endophenotype for two epilepsy syndromes, MTLE and JME. Additional studies using larger sample sizes will help validate these traits as intermediate phenotypes for future genetic mapping efforts. At present, HV in MTLE + HS appears to be the most promising neuroimaging endophenotype in epilepsy. Mapping the genetic determinants of HV in patients with MTLE + HS could unravel novel genetic variants linked to the pathogenesis of MTLE + HS. It is important to recognize, however, that the eligibility of HV as an endophenotype for MTLE + HS could be compromised by the impact of many significant environmental factors (Alhusaini et al., 2012a). Early brain insults (such as atypical FS, CNS infection, and significant head injury) are known risk factors for hippocampal damage and MTLE + HS (Scott et al., 2003, Lewis et al., 2014). In addition, the impact of epilepsy chronicity on hippocampal structure, including the effect of recurrent seizure activity, needs consideration (Fuerst et al., 2003). Nevertheless, some early insults, such as FS, could be genetically mediated and exploring their role in epilepsy predisposition is crucial (Feenstra et al., 2014). The potential limitation of environmental factors could be addressed by mapping HV genetic determinants in healthy human populations to indirectly inform on the genetic underpinnings of HV loss in MTLE + HS. An example of this approach is currently underway through collaboration between the ILAE and ENIGMA consortium (Whelan et al., 2015b).
Another important point to consider is the specificity of HV as an imaging trait for MTLE + HS. Hippocampal atrophy is associated with many other brain-related conditions such as AD (Jack et al., 1998). Further, reduced HV has been described in patients with schizophrenia (Wright et al., 2000), depression (Koolschijn et al., 2009), and post-traumatic stress disorder (Karl et al., 2006). Identifying genetic variants that influence HV is less likely to elucidate susceptibility variants unique to each brain condition, but may provide an opportunity to characterize biological pathways shared by such complex brain-related disorders, which themselves may be co-morbid. Shared underlying mechanisms between epilepsy and other neurologic and psychiatric disorders, including genetic processes, have been suggested by the observation that epileptic seizures form part of the wide phenotype of some brain-related conditions, such as autism spectrum disorder (Blackmon, 2015).
Over the last two decades, the search for neuroimaging endophenotypes has expanded. In addition to volumetric and functional QMRI, several research groups have examined cerebral cortex morphometry (e.g., cortical thickness and folding patterns), subcortical shape, and high-angular resolution DTI parameters as alternative approaches to characterize sensitive neuroimaging QTs. Many of these advanced techniques have been applied in epilepsy populations (Free et al., 1996, Sisodiya and Free, 1997, Mueller et al., 2009, Voets et al., 2011, Labate et al., 2015), but they have yet to be utilized to validate suitable endophenotypes. Alternative subclinical traits have also shown promising results. For example, the photoparoxysmal response (PPR) - an abnormal EEG trait that is provoked by intermittent photic stimulation and marked by diffuse paroxysmal discharges recorded as spike-wave complexes - is observed in up to 15% of GGEs and 40% of JME patients (Wolf and Goosses, 1986, Guerrini and Genton, 2004, Trenité, 2006). PPR has shown 100% concordance in monozygotic twins and a sibling recurrence risk of 20–30% (Doose and Waltz, 1993, Tauer et al., 2005). Interestingly, associations between PPR and candidate genes, including NEDD4-22, TRPC4 and BRD2, have previously been established (Lorenz et al., 2006, Dibbens et al., 2007, von Spiczak et al., 2010). Although a PPR gene has yet to be identified for common forms of epilepsy (Koeleman et al., 2013), this EEG trait is associated with de novo mutations in the CHD2 gene, which also confer risk for rare forms of photosensitive epilepsy (Thomas et al., 2015, Galizia et al., 2015). EEG-based topological network measures have also shown a potential genetic basis in epilepsy populations. For example, GGE patients and their asymptomatic relatives show greater mean degree (mean number of connections between nodes in a brain network) and degree distribution variance (probability distribution of connections between nodes in a brain network) compared to healthy controls in the 6–9 Hz band, suggesting possible co-segregation of abnormal brain network properties (Chowdhury et al., 2014a). Finally, evidence of a parallel profile of cognitive deficits has been reported in GGE patients and their first-degree relatives, with both groups showing impairments in nonverbal reasoning, verbal generativity, working memory and sustained attention (Chowdhury et al., 2014b).
As international collaborations such as the ILAE Consortium on Complex Epilepsies (http://www.ilae.org/Commission/class/consortium.cfm) continue to boost power to detect common genetic variants associated with common epilepsy syndromes, the traditional genome-wide genetic mapping approach may remain constrained by phenotypic heterogeneity, replicability issues and inevitable debates concerning missing heritability. This review discusses promising alternatives to the traditional clinical phenotypes historically employed by GWAS. Collective evidence from QMRI, EEG, and neuropsychology studies indicates the potential utility of several subclinical QTs as sensitive endophenotypes for particular epilepsy syndromes. Amongst these traits, QMRI-derived measures may serve as the most accessible and relevant endophenotypes for epilepsy. As the fields of neuroimaging and human genetics continue to advance, it is our hope that these intermediate phenotypes continue to be explored and, eventually, employed as useful tools in our continuing investigation of this highly complex neurological disorder.
Acknowledgments
This review was supported in part by a Consortium grant (U54 EB020403) from the NIH Institutes contributing to the Big Data to Knowledge (BD2K) Initiative, including the NIBIB.
References
- Alhusaini S., Doherty C.P., Scanlon C., Ronan L., Maguire S., Borgulya G., Brennan P., Delanty N., Fitzsimons M., Cavalleri G.L. A cross-sectional MRI study of brain regional atrophy and clinical characteristics of temporal lobe epilepsy with hippocampal sclerosis. Epilepsy Res. 2012;99:156–166. doi: 10.1016/j.eplepsyres.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Alhusaini S., Doherty C.P., Palaniyappan L., Scanlon C., Maguire S., Brennan P., Delanty N., Fitzsimons M., Cavalleri G.L. Asymmetric cortical surface area and morphology changes in mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia. 2012;53:995–1003. doi: 10.1111/j.1528-1167.2012.03457.x. [DOI] [PubMed] [Google Scholar]
- Alhusaini S., Scanlon C., Ronan L., Maguire S., Meaney J.P., Fagan A.J., Boyle G., Borgulya G., Iyer P.M., Brennan P., Costello D., Chaila E., Fitzsimons M., Doherty C.P., Delanty N., Cavalleri G.L. Heritability of subcortical volumetric traits in mesial temporal lobe epilepsy. PLoS One. 2013;8:e61880. doi: 10.1371/journal.pone.0061880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhusaini S., Whelan C.D., Doherty C.P., Delanty N., Fitzsimons M., Cavalleri G.L. Temporal cortex morphology in mesial temporal lobe epilepsy patients and their asymptomatic siblings. Cereb. Cortex. 2016;26:1234–1241. doi: 10.1093/cercor/bhu315. [DOI] [PubMed] [Google Scholar]
- Allen N.M., Mannion M., Conroy J., Lynch S.A., Shahwan A., Lynch B., King M.D. The variable phenotypes of KCNQ-related epilepsy. Epilepsia. 2014;55:e99–105. doi: 10.1111/epi.12715. [DOI] [PubMed] [Google Scholar]
- Ashbrook D.G., Williams R.W., Lu L., Stein J.L., Hibar D.P., Nichols T.E., Medland S.E., Thompson P.M., Hager R. Joint genetic analysis of hippocampal size in mouse and human identifies a novel gene linked to neurodegenerative disease. BMC Genomics. 2014;15:850. doi: 10.1186/1471-2164-15-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrini S., Milh M., Castiglioni C., Luthy K., Finelli M.J., Verstreken P. TBC1D24 genotype-phenotype correlation: epilepsies and other neurologic features. Neurology. 2016;87:77–85. doi: 10.1212/WNL.0000000000002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg A.T., Berkovic S.F., Brodie M.J., Buchhalter J., Cross J.H., van Emde B.W., Engel J., French J., Glauser T.A., Mathern G.W., Moshe S.L., Nordli D., Plouin P., Scheffer I.E. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE commission on classification and terminology, 2005–2009. Epilepsia. 2010;51:676–685. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- Berkovic S.F., Jackson G.D. ‘Idiopathic’ no more! Abnormal interaction of large-scale brain networks in generalized epilepsy. Brain. 2014;137:2400–2402. doi: 10.1093/brain/awu194. [DOI] [PubMed] [Google Scholar]
- Bernasconi N., Bernasconi A., Caramanos S.B., Antel B., Andermann F., Arnold D.L. Mesial temporal damage in temporal lobe epilepsy: a volumetric MRI study of the hippocampus, amygdala, and parahippocampal region. Brain. 2003;126:462–469. doi: 10.1093/brain/awg034. [DOI] [PubMed] [Google Scholar]
- Bernhardt B.C., Hong S., Bernasconi A., Bernasconi N. Imaging structural and functional brain networks in temporal lobe epilepsy. Front. Hum. Neurosci. 2013;7:624. doi: 10.3389/fnhum.2013.00624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betting L.E., Mory S.B., Li L.M., Lopes-Cendes I., Guerreiro M.M., Guerreiro C.A., Cendes F. Voxel-based morphometry in patients with idiopathic generalized epilepsies. NeuroImage. 2006;32:498–502. doi: 10.1016/j.neuroimage.2006.04.174. [DOI] [PubMed] [Google Scholar]
- Bis J., DeCarli C., Smith A., van der Lijn F., Crivello F., Fornage M. Common variants at 12q14 and 12q24 are associated with hippocampal volume. Nat. Genet. 2012;44:545–551. doi: 10.1038/ng.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmon K. Structural MRI biomarkers of shared pathogenesis in autism spectrum disorder and epilepsy. Epilepsy Behav. 2015;47:172–182. doi: 10.1016/j.yebeh.2015.02.017. [DOI] [PubMed] [Google Scholar]
- Blümcke I., Thom M., Aronica E., Armstrong D.D., Bartolomei F., Bernasconi A., Bernasconi N., Bien C.G., Cendes F., Coras R., Cross J.H., Jacques T.S., Kahane P., Mathern G.W., Miyata H., Moshe S.L., Oz B., Ozkara C., Perucca E., Sisodiya S.M., Wiebe S., Spreafico R. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a task force report from the ILAE commission on diagnostic methods. Epilepsia. 2013;54:1315–1329. doi: 10.1111/epi.12220. [DOI] [PubMed] [Google Scholar]
- Botstein D., Risch N. Discovering genotypes underlying human phenotypes: past successes for Mendelian disease, future approaches for complex disease. Nat. Genet. 2003;33:228–237. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- Cannon T.D., Thompson P.M., van Erp T.G., Toga A.W., Pouanen V.P., Huttunen M., Lonnqvist J., Standerskjold-Nordenstam C.G., Narr K.L., Khaledy M., Zoumalan C.I., Dail R., Kaprio J. Cortex mapping reveals regionally specific patterns of genetic and disease-specific grey-matter deficits in twins discordant for schizophrenia. Prog. Nat. Sci. USA. 2002;99:3228–3233. doi: 10.1073/pnas.052023499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Lu J., Pan H., Zhang Y., Wu H., Xu K., Liu X., Jiang Y., Bao X., Yao Z., Ding K., Lo W.H., Qiang B., Chan P., Shen Y., Wu X. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann. Neurol. 2003;54:239–243. doi: 10.1002/ana.10607. [DOI] [PubMed] [Google Scholar]
- Chowdhury F.A., Elwes R.D., Koutroumanidis M., Morris R.G., Nashef L., Richardson M.P. Impaired cognitive function in idiopathic generalized epilepsy and unaffected family members: an epilepsy endophenotype. Epilepsia. 2014;55:835–840. doi: 10.1111/epi.12604. [DOI] [PubMed] [Google Scholar]
- Chowdhury F.A., Woldman W., FitzGerald T.H.B., Elwes R.D.C., Nashef L., Terry J.R., Richardson M.P. Revealing a brain network endophenotype in families with idiopathic generalised epilepsy. PLoS One. 2014;9:e110136. doi: 10.1371/journal.pone.0110136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Zwarte S., Brouwer R., Hillegers M., Cahn W., Hulshoff Pol H., Alpert K., Wang L., Kane F., Picchioni M., Bramon E., McDonald C., Murray R., Hajek T., Alda M., Roberts G., Mitchell P., Schofield P., Fullerton J., Mwangi B., Soares J., Richter A., Gruber O., Bonvino A., Di Giorgio A., Bertolino A., Neilson E., Lawrie S., Caseras X., Fears S., Bearden C., DC G., van Erp T., Jahanshad N., Hibar D.P., Thompson P.M., Turner J., Kahn R., van Haren N. Organization for Human Brain Mapping (OHBM) Annual Meeting. Geneva, Switzerland. 2016. Brain volumes in family members of schizophrenia or bipolar patients: an ENIGMA meta-analysis. June 26–30. [Google Scholar]
- den Braber A., Bohlken M., Brouwer R., van ‘t Ent D., Kanai R., Kahn R.S., de Geus E.J., Hulshoff Pol H.E., Boomsma D.I. Heritability of subcortical brain measures: a perspective for future genome-wide association studies. NeuroImage. 2013;83:98–102. doi: 10.1016/j.neuroimage.2013.06.027. [DOI] [PubMed] [Google Scholar]
- Dibbens L.M., Ekberg J., Taylor I., Hodgson B.L., Conroy S.J., Lensink I.L., Kumar S., Zielinski M.A., Harkin L.A., Sutherland G.R., Adams D.J., Berkovic S.F., Scheffer I.E., Mulley J.C., Poronnik P. NEDD4-2 as a potential candidate susceptibility gene for epileptic photosensitivity. Genes Brain Behav. 2007;6:750–755. doi: 10.1111/j.1601-183X.2007.00305.x. [DOI] [PubMed] [Google Scholar]
- Dibbens L.M., de Vries B., Donatello S., Heron S.E., Hodgson B.L., Chintawa S., Crompton D.E., Hughes J.N., Bellows S.T., Klein K.M., Callenbach P.M.C., Corbet M.A., Gardner A.E., Kivit S., Iona X., Regan B.M., Weller C.M., Crimmins D., O'Brien T.J., Guerrero-Lopez R., Mulley J.C., Dubeau F., Licchetta L., Bisulli F., Cossette P., Thomas P.Q., Gecz J., Serratosa J., Brouwer F.O., Andermann F., Andermann E., van den Maagdenberg A.M.J.M., Pandolfo M., Berkovic S.F., Scheffer I.E. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat. Genet. 2013;45:546–551. doi: 10.1038/ng.2599. [DOI] [PubMed] [Google Scholar]
- Dickson S.P., Wang K., Krantz I., Hakonarson H., Goldstein D.B. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doose H., Waltz S. Photosensitivity—genetics and clinical significance. Neuropediatrics. 1993;24:249–255. doi: 10.1055/s-2008-1071552. [DOI] [PubMed] [Google Scholar]
- Duncan S. Brain imaging in idiopathic generalized epilepsies. Epilepsia. 2005;46:108–111. doi: 10.1111/j.1528-1167.2005.00321.x. [DOI] [PubMed] [Google Scholar]
- EPICURE Consortium, EMINet Consortium, Steffens M., Leu C., Ak R., Zara F., Striano P. Genome-wide association analysis of genetic generalized epilepsies implicates susceptibility loci 1q43, 2p16.1, 2q22.3 and 17q21.32. Hum. Mol. Genet. 2012;21:5359–5372. doi: 10.1093/hmg/dds373. [DOI] [PubMed] [Google Scholar]
- Feenstra B., Pasternak B., Geller F., Carstensen L., Wang T., Huang F., Eitson J.L., Hollegaard M.V., Svanstrom H., Vestergaard M., Hougaard D.M., Schoggins J.W., Jan L.Y., Melbye M., Hviid A. Common variants associated with general and MMR vaccine-related febrile seizures. Nat. Genet. 2014;46:1274–1282. doi: 10.1038/ng.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández G., Effenberger O., Vinz B., Steinlein O., Elger C.E., Dohrin W., Heinze H.J. Hippocampal malformation as a cause of familial febrile convulsions and subsequent hippocampal sclerosis. Neurology. 1998;50:909–917. doi: 10.1212/wnl.50.4.909. [DOI] [PubMed] [Google Scholar]
- Fisher R., Acevedo C., Arzimanoglou A., Bogacz A., Cross J.H., Elger C.E., Engel J., Jr., Forsgren L., French J.A., Glynn M., Hesdorffer D.C., Lee B.I., Mathern G.W., Moshe S.L., Perucca E., Scheffer I.E., Tomson T., Watanabe M., Wiebe S. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475–482. doi: 10.1111/epi.12550. [DOI] [PubMed] [Google Scholar]
- Franke B., Stein J.L., Ripke S., Anttila V., Hibar D.P., van Hulzen K.J. Genetic influences on schizophrenia and subcortical brain volumes: large-scale proof of concept. Nat. Neurosci. 2016;19:420–431. doi: 10.1038/nn.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Free S.L., Sisodiya S.M., Cook M.J., Fish D.R., Shorvon S.D. Three-dimensional fractal analysis of the white matter surface from magnetic resonance images of the brain. Cereb. Cortex. 1996;6:830–836. doi: 10.1093/cercor/6.6.830. [DOI] [PubMed] [Google Scholar]
- Fuerst D., Shah J., Shah A., Watson C. Hippocampal sclerosis is a progressive disorder: a longitudinal volumetric MRI study. Ann. Neurol. 2003;53:413–416. doi: 10.1002/ana.10509. [DOI] [PubMed] [Google Scholar]
- Galizia E.C., Myers C.T., Leu C., de Kovel C.G., Afrikanova T., Cordero-Maldonado M.L., Martins T.G., Jacmin M., Drury S., Krishna Chinthapalli V. CHD2 variants are a risk factor for photosensitivity in epilepsy. Brain. 2015;138:1198–1207. doi: 10.1093/brain/awv052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallentine W.B., Mikati M.A. Genetic generalized epilepsies. J. Clin. Neurophysiol. 2012;29:408–419. doi: 10.1097/WNP.0b013e31826bd92a. [DOI] [PubMed] [Google Scholar]
- Gambardella A., Marini C. Clinical spectrum of SCN1A mutations. Epilepsia. 2009;50(Suppl):20–23. doi: 10.1111/j.1528-1167.2009.02115.x. [DOI] [PubMed] [Google Scholar]
- Gershon E.S., Alliey-Rodriguez N., Liu C. After GWAS: searching for genetic risk for schizophrenia and bipolar disorder. Am. J. Psychiatry. 2011;168:253–256. doi: 10.1176/appi.ajp.2010.10091340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G. Rare and common variants: twenty arguments. Nat. Rev. Genet. 2012;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn D.C., Bearden C.E., Niendam A., Escamilla M.A. The feasibility of neuropsychological endophenotypes in the search for genes associated with bipolar affective disorder. Bipolar Disord. 2004;6:171–182. doi: 10.1111/j.1399-5618.2004.00113.x. [DOI] [PubMed] [Google Scholar]
- Glahn D.C., Thompson P.M., Blangero J. Neuroimaging endophenotypes: strategies for finding genes influencing brain structure and function. Hum. Brain Mapp. 2007;28:488–501. doi: 10.1002/hbm.20401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn D., Curran J., Winkler A., Carless M., Kent J.W., Jr., Charlesworth J.C., Johnson M.P., Goring H.H., Cole S.A., Dyer T.D., Moses E.K., Olvera R.L., Kochunov P., Duggirala R., Fox P.T., Almasy L., Blangero J. High dimensional endophenotype ranking in the search for major depression risk genes. Biol. Psychiatry. 2012;71:6–14. doi: 10.1016/j.biopsych.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman A.L., Pezawas L., Mattay V.S., Fischl B., Verchinski B.A., Zoltick B., Weinberger D.R., Meyer-Lindenberg A. Heritability of brain morphology related to schizophrenia: a large-scale automated magnetic resonance imaging segmentation study. Biol. Psychiatry. 2008;63:475–483. doi: 10.1016/j.biopsych.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Gottesman I.I., Gould T.D. The endophenotype concept in psychiatry: etymology and strategic intentions. Am. J. Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Gould T.D., Gottesman I.I. Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav. 2006;5:113–119. doi: 10.1111/j.1601-183X.2005.00186.x. [DOI] [PubMed] [Google Scholar]
- Guerrini R., Genton P. Epileptic syndromes and visually induced seizures. Epilepsia. 2004;45:14–18. doi: 10.1111/j.0013-9580.2004.451011.x. [DOI] [PubMed] [Google Scholar]
- Guo Y., Baum L.W., Sham P.C., Wong V., Ng P.W., Lui C.H., Sin N.C., Tsoi T.H., Tang C.S., Kwan J.S., Yip B.H., Xiao S.M., Thomas G.N., Lau Y.L., Yang W., Cherny S.S., Kwan P. Two-stage genome-wide association study identifies variants in CAMSAP1L1 as susceptibility loci for epilepsy in Chinese. Hum. Mol. Genet. 2012;21:1184–1189. doi: 10.1093/hmg/ddr550. [DOI] [PubMed] [Google Scholar]
- Hardies K., Weckhuysen S., De Jonghe P., Suls A. Lessons learned from gene identification studies in Mendelian epilepsy disorders. Eur. J. Hum. Genet. 2016;24:961–967. doi: 10.1038/ejhg.2015.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler G., Drevets W.C., Gould T.D., Gottesman I.I. Toward constructing an endophenotype strategy for bipolar disorders. Biol. Psychiatry. 2006;60:93–105. doi: 10.1016/j.biopsych.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Heinzen E.L., Radtke R.A., Urban T.J., Cavalleri G.L., Depondt C., Need A.C., Walley N.M., Nicoletti P., Ge D., Catarino C.B. Rare deletions at 16p13.11 predispose to a diverse spectrum of sporadic epilepsy syndromes. Am. J. Hum. Genet. 2010;86:707–718. doi: 10.1016/j.ajhg.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig I., Scheffer I.E., Mulley J., Berkovic S.F. Navigating the channels and beyond: unraveling the genetics of the epilepsies. Lancet Neurol. 2008;7:231–245. doi: 10.1016/S1474-4422(08)70039-5. [DOI] [PubMed] [Google Scholar]
- Helbig I., Mefford H.C., Sharp A.J., Guipponi M., Fichera M., Franke A., Muhle H., de Kovel C., Baker C., von Spiczak S. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat. Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibar D.P., Stein J.L., Renteria M.E., Arias-Vasquez A., Desrivières S., Jahanshad N. Common genetic variants influence human subcortical brain structures. Nature. 2015;520:224–229. doi: 10.1038/nature14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi M., Yaghootkar H., Mook‐Kanamori D.O., Sovio U., Taal H.R. New loci associated with birth weight identify genetic links between intrauterine growth and adult height and metabolism. Nat. Genet. 2013;45:76–82. doi: 10.1038/ng.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacono W.G., Malone S.M. Developmental endophenotypes: indexing genetic risk for substance abuse with the P300 brain event-related potential. Child Dev. Perspect. 2011;5:239–247. doi: 10.1111/j.1750-8606.2011.00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International League Against Epilepsy Consortium on Complex Epilepsies Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol. 2014;13:893–903. doi: 10.1016/S1474-4422(14)70171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C.R., Petersen R.C., Xu Y.C., O'Brien P.C., Waring S.C., Tangalos E.G., Smith G.E., Ivnik R.J., Thibodeau S.N., Kokmen E. Hippocampal atrophy and apolipoprotein E genotype are independently associated with Alzheimer's disease. Ann. Neurol. 1998;43:303–310. doi: 10.1002/ana.410430307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson G.D., McIntosh A.M., Briellmann R.S., Berkovic S.F. Hippocampal sclerosis in identical twins. Neurology. 1998;51:78–84. doi: 10.1212/wnl.51.1.78. [DOI] [PubMed] [Google Scholar]
- Kalachikov S., Evgrafov O., Ross B., Winawer M., Barker-Cummings C., Boneschi F.M., Choi C., Morozov P., Das K., Teplitskaya E., Yu A., Cayanis E., Penchaszadeh G., Kottmann A.H., Pedley T.A., Hauser W.A., Ottman R., Gilliam T.C. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat. Genet. 2002;330:335–341. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl A., Schaefer M., Malta L.S., Dorfel D., Rohleder N., Werner A. A meta-analysis of structural brain abnormalities in PTSD. Neurosci. Biobehav. Rev. 2006;30:1004–1031. doi: 10.1016/j.neubiorev.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Kasperaviciute D., Catarino C.B., Matarin M., Leu C., Novy J., Tostevin A. Epilepsy, hippocampal sclerosis and febrile seizures linked by common genetic variation around SCN1A. Brain. 2013;136:3140–3150. doi: 10.1093/brain/awt233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller S.S., Roberts N. Voxel-based morphometry of temporal lobe epilepsy: an introduction and review of the literature. Epilepsia. 2008;49:741–757. doi: 10.1111/j.1528-1167.2007.01485.x. [DOI] [PubMed] [Google Scholar]
- Kjeldsen M.J., Kyvik K.O., Christensen K., Friis M.L. Genetic and environmental factors in epilepsy: a population-based study of 11,900 Danish twin pairs. Epilepsy Res. 2001;44:167–178. doi: 10.1016/s0920-1211(01)00196-6. [DOI] [PubMed] [Google Scholar]
- Klassen T., David C., Goldman A., Burgess D., Chen T., Wheeler D., McPherson J., Bourquin T., Lewis L., Villasana D., Morgan M., Muzny D., Gibbs R., Noebels J. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi E., Li L.M., Lopes-Cendes I., Cendes F. Magnetic resonance imaging evidence of hippocampal sclerosis in asymptomatic first-degree relatives of patients with familial mesial temporal lobe epilepsy. Arch. Neurol. 2002;59:1891–1894. doi: 10.1001/archneur.59.12.1891. [DOI] [PubMed] [Google Scholar]
- Koeleman B.P., de Kovel C.G., Kasteleijn‐Nolst Trenité D.G. Photoparoxysmal EEG response and genetic dissection of juvenile myoclonic epilepsy. Epilepsy Behav. 2013;28:S69–S71. doi: 10.1016/j.yebeh.2012.07.016. [DOI] [PubMed] [Google Scholar]
- Koolschijn P.C.M.P., van Haren N.E.M., Lensvelt-Mulders G.J.L.M., Hulshoff Pol H.E., Kahn R.S. Brain volume abnormalities in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Hum. Brain Mapp. 2009;30:3719–3735. doi: 10.1002/hbm.20801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labate A., Cherubini A., Tripepi G., Mumoli L., Ferlazzo E., Aguglia U., Quattrone A., Gambardella A. White matter abnormalities differentiate severe from benign temporal lobe epilepsy. Epilepsia. 2015;56:1109–1116. doi: 10.1111/epi.13027. [DOI] [PubMed] [Google Scholar]
- Leu C., Coppola A., Sisodiya S.M. Progress from genome-wide association studies and copy number variants studies in epilepsy. Curr. Opin. Neurol. 2016;29:158–167. doi: 10.1097/WCO.0000000000000296. [DOI] [PubMed] [Google Scholar]
- Lewis D.V., Shinnar S., Hesdorffer D., Bagiella E., Bello J.A., Chan S. Hippocampal sclerosis after febrile status epilepticus: the FEBSTAT study. Ann. Neurol. 2014;75:178–185. doi: 10.1002/ana.24081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K., Carrete H., Jr., Lin J., Peruchi M.M., de Araujo Filho G.M., Guaranaha M.S., Guilhoto L.M., Sakamoto A.C., Yacubian E.M. Magnetic resonance spectroscopy reveals an epileptic network in juvenile myoclonic epilepsy. Epilepsia. 2009;50:1191–1200. doi: 10.1111/j.1528-1167.2008.01948.x. [DOI] [PubMed] [Google Scholar]
- Lorenz S., Taylor K.P., Gehrmann A., Becker T., Muhle H., Gresch M., Tauer U., Sander T., Steohani U. Association of BRD2 polymorphisms with photoparoxysmal response. Neurosci. Lett. 2006;400:135–139. doi: 10.1016/j.neulet.2006.02.026. [DOI] [PubMed] [Google Scholar]
- Manchia M., Cullis J., Turecki G., Rouleau G.A., Uher R., Alda M. The impact of phenotypic and genetic heterogeneity on results of genome wide association studies of complex diseases. PLoS One. 2013;8:e76295. doi: 10.1371/journal.pone.0076295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann J.J., Arango V.A., Avenevoli S., Brent D.A., Champagne F.A., Clayton P., Currier D., Dougherty D.M., Haghighi F., Hodge S.E., Kleinman J., Lehner T., McMahon F., Mościcki E.K., Oquendo M.A., Pandey G.N., Pearson J., Stanley B., Terwilliger J., Wenzel A. Candidate endophenotypes for genetic studies of suicidal behavior. Biol. Psychiatry. 2009;65:556–563. doi: 10.1016/j.biopsych.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazhari S., Price G., Dragovic M., Waters F.A., Clissa P., Jablensky A. Revisiting the suitability of antisaccade performance as an endophenotype in schizophrenia. Brain Cogn. 2011;77:223–230. doi: 10.1016/j.bandc.2011.08.006. [DOI] [PubMed] [Google Scholar]
- McCarthy M.I., Hirschhorn J.N. Genome‐wide association studies: past, present and future. Hum. Mol. Genet. 2008;17:R100–101. doi: 10.1093/hmg/ddn298. [DOI] [PubMed] [Google Scholar]
- McClellan J., King M.C. Genetic heterogeneity in human disease. Cell. 2010;151:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- McDonald C.R., Hagler D.J., Jr., Ahmadi M.E., Tecoma E., Iragui V., Dale A.M., Halgren E. Subcortical and cerebellar atrophy in mesial temporal lobe epilepsy revealed by automatic segmentation. Epilepsy Res. 2008;79:130–138. doi: 10.1016/j.eplepsyres.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Lindenberg A. Imaging genetics of schizophrenia. Dialogues Clin. Neurosci. 2010;12:449–456. doi: 10.31887/DCNS.2010.12.4/amlindenberg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mier K., Meyer-Lindenberg Neural substrates of pleiotropic action of genetic variation in COMT: a meta-analysis. Mol. Psychiatry. 2009;15:918–927. doi: 10.1038/mp.2009.36. [DOI] [PubMed] [Google Scholar]
- Miller G.A., Rockstroh B. Endophenotypes in psychopathology research: where do we stand? Clin. Psychol. 2013;9:177–213. doi: 10.1146/annurev-clinpsy-050212-185540. [DOI] [PubMed] [Google Scholar]
- Miller L.L., Pellock J.M., De Lorenzo R.J., Meyer J.M., Corey L.A. Univariate genetic analyses of epilepsy and seizures in a population-based twin study: the Virginia Twin registry. Genet. Epidemiol. 1998;15:3–49. doi: 10.1002/(SICI)1098-2272(1998)15:1<33::AID-GEPI3>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Moran N.F., Lemieux L., Kitchen N.D., Fish D.R., Shorvon S.D. Extrahippocampal temporal lobe atrophy in temporal lobe epilepsy and mesial temporal sclerosis. Brain. 2001;124:167–175. doi: 10.1093/brain/124.1.167. [DOI] [PubMed] [Google Scholar]
- Mueller S.G., Laxer K.D., Barakos J., Cheong I., Garcia P., Weiner M.W. Widespread neocortical abnormalities in temporal lobe epilepsy with and without mesial sclerosis. NeuroImage. 2009;46:353–359. doi: 10.1016/j.neuroimage.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narr K.L., Cannon T.D., Woods P.R., Thompson P.M., Kim S., Asunction D., van Erp T.G., Poutanen V.P., Huttunen M., Lonnqvist J., Standerksjold-Nordenstam C.G., Kaprio J., Mazziotta J.C., Toga A.W. Genetic contributions to altered callosal morphology in schizophrenia. J. Neurosci. 2002;22:3720–3729. doi: 10.1523/JNEUROSCI.22-09-03720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal D.K., Strug L.J. The genetics of common epilepsies: common or distinct? Lancet Neurol. 2014;13:859–860. doi: 10.1016/S1474-4422(14)70124-3. [DOI] [PubMed] [Google Scholar]
- Peper J.S., Brouwer R.M., Boomsma D.I., Kahn R.S., Hulshoff Pol H.E. Genetic influences on human brain structure: a review of brain imaging studies in twins. Hum. Brain Mapp. 2007;28:464–473. doi: 10.1002/hbm.20398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potkin S.G., Guffanti G., Lakatos A., Turner J.A., Kruggel F., Fallon J.H., Saykin A.J., Orro A., Lupoli S., Salvi E., Weiner M., Macciardi F. Alzheimer's disease neuroimaging initiative. Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer's disease. PLoS One. 2009;4:e6501. doi: 10.1371/journal.pone.0006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard J.K., Cox N.J. The allelic architecture of human disease genes: common disease–common variant… or not? Hum. Mol. Genet. 2002;11:2417–2423. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- Reich D.E., Lander E.S. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- Roussotte F.F., Daianu M., Jahanshad N., Leonardo C.D., Thompson P.M. Neuroimaging and genetic risk for Alzheimer's disease and addiction-related degenerative brain disorders. Brain Imaging Behav. 2014;8:217–233. doi: 10.1007/s11682-013-9263-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon C., Ronan L., Doherty C.P., Cavalleri G.L., Tirupati A.S., Maguire S., Delanty N., Iyer P.M., Chaila E., Fitzsimons MRI-based brain structure volumes in temporal lobe epilepsy patients and their unaffected siblings: a preliminary study. J. Neuroimaging. 2013;23:64–70. doi: 10.1111/j.1552-6569.2012.00736.x. [DOI] [PubMed] [Google Scholar]
- Scott R.C., King M.D., Gadian D.G., Neville B.G.R., Connelly A. Hippocampal abnormalities after prolonged febrile convulsion: a longitudinal MRI study. Brain. 2003;126:2551–2559. doi: 10.1093/brain/awg262. [DOI] [PubMed] [Google Scholar]
- Shen L., Thompson P., Potkin S., Bertram L., Farrer L., Foroud T. Genetic analysis of quantitative phenotypes in AD and MCI: imaging, cognition and biomarkers. Brain Imaging Behav. 2014;8:183–207. doi: 10.1007/s11682-013-9262-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodiya S.M., Free S.L. Disproportion of cerebral surface areas and volumes in cerebral dysgenesis. MRI-based evidence for connectional abnormalities. Brain. 1997;120:271–281. doi: 10.1093/brain/120.2.271. [DOI] [PubMed] [Google Scholar]
- Speed D., O'Brien T.J., Palotie A., Shkura K., Marson A.G., Balding D.J., Johnson M.R. Describing the genetic architecture of epilepsy through heritability analysis. Brain. 2014;137:2680–2689. doi: 10.1093/brain/awu206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprooten E., Brumbaugh M.S., Knowles E.E., McKay R., Lewis J., Barrett J., Landau S., Cyr L., Kochunov P., Winkler A.M., Pearlson G.D., Glahn D. Reduced white matter integrity in sibling pairs discordant for bipolar disorder. Am. J. Psychiatry. 2013;170:1317–1325. doi: 10.1176/appi.ajp.2013.12111462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprooten E., Barrett J., McKay R., Knowles E.E., Mathias S.R., Winkler A.M., Brumbaugh M.S., Landau S., Cyr L., Kochunov P., Glahn D. A comprehensive tractography study of patients with bipolar disorder and their unaffected siblings. Hum. Brain Mapp. 2016 doi: 10.1002/hbm.23253. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein J., Medland S., Vasquez A., Hibar D., Senstad R., Winkler A. Identification of common variants associated with human hippocampal and intracranial volumes. Nat. Genet. 2012;44:552–561. doi: 10.1038/ng.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striano P., Weber Y.G., Toliat M.R., Schubert J., Leu C., Chaimana R., Baulac S., Guerrero R., LeGuern E., Lehesjoki A.E., Polvi A., Robbiano A., Serratosa J.M., Guerrini R., Nürnberg P., Sander T., Zara F., Lerche H., Marini C., EPICURE Consortium GLUT1 mutations are a rare cause of familial idiopathic generalized epilepsy. Neurology. 2012;78:557–562. doi: 10.1212/WNL.0b013e318247ff54. [DOI] [PubMed] [Google Scholar]
- Suemitsu L.A., Yasuda C.L., Morita M.E., Beltramini G.C., Coan A.C., Bergo F., Lopes‐Cendes I., Cendes F. Longitudinal analysis of hippocampal T2 relaxometry in FMTLE. Epilepsy Behav. 2014;36:154–158. doi: 10.1016/j.yebeh.2014.05.023. [DOI] [PubMed] [Google Scholar]
- Tauer U., Lorenz S., Lenzen K.P., Heils A., Muhle H., Gresch M., Neubauer B.A., Waltz S., Rudolf G., Mattheisen M., Strauch K., Numberg P., Schmitz B., Stephani U., Sander T. Genetic dissection of photosensitivity and its relation to idiopathic generalized epilepsy. Ann. Neurol. 2005;57:866–873. doi: 10.1002/ana.20500. [DOI] [PubMed] [Google Scholar]
- Tepest R., Wang L., Miller M.I., Falkai P., Csernansky J.G. Hippocampal deformities in the unaffected siblings of schizophrenia subjects. Biol. Psychiatry. 2003;54:1234–1240. doi: 10.1016/s0006-3223(03)00702-9. [DOI] [PubMed] [Google Scholar]
- Thomas R.H., Zhang L.M., Carvill G.L., Archer J.S., Heavin S.B., Mandelstam S.A., Craiu D., Berkovic S.F., Gill D.S., Mefford H.C., Scheffer I.E., EuroEPINOMICS RES Consortium CHD2 myoclonic encephalopathy is frequently associated with self-induced seizures. Neurology. 2015;84:951–958. doi: 10.1212/WNL.0000000000001305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson P., Stein J., Medland S., Hibar D., Vasquez A., Rentería M. The ENIGMA Consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain Imaging Behav. 2014;8:153–182. doi: 10.1007/s11682-013-9269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenité D.G. Photosensitivity, visually sensitive seizures and epilepsies. Epilepsy Res. 2006;70:S269–S279. doi: 10.1016/j.eplepsyres.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Tsai M.H., Pardoe H.R., Perchyonok Y., Fitt G.J., Scheffer I.E., Jackson G.D., Berkovic S.F. Etiology of hippocampal sclerosis: evidence for a predisposing familial morphologic anomaly. Neurology. 2013;81:144–149. doi: 10.1212/WNL.0b013e31829a33ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets N.L., Bernhardt B.C., Kim H., Yoon U., Bernasconi N. Increased temporolimbic cortical folding complexity in temporal lobe epilepsy. Neurology. 2011;76:138–144. doi: 10.1212/WNL.0b013e318205d521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmar C., O'Muircheartaigh J., Barker G.J., Symms M.R., Thompson P., Kumari V., Duncan J.S., Janz D., Richardson M.P., Koepp M.J. Motor system hyperconnectivity in juvenile myoclonic epilepsy: a cognitive functional magnetic resonance imaging study. Brain. 2011;134:1710–1719. doi: 10.1093/brain/awr098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Spiczak S., Muhle H., Helbig I., de Kovel C.G., Hampe J., Gaus V., Koeleman B.P., Lindhout D., Schreiber S., Sander T., Stephani U. Association study of TRPC4 as a candidate gene for generalized epilepsy with photosensitivity. Neruomol. Med. 2010;12:292–299. doi: 10.1007/s12017-010-8122-x. [DOI] [PubMed] [Google Scholar]
- Walters J.T., Owen M.J. Endophenotypes in psychiatric genetics. Mol. Psychiatry. 2007;12:886–890. doi: 10.1038/sj.mp.4002068. [DOI] [PubMed] [Google Scholar]
- Wandschneider B., Centeno M., Vollmar C., Symms M., Thompson P.J., Duncan J.S., Koepp M.J. Motor co-activation in siblings of juvenile myoclonic epilepsy patients: an imaging endophenotype? Brain. 2014;137:2469–2479. doi: 10.1093/brain/awu175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan C.D., Alhusaini S., O'Hanlon E., Cheung M., Iyer P.M., Meaney J.F., Fagan A.J., Boyle G., Delanty N., Doherty C.P., Cavalleri G.L. White matter alterations in patients with MRI-negative temporal lobe epilepsy and their asymptomatic siblings. Epilepsia. 2015;56:1551–1561. doi: 10.1111/epi.13103. [DOI] [PubMed] [Google Scholar]
- Whelan C.D., Speed D., deKovel C., Bradfield J., Hongsheng G., Leu C., ILAE Consortium on Complex Epilepsies, DP H., Stein J., Johnson M., Sisodiya S., Goldstein D., Delanty N., Medland S., Franke B., Thompson P.M., Cavalleri G.L. Organization for Human Brain Mapping (OHBM) Annual Meeting, Honolulu, Hawaii, USA. 2015. Polygenic contributions of ENIGMA2 hippocampal SNPs in 8,835 epilepsy patients and 29,037 controls. June 14–18. [Google Scholar]
- Wieser H.G., ILAE Commission on Neurosurgery of epilepsy ILAE Commission report: mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia. 2004;45:695–714. doi: 10.1111/j.0013-9580.2004.09004.x. [DOI] [PubMed] [Google Scholar]
- Woermann F.G., Free S.L., Koepp M.J., Sisodiya S.M., Duncan J.S. Abnormal cerebral structure in juvenile myoclonic epilepsy demonstrated with voxel-based analysis of MRI. Brain. 1999;122:2101–2108. doi: 10.1093/brain/122.11.2101. [DOI] [PubMed] [Google Scholar]
- Wolf P., Goosses R. Relation of photosensitivity to epileptic syndromes. J. Neurol. Neurosurg. Psychiatry. 1986;49:1386–1391. doi: 10.1136/jnnp.49.12.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray N.R., Purcell S.M., Visscher P.M. Synthetic associations created by rare variants do not explain most GWAS results. PLoS Biol. 2011;9:e1000579. doi: 10.1371/journal.pbio.1000579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright I.C., Rabe-Hesketh S., Woodruff P.W., David P.W., David A.S., Murray R.M., Bullmore E.T. Meta-analysis of regional brain volumes in schizophrenia. Am. J. Psychiatry. 2000;157:16–25. doi: 10.1176/ajp.157.1.16. [DOI] [PubMed] [Google Scholar]
- Yang J., Benyamin B., McEvoy B.P., Gordon S., Henders A.K., Nyholt D.R., Madden P.A., Heath A.C., Martin N.G., Montgomery G.W., Goddard M.E., Visscher P.M. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitlen N., Kraft P. Heritability in the genome-wide association era. Hum. Genet. 2012;131:1644–1664. doi: 10.1007/s00439-012-1199-6. [DOI] [PMC free article] [PubMed] [Google Scholar]