

Abstract
Heart failure is a multifactorial disease with poor prognosis. There are many hypotheses regarding the cause of heart failure. Leading among them are the hemodynamic and the neuro-hormonal hypotheses. Although the energy depletion hypothesis has been fairly recent, there is evidence suggesting that declining bioenergy plays a major role in heart failure. This review explored the myocardial energy depletion hypothesis from the role of micronutrients in correcting and alleviating symptoms of heart failure. Even though focus was on key nutrients such as coenzyme Q10, thiamine, riboflavin, L-carnitine, and taurine, emphasis was on the combined effect of multiple micronutrients as a whole. Search from databases from 2000 to 2015 produced four clinical studies using multiple micronutrients on heart failure. Evidence from the studies show that using high doses of multiple micronutrients may have positive effects on heart failure and simultaneously support the myocardial energy depletion hypothesis.
Keywords: Heart failure, myocardial energetics, adenosine triphosphate, Krebs cycle
Introduction
Heart failure (HF) is a progressive, complex, end-stage disease condition of all maladies of the heart resulting in loss of function of the cardiac myocytes. As a consequence, heart-pumping function weakens impairing blood circulation and the body’s cells are unable to receive enough oxygen and nutrients to meet the body’s needs. This results in symptoms of fatigue and shortness of breath, and for an affected person this makes everyday activities such as walking, climbing stairs or carrying groceries difficult.
Due to its high prevalence, and with estimated cases of over 23 million worldwide [1], a steady increase in HF diagnosis, poor prognosis, and high cost, HF has become a major societal burden [2]. Fifty percent of HF patients die within five years from the initial diagnosis [3].
HF is a multifactorial disease and its various mechanisms have been investigated from a variety of perspectives. These pathogenetic mechanisms (also referred to as models) include increased hemodynamic overload, ischemic-related dysfunction, ventricular remodeling, excessive neuro-hormonal stimulation, abnormal myocyte calcium cycling, excessive or inadequate accumulation of extracellular matrix (fibrosis), accelerated apoptosis, and genetic mutations [4].
Although there are many hypotheses regarding the cause of HF, and with the hemodynamic and the neuro-hormonal as the two leading ones, there is no single unifying pathogenetic theory explaining HF. There are different concepts regarding the underlying causes of the disease, however consensus regarding the definition of HF is lacking [5]. The available evidence suggests that declining bioenergy plays a major role in the failing heart. Accordingly, current conventional treatments for HF include energy-sparing medications like beta-receptor blockers, ACE inhibitors, or angiotensin II blockers, but these are demerits.
Treatment for HF includes pharmacological agents (diuretics, beta-blockers, ACE inhibitors, and neuro-hormonal antagonists), as well as mechanical devices (left ventricular assist devices) and, finally, transplantation for end-stage HF. These treatment strategies are designed to increase inotropic function of the heart or reduce one or more compensating mechanisms. They are not intended to assure functional recovery or the regeneration of normal cardiac myocytes. This perhaps can be an underlying reason why HF has such poor prognosis.
Therefore, we propose that the most promising therapeutic strategy in achieving partial or total functional recovery of cardiac myocytes should involve the enhancement of bio-energy metabolism in the heart, the myocardial energetics. The concept of the failing heart due to energy depletion was first proposed by Herrmann and Decherd in 1939 [6], but it was not adequately investigated. Our aim is to explore the myocardial energy depletion hypothesis/model of HF from the role of micronutrients in correcting and alleviating symptoms of HF.
Hemodynamic hypothesis
From the beginning, the HF syndrome has been considered as a hemodynamic disorder. Thus, it was defined as a pathologic state where the heart muscle fails to pump blood at a rate to meet the requirements of the metabolizing tissues during ordinary activity [7]. In addition, it was defined as the failure of the heart to deliver oxygen at a rate commensurate with requirements of the metabolizing tissues despite normal filling pressure, or only at the expense of increased filling pressure [8]. HF is initiated by an event, most often myocardial infarction, after a prolonged ischemia. This results in death of cardiac myocytes and eventually diminished pumping capacity of the heart muscle, and is clinically viewed as left ventricular dysfunction.
Based on this hemodynamic concept, the pathophysiology of HF is understood from the perspectives of cardiac contractility, preload, afterload, volume, flow, and the left ventricular ejection fraction. Therefore, treatment is tailored to increase cardiac output by reducing the systemic and pulmonary vascular pressure. Whether it is inotropic, chronotropic, vasodilation or diuretics, the pharmacotropic agents like beta-blockers, digoxin, enalapril, losartan, furosemide, spironolactone, and the like, are aimed to correct the symptoms of hemodynamic abnormalities. Consequently, symptoms like high blood pressure, fatigue, angina, pulmonary edema, and edema of the lower extremities may be partially or fully alleviated and the patient can gain temporary respite. However, despite increase in contractility, vasodilation and reduced ischemia, the disease continues and ultimately the patient dies.
Neuro-hormonal hypothesis
The inability to fully explain why HF continues to progress led researchers to look at the pathogenesis of HF from the role of biologically active molecules which initially contribute to alleviating HF symptoms, but later exert toxic effects hastening the degeneration of HF. This mechanism is known as the neuro-hormonal model.
When a myocardial infarction event occurs, cardiac dysfunction and decreased cardiac output lead to activation of several neuro-hormonal mechanisms to offset the diminished heart contractions, such as activation of the sympathetic nervous system, and increased sodium and water retention [9]. As a result, natriuretic peptides, prostaglandins, and nitric oxide molecules are released to enhance vasodilation [10,11] in order to improve blood flow.
These neuro-hormonal mechanisms are initially adaptive, compensatory and beneficial, and patients remain asymptomatic, but when sustained they eventually become deleterious and patients become overly symptomatic [12]. After some time the continuous excessive secretion of biologically active molecules is capable of exerting toxic effects on the heart [13]. Some of these proteins that are known to contribute to the disease progression in HF are norepinephrine, angiotensin II, endothelin, aldosterone, and tumor necrosis factor. Their long-term detrimental effects include overgrowth of cardiac myocytes, fibroblast hyperplasia, myocyte damage/myopathy, fetal gene induction, apoptosis of myocytes, pro-arrhythmic effects, vasoconstriction [12], and increased cardiac energy expenditure [14,15].
Energy depletion hypothesis
The heart has the highest resting energy expenditure than any other organ in the body and has a constant high demand for bio-energy to sustain its workload. The requirement of biochemical energy to support excitation, contraction, relaxation and the molecular synthesis and degradation is absolute. Its production relies mainly on the aerobic oxidation of substrates such as fatty acids, glucose, lactate, and ketone bodies, converting them into energy units called adenosine triphosphate (ATP). The two main substrates for the heart muscle energy generation are fatty acids and glucose (Figure 1).
Figure 1.
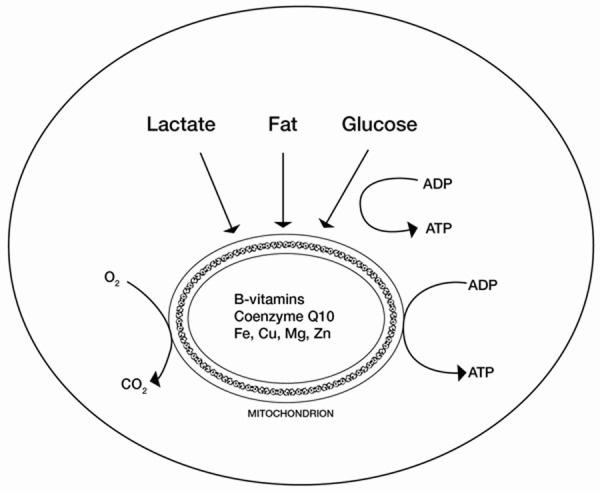
Schematic representation of the ATP synthesis sources and key micronutrients utilized in mitochondrial electron transport system.
Under normal physiological conditions, the heart relies mainly on fatty acids as substrates for beta-oxidation that contributes 60% to 70% of total ATP production, while the remaining 30% to 40% is provided by glucose and lactate oxidation [16]. Fatty acid oxidation produces more ATP per molecule oxidized than glucose oxidation. However, fatty acid oxidation requires more oxygen per molecule of ATP produced. The ratio of useful energy produced to oxygen consumed is defined as mechanical efficiency. The complete oxidation of 1 fatty acid molecule generates 105 molecules of ATP and consumes 46 atoms of oxygen, whereas the complete oxidation of 1 molecule of glucose generates 31 molecules of ATP and consumes 12 atoms of oxygen [17]. Therefore, fatty acid oxidation is less efficient than glucose oxidation with regards to ATP production based on oxygen consumption.
In ischemic heart disease and HF, oxygen supply is diminished as a result of insufficient tissue perfusion. Therefore, mechanical efficiency is reduced. To compensate, the production of ATP in the myocardium switches from aerobic to anaerobic mode, and from beta-oxidation to glycolysis as the latter yields 11% to 13% more ATP per unit of consumed oxygen [18,19]. In chronic HF and cardiac hypertrophy, there is a switch in primary substrate utilization from fatty acids to glucose [20] for supporting mechanical efficiency and to conserve energy.
During the initial acute phase of HF, metabolic adaptation and compensation enable the heart to function without compromising mechanical efficiency. Reduction in myocardial oxygen consumption and contractile function are matched [21]. As HF becomes more chronic and the process of remodeling progresses towards an uncompensated state, metabolic adaptation becomes insufficient and the capacity to oxidize glucose decreases along with decreased mechanical efficiency [22]. In HF the ATP is about 25% to 30% lower than its normal values [23,24].
Energy depletion caused by drugs
In cardiotonic medications, it has been established that vasodilation therapy may reduce myocardial oxygen demand, whereas inotropic stimulation therapy may increase myocardial oxygen demand. Inotropic stimulation consumes energy. Therefore in chronic ischemic conditions and compromised oxygen supply, inotropes may render the failing heart in a more disadvantaged condition, contributing to increased rather than decreased heart failure mortality.
In terms of cardiac output, inotropic agents are known to increase energetic costs of non-mechanical work, which is often referred to as the oxygen-wasting effect [25]. In their study of 30 patients in varying degrees of left ventricular dysfunction, Hayashi and colleagues [26] showed that the oxygen cost of contractility in the severe left ventricular group was significantly greater than in the groups with milder left ventricular dysfunction. Increasing contractility through inotropic agents means increasing oxygen consumption for every beat of the heart.
Beta-blockers are generally considered to be energy sparing in myocardial energetics, however with certain limitations. The study of Hawkins and colleagues [27] showed that metoprolol has a beneficial effect on cardiac efficiency of subjects on mild exercise intensity. However, in subjects on extensive exercise intensity, the cardiac work and calculated cardiac efficiency were reduced. Therefore, caution should be advised for patients with ischemic heart disease who are on beta-blockers and on high intensity exercise, because instead of energy sparing this treatment may hasten energy depletion. Use of ACE inhibitors in patients with impaired cardiac function has been associated with an increase in insulin-like growth hormone factor-1 (IGF-1). However, high levels of IGF-1 predict cardiovascular mortality [28]. It has also been documented in an animal study that IGF-1 increases autophagy and cell death in induced starvation [29].
Nutrients in cardiac metabolism
The contracting heart requires biological fuel in the form of energy units (ATP). As such, it is only logical to analyze the role of nutrients in cardiac metabolism from a reductionist approach. Since the postulation of Herrmann and Dechard in 1939 on energy depletion of the failing heart, the next few decades saw much research on energy metabolism of the heart. However, progress has been slow and almost came to a halt by the first decade of the 21st century.
First, this was because of different views on the cause of HF syndrome. Second, when the neuro-hormonal hypothesis of the cause of HF became more established, pharmacological research mainly targeted the development of drugs modulating substrate utilization as a means to economize and maximize energy production in the failing heart. Studies on nutrients in cardiac metabolism have been sparse and confined mainly to a single nutrient or a combination of a few nutrients. Even much less has been done in addressing cardiovascular endpoints with micronutrient-based approaches.
As discussed earlier, the main energy source of ATP for the heart muscle is fatty acids and carbohydrates. However, the conversion of these macronutrients to biological energy requires micronutrients such as coenzyme Q10 (CoQ10), thiamine, riboflavin, L-carnitine, taurine, and other amino acids that function as essential cofactors for energy production, energy transfer, and maintenance of the physiological heart function [30]. It has been observed that patients with chronic HF do not meet dietary reference values for energy and micronutrients [31]. It was also suggested that nutritional therapy of heart failure should be directed to the replacement of L-carnitine, CoQ10, taurine, antioxidants, and thiamine [32].
Most of the earlier studies on HF and micronutrients have been conducted with a single nutrient or with two-nutrient compounds. However, there have been suggestions to investigate the effects of multiple nutrients on HF [30,33,34]. The argument against the use of multiple nutrients is that if improvement is observed, the identification of the active compound and its underlying mechanism will be unclear. Witte and colleagues have argued that using a single nutrient to correct one deficiency when multiple deficiencies exist may be ineffective and could be harmful by causing the accumulation of harmful metabolic products [35]. Soukoulis and colleagues further suggested that using a single micronutrient might potentially shift one metabolic pathway in the energy cascade to another [34], and that correcting one deficiency may unmask other of the many deficiencies present. Indeed, considering that the production and utilization of ATP in the myocyte pathways involves a number of cofactors and intermediary metabolites in energy substrate metabolism, such as CoQ10, thiamine, riboflavin, L-carnitine, taurine and amino acids, it would be appropriate to use multiple nutrients to address multiple nutrient deficiencies especially in HF syndrome. Taking into account a high safety record of micronutrient supplementation and a low risk of their overdosing, this approach does not carry a risk of detrimental side effects as it is in case of pharmaceutical drugs.
Studies on heart failure using multi micronutrients
A thorough search through the Cochrane register of controlled trials, Medline, Pubmed, Embase, and Google Scholar databases from 2000 to 2015 revealed only a few studies evaluating HF from a multiple micronutrient approach. These studies are summarized in Table 1.
Table 1.
Clinical studies using multiple micronutrients on heart failure
| Study/Study/First Author | Study Design | Subjects | Endpoints | Intervention/Main Micronutrients | Main Outcome |
|---|---|---|---|---|---|
| Jeejeebhoy et al. (2002). [36] | RCT | n = 41. | Primary: Myocardial levels of carnitine, CoQ10, and taurine. | Daily single dose: | Significant increase in myocardial levels of carnitine, CoQ10, and taurine. |
| HF with IHD | Secondary: LVEDV and LVESV. | Carnitine 3 mg. | Significant decrease in LVEDV and LVESV. | ||
| EF ≤ 40% | CoQ10 150 mg. | No significant change in EF. | |||
| Age: 62 ± 11 | Taurine 3 mg. | No significant difference in cystokine levels. | |||
| Thiamine 25 mg. | Significant improvement in QOL scores. | ||||
| Riboflavin 3 mg. | |||||
| Ascorbate 250 mg. | |||||
| 30 to 45 days. | |||||
| Witte et al. (2005). [35] | RCT | n = 30 | LV function. | Daily single dose: | Significant decrease in LVEDV and LVESV. |
| HF with IHD | Proinflammatory cystokines. | CoQ10 150 mg. | Significant increase in EF. | ||
| EF ≤ 35% | QOL. | Thiamine 200 mg. | No significant difference in cystokine level. | ||
| Age: 75.4 ± 0.7 | Riboflavin 2 mg. | Significant improvement in QOL scores. | |||
| Ascorbate 500 mg. | |||||
| 9 months. | |||||
| McKeag et al. (2014). [37] | RCT | n = 74 | Primary: LVEF. | Daily single dose: | No significant difference in LVEF. |
| HF with IHD | Secondary: QOL, 6MWT, NT-proBNP, Proinflammatory cystokines. | Thiamine 1.5 mg. | No significant difference in QOL score. | ||
| EF ≤ 38% | Riboflavin 1.6 mg. | No significant difference in 6MWT, NT-proBNP, and cystokine levels. | |||
| Age: 65.8 ± 9.4 | Ascorbate 60 mg. | ||||
| 12 months. | |||||
| Wong et al. (2015). [38] | Prospec-tive case series. | n = 12 | MLHFQ total score. | Daily three doses: | Significant decrease in MLHFQ total score. |
| HF with IHD | MLHFQ symptoms score. | Carnitine 195 mg. | Significant decrease in MLHFQ symptoms score. | ||
| Age: 57.6 ± 9 | CoQ10 27 mg. | Improvement in NYHA classification. | |||
| Taurine 200 mg. | |||||
| Thiamine 22 mg. | |||||
| Riboflavin 22 mg. | |||||
| Ascorbate 2,450 mg. | |||||
| Lysine 1,110 mg. | |||||
| Proline 110 mg. | |||||
| Arginine 790 mg. | |||||
| 4 to 8 months. |
EF: ejection fraction; LVEDV: left ventricular end-diastolic volume; LVEF: left ventricular ejection fraction; LVESV: left ventricular end-systolic volume; MLHFQ: Minnesota Living with Heart Failure Questionnaire; NT-proBNP: N terminal prohormone brain natriuretic peptide; QOL: quality of life; RCT: randomized controlled trial; 6MWT: 6 minute walk test.
Of the four studies selected, three were randomized controlled trials (RCT), while one was a prospective case series. Even though all four studies used multiple micronutrients, the focus in this review is primarily on L-carnitine, CoQ10, thiamine, riboflavin, and taurine, because the nutrients are known metabolites in the pathways of energy production, oxidative defense, and myocardial calcium balance. There is evidence that L-carnitine [39], CoQ10 [40], taurine [41], and thiamine [42] have a positive effect on HF. There is also evidence of a high prevalence of deficiencies in riboflavin and other B vitamins among patients with HF [43].
Three of the four studies selected showed positive results of micronutrient administration while one showed negative results. The difference in the study outcomes could be explained by differences in micronutrient doses used in the studies. As such, the three studies with positive results used much higher dosages than the study with negative results. In addition, the studies with positive results used CoQ10 and L-carnitine, except for the positive outcome of the study by Witte and colleagues that used only CoQ10 but no amino acids.
Significance of micronutrient dosages
In all previous studies on HF and myocardial energetics, there has been no mention of the significance of dosage of micronutrients. In describing myocardial protein degradation as a consequence of nutrient deprivation, Taegtmeyer and Lubrano [44] proposed restoring fuel homeostasis in order to restore the cycle of protein turnover. The rationale is that there will be substrate deprivation in chronic HF. When there is nutrient deprivation, intracellular concentration of ATP will decline while adenosine monophosphate (AMP) levels rise, resulting in the activation of the enzyme, AMP-activated protein kinase (AMPK). The activation of AMPK results in providing energy by fueling the Krebs cycle with amino acids [45]. AMPK allows for the liberation and recycle of nutrients from the myocytes [46] which maintain a continuous catabolic state for the failing heart.
Refueling the failing heart with metabolites and amino acids supports generation of myocardial energy and it may solve half of the problem. Another therapeutic target should involve arresting the chronic catabolic state and cardiac cachexia. It is reasonable to assume that this can be achieved with supplementation of the necessary nutrients in high dosages. A low micronutrient dosage merely helps maintain basic metabolic needs, but in chronic HF appropriate higher dosages of nutrients are necessary to see therapeutic effect. The positive results of those studies highlighted in Table 1 provide supporting evidence indicating the significance of using high dosages of multiple micronutrients in achieving therapeutic effects in HF.
Under conditions of ischemic HF and starvation, autophagic activity becomes increased in order to support cellular energy and function by mobilizing endogenous nutritional sources [47]. Organelles and large cellular components are degraded into amino acids, fatty acids, sugars, and nucleotides to be rechanneled for the myocardial energetic and Krebs cycle pathways. AMPK is activated as a mediator for autophagy in the recycling of myocardial energetics [47]. As a remedial measure, it would be appropriate to incorporate a high dosage of micronutrient supplementation to mitigate potential damage caused by chronic HF. For the improved nutrition of the heart, Taegtmeyer and colleagues have proposed that it should go beyond the mere supply of energy-substrates, but also include the supply of amino acids and micronutrients [48].
The role of amino acids
The increased autophagic activity in patients with chronic HF leading to a hyper catabolic state gives an indication of the important role of amino acids in this disease syndrome. In cardiac metabolism, amino acids are necessary for protein synthesis and as intermediate metabolites in energy cycle pathways. In the myocytes, amino acids also act as cofactors or coenzymes for intracellular pathways of energy conversion. In has been mentioned earlier that the heart requires a large and constant supply of amino acids for protein synthesis and also as intermediates for energy production in the Krebs cycle.
In HF, amino acids have a close relationship with the growth hormone (GH)/insulin-like growth hormone factor-1 (IGF-1) axis. Patients with HF are usually deficient in GH and IGF-1 [49,50]. When absorbed into the portal system, amino acids stimulate release of IGF-1 and that in turn induces the activation of GH and therefore promotes anabolic metabolism. IGF-1 promotes collagen synthesis by fibroblasts, while GH increases collagen deposition rate in the heart [51,52]. Thus, amino acids as a group have their important role in maintaining cardiac functions. However, this seems to be a paradox because high levels of IGF-1 predicts cardiovascular mortality [28] and increases autophagy [29]. The possible explanation for this is that IGF-1 during the initial stage of HF is compensatory. When the disease syndrome progresses without a sufficient supply of nutrients to support energy production, the pathology deepens with manifestations in wet beriberi, left ventricular hypertrophy, cachexia, and extreme lethargy. Studies using high daily dosages (8 grams) of the amino acid mixtures in HF, and measuring different outcomes and different endpoints, have produced positive results [53-55]. Single amino acids have been evaluated separately on HF with different endpoints.
L-carnitine has been widely studied in HF and many studies have brought positive results [56,60]. The dosage of L-carnitine used in these studies ranges from 1.5 grams to a high dosage of 6 grams daily. These dosages seem to be well tolerated. Carnitine is not an essential amino acid. Its biosynthesis takes place in the liver and kidneys from the two amino acids, lysine and methionine [61], and it requires ascorbic acid as a cofactor [62,63]. L-carnitine plays an important role in transporting fatty acid into the mitochondria [64] and, by improving the coupling between glycolysis and glucose oxidation, it improves the utilization of pyruvate in the Krebs cycle [65].
Taurine functions as an antioxidant and a regulator of intracellular calcium homeostasis [41,66] protecting the myocytes from calcium overload, as in the case in HF. Calcium overload can lead to injury of the myocytes. Studies on the effects of taurine in HF have also produced positive results [41,67-69]. A daily dosage of 1.5 to 6 grams of taurine was used in these studies, and it was well tolerated.
Other than the one study mentioned in Table 1 [38], there are no studies involving L-lysine whether used individually or as a component of multiple nutrients in HF. However, this does not mean that its role in HF should be relegated. In the human cells, with a constant flux of biological materials, lysine acetylation plays an important role in a dynamic and reversible post-translational modification of proteins involved in cellular processes including genetic regulations, transcription, survival, apoptosis, and mitochondrial-derived processes in acetyl coenzyme A formation [70,71]. Almost every enzyme in glycolysis, glucose and fatty acid oxidation are acetylated at least on one lysine residue. Therefore, as lysine is an essential amino acid, and with such myriad cellular processes, the need for its constant supply in the diet or through nutritional supplementation cannot be emphasized enough. In chronic HF, matrix metalloproteinases (MMPs), and other proteolytic enzymes, are overly expressed while tissue inhibitors of MMPs (TIMPs) are under expressed [72]. The disparity in expression between MMPs and TIMPs levels, which favors MMPs activation and proteolysis, contributes to left ventricular remodeling process. While mainstream research has yet to discover a novel strategy to normalize the balance between MMPs and TIMPs, in 1992 Rath and Pauling postulated that L-lysine is a natural inhibitor of proteolysis by MMPs and plasmin [73].
Coenzyme Q10
Coenzyme Q10 (CoQ10), also known as ubiquinone, is present in high concentrations in the mitochondria of the myocardium, liver, and kidneys. It has a vital role in myocardial energetics and the protection of the myocytes. CoQ10, which acts as a mobile electron carrier in the mitochondria, is a cofactor in mitochondrial oxidative phosphorylation leading to ATP production [74]. In addition, it also acts as an antioxidant protecting the cell membrane against oxidation [75] and inhibiting the peroxidation of lipids and lipoproteins [76,77]. Over the last few decades, there have been numerous studies with CoQ10 supplementation in HF each using different study designs, dosages, time frame and outcome measures, and often bringing inconclusive results. Of importance are three meta-analyses [78-80] that showed positive results of CoQ10 administration in surrogate endpoints. The meta-analyses of Sander and colleagues [78] and Fotino and colleagues [80] showed significant improvement in ejection fraction using CoQ10 in dosage ranges from 60 to 300 mg/day. Whereas the meta-analysis of Soja and colleagues [79] showed significant improvement in stroke volume, ejection fraction, cardiac output, cardiac index, and end-diastolic volume index.
The B vitamins
The B vitamins are a group of water-soluble vitamins that are important coenzymes in the Krebs cycle and are necessary for ATP production. Of the eight B vitamins, thiamine (vitamin B1) has a direct role in myocardial energetics as a coenzyme in converting carbohydrates to energy. Thiamine is a form of thiamine pyrophosphate and serves as a cofactor of the pyruvate dehydrogenase and transketolase, which are both mediators in energy substrate metabolism [81]. Thiamine is an essential vitamin, therefore it cannot be synthesized endogenously and, as a water soluble vitamin it can be stored only in small quantities. In the clinical trial by Shimon and colleagues [82], patients given 200 mg/day of thiamine showed significant improvements in diuresis and ejection fraction. A recent clinical trial using a high thiamine dosage of 300 mg/day also showed significant improvements in the ejection fraction [42].
Riboflavin (vitamin B2) and pyridoxine (vitamin B6) also play important roles in carbohydrate energy metabolism. Deficiency of riboflavin and pyridoxine has been documented in HF patients [43]. Being water-soluble and essential, these vitamins are subjected to renal loss, have limited tissue storage, and are dependent on dietary intake. Although these B vitamins are known to play an important role in energy metabolism, data to support an effect in HF are lacking.
Conclusion and future direction
Over the last few decades, there have been many clinical studies using mono or multiple micronutrients showing significant positive effects in HF using different surrogate endpoints. These studies used a high dosage of the micronutrients and patients seemed to tolerate well. Taking into account that HF is a syndrome of a chronic failing organ involving multi-faceted pathways, treating this disease with a single nutrient may show a temporary positive effect, but the disease as a whole continues. On the other hand, treatments that involve multiple micronutrients may produce better results, but if key micronutrients are applied in low, insufficient doses the positive symptomatic effect may not be immediately demonstrated. Current recommended daily allowances (RDA) for micronutrients are designed to maintain health in a healthy person. However, restoring health in a chronic disease condition may require therapeutic micronutrient doses which are much higher than the RDA. Clinical evidence coming from the studies presented in Table 1 substantiates the use of multiple micronutrient supplementation in high dosages as an adjunctive therapy for chronic HF.
The case for myocardial bioenergetics as a model for HF has been well established. Current interest has been on modulation of substrates to maximize energy production or energy sparing. Despite research advances in understanding the role of substrate metabolism and pharmacological cardio tonics, morbidity and mortality in HF remain high.
Perhaps it is time that research returns to the basics and refocuses on myocardial energetics with its special emphasis on the role of specific micronutrient combinations. It is a matter of urgency for HF therapy especially if such a repertoire of therapeutics includes safe and affordable micronutrients.
More long-term studies in HF are warranted with larger patient populations and vigorous designs, using high dosage multiple micronutrients with different surrogate and cardiovascular endpoints.
Disclosure of conflict of interest
None.
References
- 1.McMurray JJ, Petrie MC, Murdoch DR, Davie AP. Clinical epidemiology of heart failure: public and private health burden. Eur Heart J. 1998;19(Suppl P):P9–16. [PubMed] [Google Scholar]
- 2.Bloom DE, et al. The global economic burden of noncommunicable diseases. Geneva: world economic forum 2011 3 Dec 2014. [Google Scholar]
- 3.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braunwald E. Heart failure. JACC Heart Fail. 2013;1:1–20. doi: 10.1016/j.jchf.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Coronel R, de Groot JR, van Lieshout JJ. Defining heart failure. Cardiovasc Res. 2001;50:419–22. doi: 10.1016/s0008-6363(01)00284-x. [DOI] [PubMed] [Google Scholar]
- 6.Herrmann G, Decherd GM. The chemical nature of heart failure. Ann Intern Med. 1939;12:1233–1244. [Google Scholar]
- 7.Braunwald E, Ross J Jr, Sonnenblick EH. Mechanisms of contraction of the normal and failing heart. N Engl J Med. 1967;277:1012–22. doi: 10.1056/NEJM196711092771907. concl. [DOI] [PubMed] [Google Scholar]
- 8.McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Gomez-Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popescu BA, Rønnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A ESC Committee for Practice Guidelines. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2012;33:1787–847. doi: 10.1093/eurheartj/ehs104. [DOI] [PubMed] [Google Scholar]
- 9.Eisenhofer G, Friberg P, Rundqvist B, Quyyumi AA, Lambert G, Kaye DM, Kopin IJ, Goldstein DS, Esler MD. Cardiac sympathetic nerve function in congestive heart failure. Circulation. 1996;93:1667–76. doi: 10.1161/01.cir.93.9.1667. [DOI] [PubMed] [Google Scholar]
- 10.Dzau VJ, Packer M, Lilly LS, Swartz SL, Hollenberg NK, Williams GH. Prostaglandins in severe congestive heart failure. Relation to activation of the renin-angiotensin system and hyponatremia. N Engl J Med. 1984;310:347–52. doi: 10.1056/NEJM198402093100603. [DOI] [PubMed] [Google Scholar]
- 11.Dzau VJ, Colucci WS, Hollenberg NK, Williams GH. Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation. 1981;63:645–51. doi: 10.1161/01.cir.63.3.645. [DOI] [PubMed] [Google Scholar]
- 12.Mann DL, Bristow MR. Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation. 2005;111:2837–49. doi: 10.1161/CIRCULATIONAHA.104.500546. [DOI] [PubMed] [Google Scholar]
- 13.Tan LB, Jalil JE, Pick R, Janicki JS, Weber KT. Cardiac myocyte necrosis induced by angiotensin II. Circ Res. 1991;69:1185–95. doi: 10.1161/01.res.69.5.1185. [DOI] [PubMed] [Google Scholar]
- 14.Katz AM. Cardiomyopathy of overload. A major determinant of prognosis in congestive heart failure. N Engl J Med. 1990;322:100–10. doi: 10.1056/NEJM199001113220206. [DOI] [PubMed] [Google Scholar]
- 15.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–45. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 16.Stanley WC, Chandler MP. Energy metabolism in the normal and failing heart: potential for therapeutic interventions. Heart Fail Rev. 2002;7:115–30. doi: 10.1023/a:1015320423577. [DOI] [PubMed] [Google Scholar]
- 17.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–58. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 18.Klein LJ, Visser FC, Knaapen P, Peters JH, Teule GJ, Visser CA, Lammertsma AA. Carbon-11 acetate as a tracer of myocardial oxygen consumption. Eur J Nucl Med. 2001;28:651–68. doi: 10.1007/s002590000472. [DOI] [PubMed] [Google Scholar]
- 19.Essop MF, Opie LH. Metabolic therapy for heart failure. Eur Heart J. 2004;25:1765–8. doi: 10.1016/j.ehj.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 20.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–51. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 21.Knaapen P, Germans T, Knuuti J, Paulus WJ, Dijkmans PA, Allaart CP, Lammertsma AA, Visser FC. Myocardial energetics and efficiency: current status of the noninvasive approach. Circulation. 2007;115:918–27. doi: 10.1161/CIRCULATIONAHA.106.660639. [DOI] [PubMed] [Google Scholar]
- 22.Leong HS, Brownsey RW, Kulpa JE, Allard MF. Glycolysis and pyruvate oxidation in cardiac hypertrophy--why so unbalanced? Comp Biochem Physiol A Mol Integr Physiol. 2003;135:499–513. doi: 10.1016/s1095-6433(03)00007-2. [DOI] [PubMed] [Google Scholar]
- 23.Starling RC, Hammer DF, Altschuld RA. Human myocardial ATP content and in vivo contractile function. Mol Cell Biochem. 1998;180:171–7. [PubMed] [Google Scholar]
- 24.Nascimben L, Ingwall JS, Pauletto P, Friedrich J, Gwathmey JK, Saks V, Pessina AC, Allen PD. Creatine kinase system in failing and nonfailing human myocardium. Circulation. 1996;94:1894–901. doi: 10.1161/01.cir.94.8.1894. [DOI] [PubMed] [Google Scholar]
- 25.Vanoverschelde JL, Wijns W, Essamri B, Bol A, Robert A, Labar D, Cogneau M, Michel C, Melin JA. Hemodynamic and mechanical determinants of myocardial O2 consumption in normal human heart: effects of dobutamine. Am J Physiol. 1993;265:H1884–92. doi: 10.1152/ajpheart.1993.265.6.H1884. [DOI] [PubMed] [Google Scholar]
- 26.Hayashi Y, Takeuchi M, Takaoka H, Hata K, Mori M, Yokoyama M. Alteration in energetics in patients with left ventricular dysfunction after myocardial infarction: increased oxygen cost of contractility. Circulation. 1996;93:932–9. doi: 10.1161/01.cir.93.5.932. [DOI] [PubMed] [Google Scholar]
- 27.Hawkins MN, Barnes Q, Purkayastha S, Eubank W, Ogoh S, Raven PB. The effects of aerobic fitness and beta1-adrenergic receptor blockade on cardiac work during dynamic exercise. J Appl Physiol (1985) 2009;106:486–93. doi: 10.1152/japplphysiol.90795.2008. [DOI] [PubMed] [Google Scholar]
- 28.Chisalita SI, Dahlström U, Arnqvist HJ, Alehagen U. Increased IGF1 levels in relation to heart failure and cardiovascular mortality in an elderly population: impact of ACE inhibitors. Eur J Endocrinol. 2011;165:891–8. doi: 10.1530/EJE-11-0584. [DOI] [PubMed] [Google Scholar]
- 29.Aki T, Yamaguchi K, Fujimiya T, Mizukami Y. Phosphoinositide 3-kinase accelerates autophagic cell death during glucose deprivation in the rat cardiomyocyte-derived cell line H9c2. Oncogene. 2003;22:8529–35. doi: 10.1038/sj.onc.1207197. [DOI] [PubMed] [Google Scholar]
- 30.Witte KK, Clark AL. Micronutrients and their supplementation in chronic cardiac failure. An update beyond theoretical perspectives. Heart Fail Rev. 2006;11:65–74. doi: 10.1007/s10741-006-9194-4. [DOI] [PubMed] [Google Scholar]
- 31.Hughes CM, Woodside JV, McGartland C, Roberts MJ, Nicholls DP, McKeown PP. Nutritional intake and oxidative stress in chronic heart failure. Nutr Metab Cardiovasc Dis. 2012;22:376–82. doi: 10.1016/j.numecd.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 32.Jeejeebhoy KN, Sole MJ. Nutrition and the heart. Clinical Nutrition. 2001;20:181–186. [Google Scholar]
- 33.Lee JH, Jarreau T, Prasad A, Lavie C, O’Keefe J, Ventura H. Nutritional assessment in heart failure patients. Congest Heart Fail. 2011;17:199–203. doi: 10.1111/j.1751-7133.2011.00239.x. [DOI] [PubMed] [Google Scholar]
- 34.Soukoulis V, Dihu JB, Sole M, Anker SD, Cleland J, Fonarow GC, Metra M, Pasini E, Strzelczyk T, Taegtmeyer H, Gheorghiade M. Micronutrient deficiencies an unmet need in heart failure. J Am Coll Cardiol. 2009;54:1660–73. doi: 10.1016/j.jacc.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 35.Witte KK, Nikitin NP, Parker AC, von Haehling S, Volk HD, Anker SD, Clark AL, Cleland JG. The effect of micronutrient supplementation on quality-of-life and left ventricular function in elderly patients with chronic heart failure. Eur Heart J. 2005;26:2238–44. doi: 10.1093/eurheartj/ehi442. [DOI] [PubMed] [Google Scholar]
- 36.Jeejeebhoy F, Keith M, Freeman M, Barr A, McCall M, Kurian R, Mazer D, Errett L. Nutritional supplementation with MyoVive repletes essential cardiac myocyte nutrients and reduces left ventricular size in patients with left ventricular dysfunction. Am Heart J. 2002;143:1092–100. doi: 10.1067/mhj.2002.121927. [DOI] [PubMed] [Google Scholar]
- 37.McKeag NA, McKinley MC, Harbinson MT, Noad RL, Dixon LH, McGinty A, Neville CE, Woodside JV, McKeown PP. The effect of multiple micronutrient supplementation on left ventricular ejection fraction in patients with chronic stable heart failure: a randomized, placebo-controlled trial. JACC Heart Fail. 2014;2:308–17. doi: 10.1016/j.jchf.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 38.Wong AP, Mohamed AL, Niedzwiecki A. The effect of multiple micronutrient supplementation on quality of life in patients with symptomatic heart failure secondary to ischemic heart disease: a prospective case series clinical study. Am J Cardiovasc Dis. 2015;5:146–52. [PMC free article] [PubMed] [Google Scholar]
- 39.Ueland T, Svardal A, Øie E, Askevold ET, Nymoen SH, Bjørndal B, Dahl CP, Gullestad L, Berge RK, Aukrust P. Disturbed carnitine regulation in chronic heart failure--increased plasma levels of palmitoyl-carnitine are associated with poor prognosis. Int J Cardiol. 2013;167:1892–9. doi: 10.1016/j.ijcard.2012.04.150. [DOI] [PubMed] [Google Scholar]
- 40.Alehagen U, Johansson P, Björnstedt M, Rosén A, Dahlström U. Cardiovascular mortality and N-terminal-proBNP reduced after combined selenium and coenzyme Q10 supplementation: a 5-year prospective randomized double-blind placebo-controlled trial among elderly Swedish citizens. Int J Cardiol. 2013;167:1860–6. doi: 10.1016/j.ijcard.2012.04.156. [DOI] [PubMed] [Google Scholar]
- 41.Azuma J, Sawamura A, Awata N. Usefulness of taurine in chronic congestive heart failure and its prospective application. Jpn Circ J. 1992;56:95–9. doi: 10.1253/jcj.56.95. [DOI] [PubMed] [Google Scholar]
- 42.Schoenenberger AW, Schoenenberger-Berzins R, der Maur CA, Suter PM, Vergopoulos A, Erne P. Thiamine supplementation in symptomatic chronic heart failure: a randomized, double-blind, placebo-controlled, cross-over pilot study. Clin Res Cardiol. 2012;101:159–64. doi: 10.1007/s00392-011-0376-2. [DOI] [PubMed] [Google Scholar]
- 43.Keith ME, Walsh NA, Darling PB, Hanninen SA, Thirugnanam S, Leong-Poi H, Barr A, Sole MJ. B-vitamin deficiency in hospitalized patients with heart failure. J Am Diet Assoc. 2009;109:1406–10. doi: 10.1016/j.jada.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 44.Taegtmeyer H, Lubrano G. Rethinking cardiac metabolism: metabolic cycles to refuel and rebuild the failing heart. F1000Prime Rep. 2014;6:90. doi: 10.12703/P6-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim AS, Miller EJ, Young LH. AMP-activated protein kinase: a core signalling pathway in the heart. Acta Physiol (Oxf) 2009;196:37–53. doi: 10.1111/j.1748-1716.2009.01978.x. [DOI] [PubMed] [Google Scholar]
- 46.Baskin KK, Rodriguez MR, Kansara S, Chen W, Carranza S, Frazier OH, Glass DJ, Taegtmeyer H. MAFbx/Atrogin-1 is required for atrophic remodeling of the unloaded heart. J Mol Cell Cardiol. 2014;72:168–76. doi: 10.1016/j.yjmcc.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–8. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taegtmeyer H, Harinstein ME, Gheorghiade M. More than bricks and mortar: comments on protein and amino acid metabolism in the heart. Am J Cardiol. 2008;101:3E–7E. doi: 10.1016/j.amjcard.2008.02.064. [DOI] [PubMed] [Google Scholar]
- 49.Kontoleon PE, Anastasiou-Nana MI, Papapetrou PD, Alexopoulos G, Ktenas V, Rapti AC, Tsagalou EP, Nanas JN. Hormonal profile in patients with congestive heart failure. Int J Cardiol. 2003;87:179–83. doi: 10.1016/s0167-5273(02)00212-7. [DOI] [PubMed] [Google Scholar]
- 50.Jankowska EA, Biel B, Majda J, Szklarska A, Lopuszanska M, Medras M, Anker SD, Banasiak W, Poole-Wilson PA, Ponikowski P. Anabolic deficiency in men with chronic heart failure: prevalence and detrimental impact on survival. Circulation. 2006;114:1829–37. doi: 10.1161/CIRCULATIONAHA.106.649426. [DOI] [PubMed] [Google Scholar]
- 51.Li Q, Li B, Wang X, Leri A, Jana KP, Liu Y, Kajstura J, Baserga R, Anversa P. Overexpression of insulin-like growth factor-1 in mice protects from myocyte death after infarction, attenuating ventricular dilation, wall stress, and cardiac hypertrophy. J Clin Invest. 1997;100:1991–9. doi: 10.1172/JCI119730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mayoux E, Ventura-Clapier R, Timsit J, Béhar-Cohen F, Hoffmann C, Mercadier JJ. Mechanical properties of rat cardiac skinned fibers are altered by chronic growth hormone hypersecretion. Circ Res. 1993;72:57–64. doi: 10.1161/01.res.72.1.57. [DOI] [PubMed] [Google Scholar]
- 53.Aquilani R, Opasich C, Gualco A, Verri M, Testa A, Pasini E, Viglio S, Iadarola P, Pastoris O, Dossena M, Boschi F. Adequate energy-protein intake is not enough to improve nutritional and metabolic status in muscle-depleted patients with chronic heart failure. Eur J Heart Fail. 2008;10:1127–35. doi: 10.1016/j.ejheart.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 54.Aquilani R, Viglio S, Iadarola P, Opasich C, Testa A, Dioguardi FS, Pasini E. Oral amino acid supplements improve exercise capacities in elderly patients with chronic heart failure. Am J Cardiol. 2008;101:104E–110E. doi: 10.1016/j.amjcard.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 55.Scognamiglio R, Testa A, Aquilani R, Dioguardi FS, Pasini E. Impairment in walking capacity and myocardial function in the elderly: is there a role for nonpharmacologic therapy with nutritional amino acid supplements? Am J Cardiol. 2008;101:78E–81E. doi: 10.1016/j.amjcard.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 56.Mancini M, Rengo F, Lingetti M, Sorrentino GP, Nolfe G. Controlled study on the therapeutic efficacy of propionyl-L-carnitine in patients with congestive heart failure. Arzneimittelforschung. 1992;42:1101–4. [PubMed] [Google Scholar]
- 57.Iliceto S, Scrutinio D, Bruzzi P, D’Ambrosio G, Boni L, Di Biase M, Biasco G, Hugenholtz PG, Rizzon P. Effects of L-carnitine administration on left ventricular remodeling after acute anterior myocardial infarction: the L-Carnitine Ecocardiografia Digitalizzata Infarto Miocardico (CEDIM) Trial. J Am Coll Cardiol. 1995;26:380–7. doi: 10.1016/0735-1097(95)80010-e. [DOI] [PubMed] [Google Scholar]
- 58.Anand I, Chandrashekhan Y, De Giuli F, Pasini E, Mazzoletti A, Confortini R, Ferrari R. Acute and chronic effects of propionyl-L-carnitine on the hemodynamics, exercise capacity, and hormones in patients with congestive heart failure. Cardiovasc Drugs Ther. 1998;12:291–9. doi: 10.1023/a:1007721917561. [DOI] [PubMed] [Google Scholar]
- 59.Löster H, Miehe K, Punzel M, Stiller O, Pankau H, Schauer J. Prolonged oral L-carnitine substitution increases bicycle ergometer performance in patients with severe, ischemically induced cardiac insufficiency. Cardiovasc Drugs Ther. 1999;13:537–46. doi: 10.1023/a:1007883822625. [DOI] [PubMed] [Google Scholar]
- 60.Rizos I. Three-year survival of patients with heart failure caused by dilated cardiomyopathy and L-carnitine administration. Am Heart J. 2000;139:S120–3. doi: 10.1067/mhj.2000.103917. [DOI] [PubMed] [Google Scholar]
- 61.Steiber A, Kerner J, Hoppel CL. Carnitine: a nutritional, biosynthetic, and functional perspective. Mol Aspects Med. 2004;25:455–73. doi: 10.1016/j.mam.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 62.Rebouche CJ. Ascorbic acid and carnitine biosynthesis. Am J Clin Nutr. 1991;54:1147S–1152S. doi: 10.1093/ajcn/54.6.1147s. [DOI] [PubMed] [Google Scholar]
- 63.Dunn WA, Rettura G, Seifter E, Englard S. Carnitine biosynthesis from gamma-butyrobetaine and from exogenous protein-bound 6-N-trimethyl-L-lysine by the perfused guinea pig liver. Effect of ascorbate deficiency on the in situ activity of gamma-butyrobetaine hydroxylase. J Biol Chem. 1984;259:10764–70. [PubMed] [Google Scholar]
- 64.Arsenian MA. Carnitine and its derivatives in cardiovascular disease. Prog Cardiovasc Dis. 1997;40:265–86. doi: 10.1016/s0033-0620(97)80037-0. [DOI] [PubMed] [Google Scholar]
- 65.Schönekess BO, Allard MF, Lopaschuk GD. Propionyl L-carnitine improvement of hypertrophied heart function is accompanied by an increase in carbohydrate oxidation. Circ Res. 1995;77:726–34. doi: 10.1161/01.res.77.4.726. [DOI] [PubMed] [Google Scholar]
- 66.Schaffer SW, Kramer J, Chovan JP. Regulation of calcium homeostasis in the heart by taurine. Fed Proc. 1980;39:2691–4. [PubMed] [Google Scholar]
- 67.Azuma J, Hasegawa H, Sawamura A, Awata N, Ogura K, Harada H, Yamamura Y, Kishimoto S. Therapy of congestive heart failure with orally administered taurine. Clin Ther. 1983;5:398–408. [PubMed] [Google Scholar]
- 68.Azuma J, Sawamura A, Awata N, Ohta H, Hamaguchi T, Harada H, Takihara K, Hasegawa H, Yamagami T, Ishiyama T, et al. Therapeutic effect of taurine in congestive heart failure: a double-blind crossover trial. Clin Cardiol. 1985;8:276–82. doi: 10.1002/clc.4960080507. [DOI] [PubMed] [Google Scholar]
- 69.Beyranvand MR, Khalafi MK, Roshan VD, Choobineh S, Parsa SA, Piranfar MA. Effect of taurine supplementation on exercise capacity of patients with heart failure. J Cardiol. 2011;57:333–7. doi: 10.1016/j.jjcc.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 70.Anderson KA, Hirschey MD. Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 2012;52:23–35. doi: 10.1042/bse0520023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Newman JC, He W, Verdin E. Mitochondrial protein acylation and intermediary metabolism: regulation by sirtuins and implications for met abolic disease. J Biol Chem. 2012;287:42436–43. doi: 10.1074/jbc.R112.404863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Spinale FG. Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ Res. 2002;90:520–30. doi: 10.1161/01.res.0000013290.12884.a3. [DOI] [PubMed] [Google Scholar]
- 73.Rath M, Pauling L. Plasmin-induced proteolysis and the role of apoprotein(a), lysine, and synthetic lysine analogs. J Orthomol Med. 1992;7:17–23. [Google Scholar]
- 74.Florkowski CM, Molyneux SL, Young JM. Coenzyme Q10 and congestive heart failure; an evolving evidence base. Kardiol Pol. 2015;73:73–9. doi: 10.5603/KP.2015.0015. [DOI] [PubMed] [Google Scholar]
- 75.Mellors A, Tappel AL. The inhibition of mitochondrial peroxidation by ubiquinone and ubiquinol. J Biol Chem. 1966;241:4353–6. [PubMed] [Google Scholar]
- 76.Mellors A, Tappel AL. Quinones and quinols as inhibitors of lipid peroxidation. Lipids. 1966;1:282–4. doi: 10.1007/BF02531617. [DOI] [PubMed] [Google Scholar]
- 77.Stocker R, Bowry VW, Frei B. Ubiquinol-10 protects human low density lipoprotein more efficiently against lipid peroxidation than does alpha-tocopherol. Proc Natl Acad Sci U S A. 1991;88:1646–50. doi: 10.1073/pnas.88.5.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sander S, Coleman CI, Patel AA, Kluger J, White CM. The impact of coenzyme Q10 on systolic function in patients with chronic heart failure. J Card Fail. 2006;12:464–72. doi: 10.1016/j.cardfail.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 79.Soja AM, Mortensen SA. Treatment of congestive heart failure with coenzyme Q10 illuminated by meta-analyses of clinical trials. Mol Aspects Med. 1997;18(Suppl):S159–68. doi: 10.1016/s0098-2997(97)00042-3. [DOI] [PubMed] [Google Scholar]
- 80.Fotino AD, Thompson-Paul AM, Bazzano LA. Effect of coenzyme Q(1)(0) supplementation on heart failure: a meta-analysis. Am J Clin Nutr. 2013;97:268–75. doi: 10.3945/ajcn.112.040741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Voskoboyev AI, Ostrovsky YM. Thiamin pyrophosphokinase: structure, properties, and role in thiamin metabolism. Ann N Y Acad Sci. 1982;378:161–76. doi: 10.1111/j.1749-6632.1982.tb31195.x. [DOI] [PubMed] [Google Scholar]
- 82.Shimon I, Almog S, Vered Z, Seligmann H, Shefi M, Peleg E, Rosenthal T, Motro M, Halkin H, Ezra D. Improved left ventricular function after thiamine supplementation in patients with congestive heart failure receiving long-term furosemide therapy. Am J Med. 1995;98:485–90. doi: 10.1016/s0002-9343(99)80349-0. [DOI] [PubMed] [Google Scholar]