

Abstract
Abstract: Reactive oxygen species (ROS) generation has been implicated in many pathologies including ischemia/reperfusion (I/R) injury. This led to multiple studies on antioxidant therapies to treat cardiovascular diseases but paradoxically, results have so far been mixed as ROS production can be beneficial as a signaling mechanism and in cardiac protection via preconditioning interventions. We investigated whether the differential impact of increased ROS in injury as well as in protection could be explained by their site of production on the mitochondrial electron transport chain. Using amplex red to measure ROS production, we found that mitochondria isolated from hearts after I/R produced more ROS than non-ischemic when complex I substrate (glutamate/malate) was used. Interestingly, the substrates of complex II (succinate) and ubiquinone (sn-glycerol 3-phosphate, G3P) produced less ROS in mitochondria from I/R hearts compared to normal healthy hearts. The inhibitors of complex I (rotenone) and complex III (antimycin A) increased ROS production when glutamate/malate and G3P were used; in contrast, they reduced ROS production when the complex II substrate was used. Mitochondrial calcium retention capacity required to induce mitochondrial permeability transition pore (mPTP) opening was measured using calcium green fluorescence and was found to be higher when mitochondria were treated with G3P and succinate compared to glutamate/malate. Furthermore, Langendorff hearts treated with glutamate/malate exhibited reduced cardiac functional recovery and increased myocardial infarct size compared to hearts treated with G3P. Thus, ROS production by the stimulated respiratory chain complexes I and III has opposite roles: cardio-deleterious when produced in complex I and cardio-protective when produced in complex III. The mechanism of these ROS involves the inhibition of the mPTP opening, a key event in cell death following ischemia/reperfusion injury.
Keywords: Reactive oxygen species, electron transport chain, ischemia, reperfusion, mitochondrial permeability transition pore
Introduction
Oxidative stress generated by dysfunctional mitochondria in ischemia/reperfusion (I/R) injury contributes to heart failure and other major cardiovascular diseases [1-3]. The generation of reactive oxygen species (ROS) and their by-products by the defective mitochondrial electron transport chain (ETC) activates cytotoxic mechanisms responsible for cell death via apoptosis and necrosis [1]. Cardiovascular research has for a long time been focused on antioxidant therapies like vitamin E and CoQ10 based on promising experiments but clinical trials were inconclusive at best, showed no improvement versus placebo, and were even harmful in some trials [2-6]. Speculation about why these trials failed includes questioning the doses used, disease progression at the time of administration, length of the trials as well as the antioxidants tested, but a more plausible argument has been the recognition that such therapies need to be more targeted towards mitochondria and the respiratory chain, which are the main sources of ROS in cardiac cells [2,7]. From this focus, new treatments have been proposed such as MitoQ which has shown positive results in experiments but unfortunately again, the clinical data is not definitive [8,9].
Adding to the confusing data, ROS are also known to be second-messenger molecules involved in growth factor and cytokine signaling, initiation of mitogen-activated protein kinase (MAPK) pathways and communication vital for cell adhesion [10]. In particular, ROS are known to be upregulated in ischemic and pharmacological preconditioning studies to protect the myocardium against I/R injury by activating pathways involving p38 MAPK, MAPKAP kinase 2 and NFκB [11,14]. Paradoxically, ROS inhibition has been shown to induce cardioprotection against I/R injury in post-conditioning studies [15,16], but ROS upregulation has also been observed as being necessary to activate mechanisms for limiting reperfusion injury [17]. In all, there still has yet to be proposed a unified theory that satisfactorily explains both the cardio-beneficial and cardio-deleterious effects of ROS in I/R.
As many studies have shown the opposing ROS impacts on cardiac tissue, it has become common to attribute the differences in outcomes of protection or damage to the quantity of ROS produced by mitochondria. In this theory, low quantities of ROS are responsible for the cell survival mechanisms while excess amounts lead to cell and tissue death. Appealing as this may be, it fails to account for the fact that an increase in ROS is observed in both protection and damage, thus, despite low/physiological quantities of ROS being generated naturally for signaling purposes, abnormal quantities are needed in both diseased and protected states. To this end, we proposed an extension to the quantity theory that narrows down the focus to the mitochondrial respiratory chain. We hypothesized that it is not only the quantity of ROS that determines the outcome, but it is the quantity and specific ETC source of that ROS that matters.
In this study, our focus was to determine whether ROS generated by mitochondrial ETC complexes I and III (Figure 1), the main sources of ROS, had differential impacts on the myocardium when sequentially stimulated. We provide evidence suggesting that ROS from complex III mediates cardio-protective effects of delaying the opening of the mitochondrial permeability transition pore (mPTP) in isolated mitochondria, as well as improving functional recovery and reducing infarct size in isolated hearts following I/R stress. Oppositely, our data suggests that ROS from complex I is damaging to mitochondria and whole-heart tissue as judged by the same measures of transition pore opening, functional recovery and infarct size after I/R.
Figure 1.
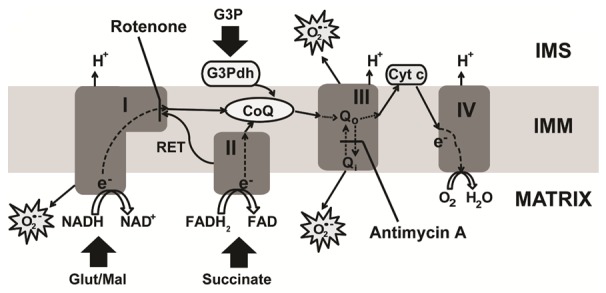
The electron transport chain within the inner mitochondrial membrane (IMM). Illustration showing electron (e-) transport chain from complex I to IV (dashed and solid lines) and highlighting different sites of reactive oxygen species (ROS) generation as well as indicating the sites of inhibitor action. Glutamate/malate (Glut/Mal) is issued as substrate of complex I; Succinate, as substrate of complex II; and sn-glycerol 3-phosphate (G3P) as, activator of the CoQ pool. G3Pdh: glycerol 3-phosphate dehydrogenase; RET: reverse electron transfer; Cyt c: cytochrome c; CoQ: ubiquinone; and IMS: intermembrane space.
Materials and methods
Animals
Male adult mice (C57BL/6J), 8-16 weeks old, were used. Protocols received approval from the UT Health Science Center at San Antonio Institutional Animal Care and Use Committee. The protocols conformed to the Guide for the Care and Use of Laboratory Animals: Eighth Edition published by the National Research Council. The investigation conformed to the Guide for the Care and Use of Laboratory Animals, published by the US National Institute of Health (NIH Publication No. 85-23, revised 1996).
Langendorff preparation and heart perfusion
All materials used were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise stated. Mice were anesthetized by intraperitoneal injection of pentobarbital (60 mg/kg), and heparin (200 UI/kg) was used to prevent blood coagulation. Hearts were surgically removed and immediately arrested in cold (4°C) Krebs Henseleit bicarbonate buffer (KH) solution (mM): glucose 11, NaCl 118, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25 and CaCl2 3, pH 7.4. The aorta was rapidly cannulated and the heart was retrograde-perfused at a constant rate (3 ml/min) in the Langendorff mode using KH buffer bubbled with 95% O2/5% CO2 at 37°C. After 30 minutes of equilibration, global normothermic ischemia was induced by stopping KH buffer supply to the aorta for 30 minutes followed by 20 minutes of reperfusion for mitochondrial isolation. For myocardial infarct size imaging studies, hearts were reperfused for 120 minutes to allow for the development of I/R damage. We preferentially considered good hearts the ones that reached a minimum left ventricular developed pressure (LVDP) of 80 mmHg at the end of the basal perfusion (before ischemia). In the mouse model, this I/R protocol typically results in ~50% of infarct size. Sham hearts were not subjected to I/R but only were perfused for the same duration as the I/R protocol. To determine the role of the stimulation of different ETC complexes in myocardial infarction and cardiac functional recovery, hearts were perfused with the KH buffer supplemented with 3 mM of each of the substrates, glutamate/malate, succinate or sn-glycerol 3-phosphate.
Functional measurements
The heart function was recorded throughout the experiments using a catheter (1.4F SPR-671; Millar Instruments, Colorado Springs, CO) connected to a MAC LAB (Powerlab) acquisition system from ADInstruments (Sydney, Australia) as previously described in our articles [18,19]. The catheter was directly inserted into the left ventricle (LV) after a left atrial incision was made to expose the mitral annulus; the LV end-systolic pressure (LVSP), the LV end-diastolic pressure (LVEDP), and heart rate (HR) were directly obtained from LabChart5.5 (ADInstruments) software as described in our article [20]. The LV developed pressure (LVDP = LVSP - LVEDP) and Rate-Pressure Product (RPP = LVDP × HR) were calculated from the recordings.
Myocardial infarct size
The heart was removed from the Langendorff apparatus at the end of the reperfusion period and cut into four transverse slices parallel to the atrio-ventricular groove as previously described [18]. Slices were incubated for 7 minutes in 2% triphenyltetrazolium chloride (TTC) at 37°C followed by fixation with 4% paraformaldehyde. This staining differentiated the infarcted (pale) from viable (red) myocardial tissue. The slices were photographed using digital microscopic imaging (Moticam 5 MP camera). The area of necrosis was quantified by computerized planimetry with Adobe Photoshop CS6, and the total area of necrosis was calculated and expressed as a percentage of the total heart area.
Preparation of isolated mitochondria
Mitochondria were isolated from fresh, ischemic (20-minute reperfusion) and non-ischemic (sham) hearts at 4°C as previously described in [20]. Myocardial sections (approximately 0.15-0.22 g) were placed in isolation buffer A (mM): sucrose 70, mannitol 210, EDTA 1 and Tris-HCl 50, pH 7.4. The tissue was finely minced and homogenized in the same Buffer A (0.1 g of tissue/ml of buffer). The homogenate was centrifuged at 3,000 rpm for 3 minutes in a Galaxy 20R centrifuge (VWR, Radnor, PA); the supernatant was centrifuged at 13,000 rpm for 10 minutes. The mitochondrial pellet was resuspended in isolation Buffer B (mM): sucrose 150, KCl 50, KH2PO4 2, succinic acid 5 and Tris/HCl 20, pH 7.4). Additional isolation buffers C and D were prepared with 5 mM glutamate/malate and 5 mM sn-glyecerol 3-phosphate respectively, instead of 5 mM succinate as used in Buffer B. Protein concentration was estimated using the Bradford method assay kit (Bio-Rad, Hercules, CA).
Mitochondrial H2O2 measurement
Mitochondrial ROS generation was measured spectrofluorometrically (560 nm excitation and 590 nm emission) in 100 µg mitochondrial protein incubated in a solution containing: 20 mM Tris, 250 mM sucrose, 1 mM EGTA, 1 mM EDTA, and 0.15% bovine serum albumin adjusted to pH 7.4 at 30°C with continuous stirring. Superoxide generated in mitochondria has a short half-life and does not diffuse easily across the membranes, but the H2O2 derived from dismutation easily diffuses through the membranes [21,22]. As a result, generation of ROS can be monitored as a function of H2O2 levels. Hydrogen peroxide was measured with the H2O2-sensitive dye amplex red reagent (10 µM) (Thermo Fisher, Waltham, MA) in the presence of horseradish peroxidase (0.345 U/mL) and H2O2 levels were calculated from a calibration curve obtained from fluorescence emission intensity as a function of H2O2 concentration. The sodium salts of glutamate/malate (3 mM), succinate (3 mM) and sn-glycerol 3-phosphate (G3P) (3 mM) were used to activate complexes I and II, as well as the coenzyme Q pool, respectively. Complex I inhibitor, rotenone (2 µM) and complex III inhibitor, antimycin A from Streptomyces sp. (20 µM) were also used.
Ca2+-induced mitochondrial permeability transition
The installation of mitochondrial permeability transition pores (mPTP) was assessed following in vitro Ca2+ overload as previously described [23,24]. Free Ca2+ concentration outside the mitochondria was recorded using 0.1 µM calcium green-5N (Thermo Fisher) which binds reversibly to Ca2+, using excitation and emission wavelengths set at 500 and 530 nm, respectively. Isolated mitochondria (500 µg of protein) were suspended in 2 ml isolation Buffer B, C or D and pre-incubated for 90 seconds in a spectrofluorometer (Hitachi F-2710) set at 30°C. CaCl2 pulses (10 µmoles or 10 µL of 1 mM stock solution) were applied every 60 seconds to the cuvette leading to 20 nmol Ca2+ (per mg of mitochondrial protein). The Ca2+ pulses induce a peak of extra-mitochondrial Ca2+ concentration that returns to near-baseline levels as Ca2+ enters the mitochondrial matrix via uptake by the Ca2+ uniporter. With increasing calcium loading, the extra-mitochondrial Ca2+ concentration starts accumulating, thereby reflecting a lower capacity for mitochondrial Ca2+ uptake. This is followed by a sustained Ca2+ increase, indicating a massive release of the mitochondria Ca2+ by the mPTP opening. The Ca2+ retention capacity (CRC) was defined as the amount of Ca2+ required to trigger this massive Ca2+ release which is used here as an indicator of the mPTP sensitivity to Ca2+. CRC is expressed as nmol of CaCl2 per mg of mitochondrial protein.
Statistical analysis
The data shown in bar graphs are expressed as means with error bars that are the standard errors of the mean (± SEM) for a minimum of five independent hearts (n ≥ 5). The Student’s t-test and one-way ANOVA with post-hoc Dunnett’s, Tukey’s and Sidak’s corrections for multiple comparisons were appropriately used to assess the differences observed. Analysis was performed using Prism 6 (Graphpad Software, La Jolla, CA). A difference of p < 0.05 was considered to be statistically significant.
Results
Following I/R, stimulation of complex I increases ROS production but stimulation of complex II and CoQ decreases it
We first sought to ascertain the production of ROS by stimulated complexes in isolated mitochondria from both sham and ischemic myocardium using commonly used [25,27] de novo ETC substrates, glutamate/malate (complex I) and succinate (complex II), as well as CoQ substrate, G3P. In line with past studies, following ischemic injury, ROS production increases in complex I [28]. Activating complex I (glutamate/malate) resulted in an increase in ROS production in mitochondria isolated from I/R hearts, as compared to mitochondria isolated from non-ischemic hearts (Figure 2A). Surprisingly, treatment of I/R mitochondria with the complex II substrate, succinate or the Q-pool substrate, G3P resulted in reduced ROS production rate in I/R mitochondria when compared to non-ischemic tissue (Figure 2B, 2C). These results show opposite ROS production when complex I is activated, compared to when complex II and CoQ are activated in mitochondria isolated from ischemic hearts and in normal mitochondria. We also noted the marked difference in rates when each complex is stimulated, with succinate/complex II stimulation resulting in tremendously more ROS production than complex I and CoQ.
Figure 2.
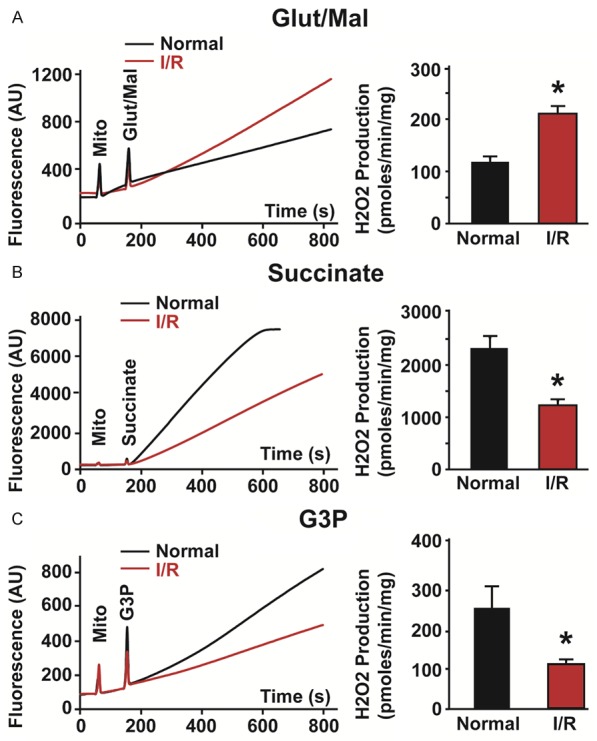
ROS production from activated ETC complexes measured as H2O2 release in normal mitochondria and mitochondria following ischemia/reperfusion (I/R) injury. Typical measurements of H2O2 release from normal (black trace) and I/R (red trace) mitochondria when complex I (3 mM glutamate/malate, Glut/Mal) (A), complex II (3 mM succinate) (B) and CoQ (3 mM sn-glycerol 3-phosphate, G3P) (C) were stimulated in the presence of amplex red and horseradish peroxidase (Left). Graphs showing how the rate of H2O2 production measured for a 60 second period after addition of substrate increased in I/R mitochondria after Glut/Mal was added but decreased after succinate and G3P were added (Right). Values are mean ± SEM; *p < 0.05 normal versus I/R mitochondria, n = 5-6/group.
Pro-apoptotic drugs increase ROS production when complex I and the Q pool are stimulated, but reduce ROS production when complex II is stimulated in healthy mitochondria
Next, we studied if pro-apoptotic drugs resulted in similar responses to complex stimulation. As shown in Figure 3, addition of complex I blocker, rotenone showed an increase in ROS generation by activated complex I and a modest increase on the activated Q cycle. The addition of antimycin A, which blocks the Q0 site of complex III led to dramatic increase in ROS generation when both complex I and CoQ were activated (Figure 3A, 3C). However, when complex II was stimulated, the addition of both inhibitors resulted in reduction of ROS generation. In fact, in the presence of rotenone, a complex I blocker and hence a blocker of reverse electron transfer (RET), ROS rate slowed down the most (succinate+rotenone, 1010 ± 139 versus succinate, 2274 ± 225 pmoles/min/mg, n = 5, p = 0.001) when compared to antimycin A (succinate+antimycin A, 1538 ± 88 pmoles/min/mg), suggesting that the electron transfer from the stimulated complex II to complex I (RET) is a larger source of ROS production than the forward transfer to complex III. Together, these findings indicate that pro-apoptosis ETC inhibitors induce opposite ROS production when complex I or II is stimulated. These results also show that these pro-apoptotic drugs that affect the ETC [29] have similar effects on ROS production to those observed following I/R injury (Figure 2), and therefore, can be used to approximate I/R effects when the different complexes are being studied.
Figure 3.
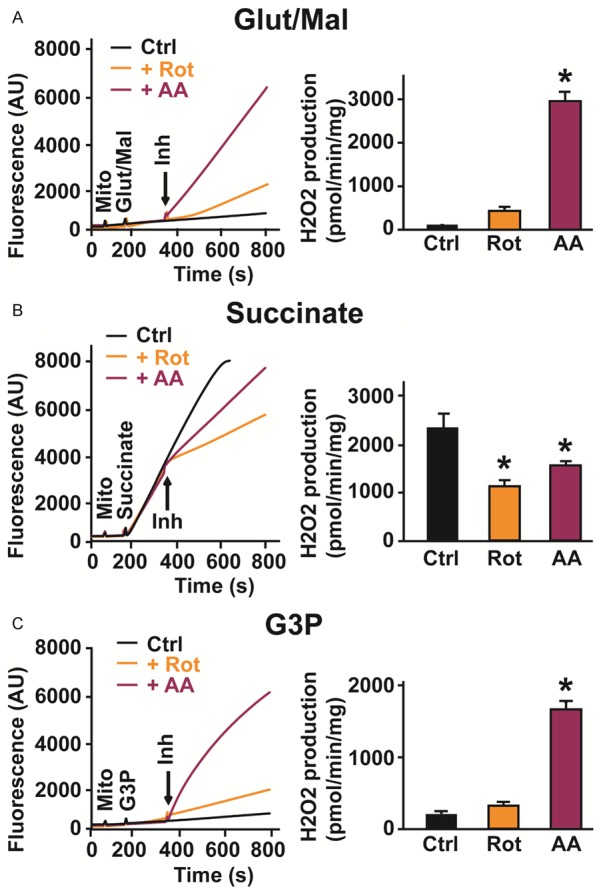
ROS production from activated ETC complexes measured as H2O2 release in normal mitochondria after insult with pro-apoptotic drugs. Typical measurements of H2O2 release in healthy mitochondria from control (black trace), with complex I inhibitor, rotenone (Rot, orange trace) and with complex III inhibitor, antimycin A (AA, purple trace) when complex I (3 mM glutamate/malate, Glut/Mal) (A), complex II (3 mM succinate) (B) and CoQ (3 mM sn-glycerol 3-phosphate, G3P) (C) were stimulated in the presence of amplex red and horseradish peroxidase (Left). Bar graphs showing changes in the rate of H2O2 production measured for a 60 second period after addition of each inhibitor (Right). Values are mean ± SEM; *p < 0.05 control versus+inhibitor, n = 5-6/group.
Activation of complex II and the Q-pool result in increased mitochondrial calcium retention capacity while activation of complex I shows a decrease in capacity
We then asked what impact the increases in ROS from each complex would have on the opening of the mPTP, a pivotal event known to cause cell death in I/R injury [30] and who’s inhibition was recently shown by our group to be necessary in cardioprotection against I/R injury via the G-protein coupled estrogen receptor 1 (GPER1) [31]. As the mPTP opening may be induced by calcium overload [32], we compared the sensitivity of mPTP opening as a function of calcium retention capacity in isolated fresh mitochondria treated with each complex’s substrate to stimulate ROS production. Figure 4 shows the time course of addition of free Ca2+ pulses in the solution surrounding mitochondria. The initial Ca2+ concentration in the buffer was set to Ca2+ contaminant that declined after adding mitochondria due to a global reduction of the solution’s optical transmittance as a result of the increase in turbidity after loading mitochondria. The following slower phase prior to the addition of the Ca2+ pulses reflects the rate of initial Ca2+ uptake by the mitochondria.
Figure 4.
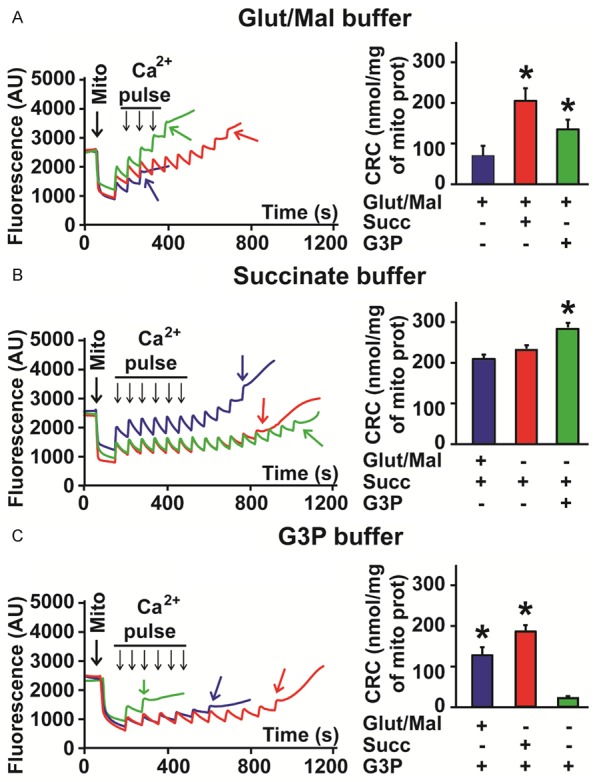
Mitochondrial Ca2+ induced opening of the mPTP for buffers of each substrate. A. Left: Example of recordings showing the calcium overload required to induce the mPTP opening (colored arrows) of isolated mitochondria re-suspended in a buffer containing 5 mM glutamate/malate (Glut/Mal) as substrate (blue trace) and after supplementation with 3 mM succinate (Succ, red trace) and 3 mM sn-glycerol 3-phosphate (G3P, green trace). Right: Bar graph showing improved calcium retention capacity (CRC) when Glut/Mal is supplemented with other substrates. Values are mean ± SEM; *p < 0.05 supplements versus control (Glut/Mal), n = 6/group. B. Left: Example of recordings showing the calcium overload required to induce the mPTP opening (colored arrows) of isolated mitochondria re-suspended in a buffer containing 5 mM succinate (Succ) as substrate (red trace) and after supplementation with 3 mM glutamate/malate (Glut/Mal, blue trace) and 3 mM sn-glycerol 3-phosphate (G3P, green trace). Right: Bar graph showing calcium retention capacity (CRC) when succinate is supplemented with other substrates. Values are mean ± SEM; *p < 0.05 supplements versus control (Succ), n = 6/group. C. Left: Example of recordings showing the calcium overload required to induce the mPTP opening (colored arrows) of isolated mitochondria re-suspended in a buffer containing 5 mM sn-glycerol 3-phosphate (G3P) as substrate (green trace) and after supplementation with 3 mM succinate (Succ, red trace) and 3 mM glutamate/malate (Glut/Mal, blue trace). Right: Bar graph showing improved calcium retention capacity (CRC) when G3P is supplemented with other substrates. Values are mean ± SEM; *p < 0.05 supplements versus control (G3P), n = 6/group.
In isolated mitochondria, using buffers of de novo ETC substrates, glutamate/malate and succinate, we found that the control mitochondrial calcium retention capacity (CRC) measurements were 73 ± 11 and 240 ± 5 nmol/mg of mitochondrial protein (n = 6), respectively. These results indicate that application of glutamate/malate, the substrate of complex I, to mitochondria increases the sensitivity of the mPTP opening to calcium overload when compared to succinate, the substrate of complex II. We also found that when glutamate/malate was used to prepare the isolation buffer, addition of either succinate (3 mM) or the ETC intermediate, G3P (3 mM) rescued the mitochondrial CRC, with succinate showing a greater recovery (glutamate/malate, 73 ± 11 versus glutamate/malate+succinate, 183 ± 24 nmol/mg of mitochondrial protein, n = 6, p = 0.002). This recovery was greater than that for G3P (glutamate/malate+succinate, 183 ± 24 versus glutamate/malate+G3P, 150 ± 20 nmol/mg of mitochondrial protein, n = 6) (Figure 4A). On the other hand, when succinate, the substrate of complex II, was used to prepare the isolation buffer, addition of glutamate/malate (3 mM), slightly decreased the CRC (succinate, 240 ± 5 versus succinate+glutamate/malate, 217 ± 11 nmol/mg of mitochondrial protein, n = 6, not significant), while the addition of G3P (3 mM) led to a statistically significant increase in CRC (succinate, 240 ± 5 nmol/mg of mitochondrial protein versus succinate+G3P, 293 ± 8 nmol/mg of mitochondrial protein, n = 6, p = 0.001) (Figure 4B). Use of the less traditional substrate, G3P as buffer (5 mM) resulted in poor CRC (G3P, 33 ± 4 nmol/mg of mitochondrial protein), which was only aided by addition of 3 mM of the more commonly used ETC substrates glutamate/malate (G3P+glutamate/malate, 120 ± 20 nmol/mg of mitochondrial protein, n = 6, p = 0.002 versus G3P control) and succinate (G3P+succinate, 193 ± 16 nmol/mg of mitochondrial protein, n = 6, p < 0.0001 versus G3P control) (Figure 4C).
Furthermore, we compared effects of ETC inhibitors rotenone and antimycin A when glutamate/malate, succinate or G3P were used as ROS substrate for mitochondrial CRC. As shown in Figure 5, addition of antimycin A resulted in mitochondrial depolarization when all three substrates were used. In fact, we found that addition of antimycin A to the cuvette containing pre-incubated mitochondria immediately led to the opening of the mPTP. Interestingly, when complex I (glutamate/malate buffer) was activated, addition of rotenone led to similar mitochondrial depolarization with immediate mPTP opening even before addition of Ca2+ pulses commenced (n = 6, p < 0.001). When CoQ (G3P buffer) was activated, addition of rotenone did not show any change in CRC (G3P+rotenone, 27 ± 4 versus G3P, 33 ± 4 nmol/mg of mitochondrial protein, n = 5-6, not significant). Interestingly, when complex II (succinate buffer) was stimulated, addition of rotenone remarkably enhanced CRC (succinate+rotenone, 267 ± 7 versus succinate, 240 ± 5 nmol/mg of mitochondrial protein, n = 6, p = 0.02) suggesting a delay of the mPTP opening. Together, these results indicate that the ROS production in complex I promotes the mPTP opening, while ROS produced in complex III induces delay of the mPTP opening and presumably may lead to cardioprotection.
Figure 5.
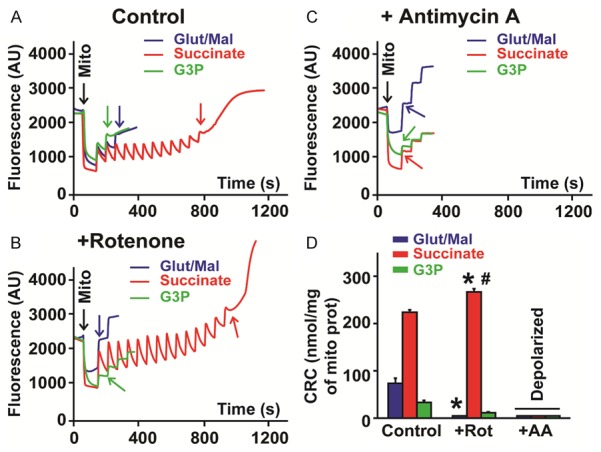
Mitochondrial Ca2+ induced opening of the mPTP for buffers of each substrate with pro-apoptotic inhibitors. A. Typical recording showing the calcium overload required to induce the mPTP opening (colored arrows) of isolated mitochondria re-suspended in a buffer containing 5 mM glutamate/malate (Glut/Mal, blue trace), 5 mM succinate (Succ, red trace) and 5 mM sn-glycerol 3-phosphate (G3P, green trace) as substrate (control). B. Typical recording showing the calcium overload required to induce the mPTP opening (colored arrows) of isolated mitochondria re-suspended in a buffer containing 5 mM glutamate/malate (Glut/Mal, blue trace), 5 mM succinate (Succ, red trace) and 5 mM sn-glycerol 3-phosphate (G3P, green trace) and treated with rotenone (Rot, 2 μM). C. Typical recording showing the calcium overload required to induce the mPTP opening (colored arrows) of isolated mitochondria re-suspended in a buffer containing 5 mM glutamate/malate (Glut/Mal, blue trace), 5 mM succinate (Succ, red trace) and 5 mM sn-glycerol 3-phosphate (G3P, green trace) and treated with antimycin A (AA, 20 μM). D. Bar graphs showing mitochondrial calcium retention capacity (CRC) measured of mitochondria re-suspended in a buffer containing each substrate and supplemented with either rotenone or antimycin A. Values are mean ± SEM; *p < 0.05 inhibitor treated versus respective control (same substrate), #p < 0.05 Succ+Rot versus other substrates+Rot, n = 6/group.
Activation of CoQ in contrast to complexes I and II improved cardiac functional recovery and reduced myocardial infarct size following I/R
Finally, we asked whether resultant differences in ROS produced by each complex could be observed in an ex vivo animal model and possibly highlight clinically relevant effects. Using Langendorff retrograde perfusion, we studied myocardial infarct size and cardiac functional recovery following I/R stress, with reduced infarct size and higher rate-pressure product (RPP) indicating cardioprotection. We compared these parameters in isolated hearts perfused with KH buffer that was enriched with each of the three substrates to stimulate ROS production. With regards to the heart regaining cardiac function following I/R, the RPP values obtained at 120 minutes of reperfusion, as a percent of RPP before ischemia, were in the order: G3P (72 ± 9%), succinate (43 ± 7%), and glutamate/malate (36 ± 1%); compared to control (36 ± 4%) (n = 5-7) (Figure 6A). As shown in Figure 6B, 6C, the resultant RPP at the end of reperfusion was mostly driven by changes in heart rate, as the LVDP readings were fairly similar across all conditions. Incitement of ROS production at complex III by activating the Q-pool with G3P resulted in smaller infarct sizes (G3P, 42 ± 2%) as compared to the control (56 ± 4%, n = 5-7, p = 0.01). However, activating complex I with glutamate/malate increased the infarct size from that exhibited by controls (glutamate/malate, 59 ± 4% versus control, 56 ± 4%, n = 5-7, not significant) (Figure 7A, 7B). Comparing G3P (complex III ROS) to glutamate/malate (complex I ROS) showed statistically significant benefits of G3P (Figures 6, 7; Infarct size: p = 0.001 and RPP: p = 0.04; n = 5-7). These results suggest that complex III ROS induce cardioprotective effects against ischemia reperfusion injury, while complex I ROS appear to promote myocardial injury.
Figure 6.
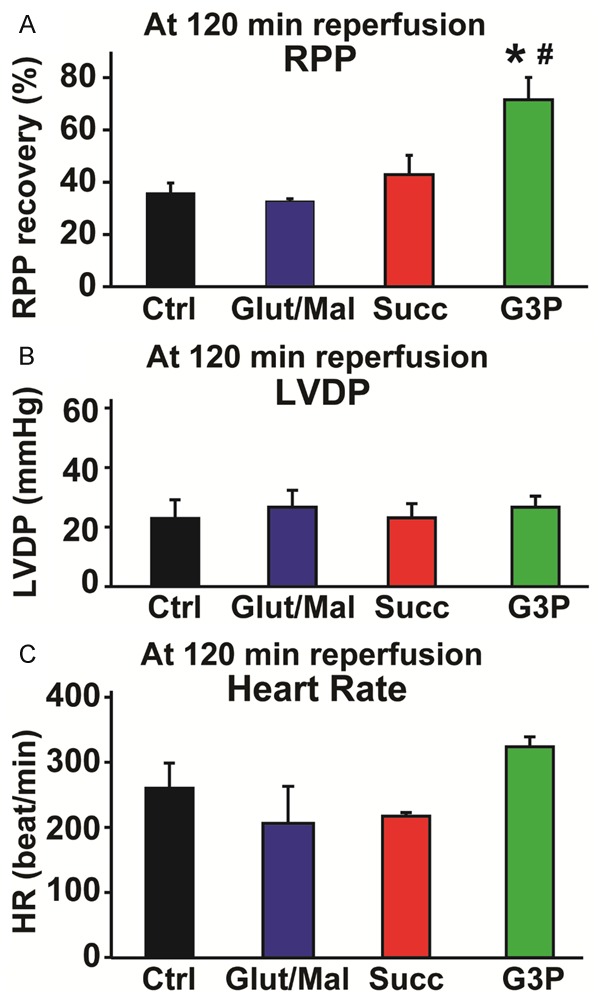
Measure of cardiac functional recovery in control and after addition of substrates after I/R. Cardiac function was recorded throughout the experiments in isolated perfused hearts treated with KH buffer for control and after addition of different substrates (3 mM). A. Bar graph showing an improvement of the cardiac functional recovery (Rate-Pressure Product, RPP) at 120 min reperfusion (end of reperfusion) in sn-glycerol 3-phosphate (G3P) group compared to control (Ctrl) and glutamate/malate (Glut/Mal) groups. B. Bar graph indicating that the left ventricular developed pressure (LVDP) at 120 min reperfusion did not change in control or the supplements of each substrate. C. Bar graph showing an improvement of the heart rate (HR) at 120 min reperfusion (end of reperfusion) in sn-glycerol 3-phosphate (G3P) group compared to control and glutamate/malate (Glut/Mal) groups. Values are mean ± SEM; *p < 0.05 substrate versus control; #p < 0.05 G3P versus Glut/Mal, n = 5-6/group.
Figure 7.
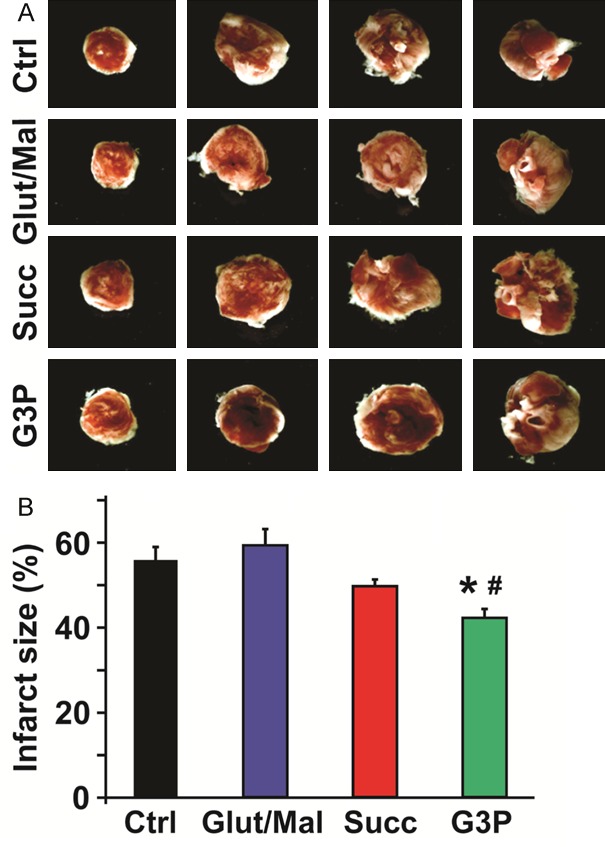
Measure of myocardial infarct size in control and after supplement of substrates after I/R. Myocardial infarct size was measured at the end of 120 min reperfusion in isolated perfused hearts treated with KH buffer for control and after addition of different substrates. A. Representative images from 4 slices of the same heart in each condition. White areas correspond to the infarcted zone. B. Bar graphs showing reduction of myocardial infarct size in sn-glycerol 3-phosphate (G3P) group compared to control (Ctrl) and glutamate/malate (Glut/Mal) groups. Values are mean ± SEM of % infarct size; *p < 0.05 substrate treated versus control; #p < 0.05 G3P versus Glut/Mal, n = 6-7/group.
Discussion
In this study, we demonstrated that not only is the quantity of ROS important, but the quantity and mitochondrial ETC source of that ROS in mitochondria is pertinent to its impact as either cardio-protective or cardio-deleterious in a heart I/R model. Specifically, we showed that ROS from complex I results in a reduction of mitochondria’s ability to sequester Ca2+ ions in healthy mitochondria, greater myocardial infarct size, and less functional recovery in the whole heart following I/R injury. Conversely, we revealed how ROS from complex III leads to an improvement in mitochondrial Ca2+ buffering, as highlighted by the delay in the mPTP opening, and reduces the overall heart infarct size while improving recovery after I/R. Our results show that stimulating complex III either from complex II or Q-pool substrates leads to ROS production that is cardio-beneficial, as opposed to that from stimulating complex I, which is detrimental to both mitochondria and the stressed heart as a whole.
ROS production at complexes I and III
The increase in production of mitochondrial ROS has been demonstrated using cell labeling methods as being a relevant process in many pathological conditions like heart attack, diabetes and cancer [33,35], and it also participates in necrosis and apoptosis associated with cardiac I/R injury (reviewed in detail in [36,37]). Complex I is a major source of electron leakage which results in ROS production in the ETC. Previous studies have debated the exact site of ROS generation on complex I [36,38], but the ubiquinone-binding domain appears to be main site during succinate-driven reverse electron transfer (RET) from complex II [39] and this also happens to be the same site at which rotenone blocks transfer of electrons to CoQ [40]. Following ischemic damage, the iron sulfur clusters proximal to this ubiquinone binding site are thought to be the locations for ROS production which result in the increase in ROS that we also observed [38] (Figure 2A). ROS production from complex I is more dependent on ΔpH than ΔΨ (membrane potential) [39,41] and thus less reliant on mitochondrial energization. Thus, I/R injury and rotenone increase ROS generation from the same region on complex I when this complex is activated by blocking electron escape, as we observed (Figures 2A and 3A).
Complex II influences ROS production at complex I (via RET) [28,29] and complex III [36] and although some evidence has suggested complex II to be a source of ROS under certain conditions [42], it is only complexes I and III that are globally accepted as main generators. Following I/R insult or application of rotenone to complex I, we observed a decrease in succinate-driven ROS (Figures 2B and 3B) which are in line with a loss of RET to complex I. For complex II, our findings further highlight its dependence on its neighboring complexes for ROS production. From this, it may be inferred that following I/R injury, activated complex II no longer influences ROS production at complexes I or III, hence the observed decrease (Figure 2); while rotenone and antimycin A also lead to diminished ROS formation as complex II’s access to the adjacent enzymatic units is diminished (Figure 3).
Complex III has two known sites of ROS production, with one releasing ROS into the intermembrane space and the other into the matrix [43]. Complex III works by transferring electrons from ubiquinol to ferricytochrome c by utilizing hemes b L and b H, and the Rieske iron-sulfur protein [36]. The contribution of complex III to ROS production in normal physiology remains uncertain with some groups suggesting it to be smaller [44,45] while others postulate it to be larger [38,46,47] than complex I involvement. However, there is consensus on complex III being a major site of ROS production after I/R [36,38]. Ischemic damage reduced ROS production from G3P stimulation (Figure 2C) contrary to the expectation of an increase. I/R disrupts complex III function [48] and G3P/CoQ stimulation probably would have been impaired from affecting ROS from this complex. Accordingly, downstream application of antimycin A, which blocks electron transfer from the Q0 site of complex III increased ROS production when complex I and complex III were stimulated (Figure 3). And despite complex III ROS relying more on ΔΨ than ΔpH [49], application of the transfer blocker, antimycin A results in ROS production. As succinate influences ROS at both complexes I and III, we opted to include G3P in order to gain further insight into ROS production from complex III. G3P, also known as L-α-glycerophosphate is thought to be an inadequate substrate by itself for studying H2O2 production in heart mitochondria [50] but when coupled with glutamate or succinate can augment ROS production at complex III. Mracek et al. have shown that glycerophosphate dehydrogenase (G3Pdh in Figure 1) is a source of ROS particularly in brown adipose tissue but that in heart mitochondria, stimulating the dehydrogenase with G3P results in the bulk of ROS being made at complex III [51]; hence, we opted to use it in our studies to influence ROS generation at complex III.
Opposing effects of ROS from complex I versus complex III
Kalogeris et al. summed up current opinion that the “double-edged sword” impact of ROS prompted by I/R may be traced back to i) the type of ROS produced ii) the amount and time when the ROS is produced and iii) the cellular and subcellular loci of creation [52]. First, reactive species known to cause damage to cells and tissue include the superoxide anion radical (O2 •-), hydroxyl radical (•OH), hydrogen peroxide (H2O2), and peroxynitrite (ONOO-) [53]. In cardiovascular tissue, the most important and abundant form of ROS is O2 •- [54] and within this tissue, it is known that mitochondria, and specifically the respiratory chain are the dominant sources of O2 •- [2,7,36]. With regards to the heart, I/R damage results in increased O2 •- production by both subsarcolemmal and interfibrillary mitochondria [55,56], which were the focus of our study.
Second, the amount of ROS and when it is produced are critical to the effect it has. The amount of ROS following I/R overwhelms the hitherto, tightly-regulating antioxidant machinery of the cell and the ROS can thus attack DNA, proteins and subcellular structures [52]. In our experiments, we used the ETC substrates glutamate/malate, succinate and G3P, at non-physiological concentrations of 3 mM to instigate this increase in ROS. As evidenced by the increase in ROS in pre- and postconditioning, the time when ROS increase commences is also vital [11-14,17]. During ischemia, cells are starved of both nutrients and oxygen and when reperfusion begins, it is the accumulation of Ca2+ in the matrix, depletion of adenine nucleotide, increased inorganic phosphate concentration, and oxidative stress that leads to the opening of mPTP [32] whose effects include depolarizing mitochondria, uncoupling the ETC, allowing for efflux of Ca2+ and intermembrane proteins such as the pro-apoptotic cytochrome c, and causing swelling of the mitochondrial matrix [57].
Thirdly, the cellular/subcellular location of ROS generation is pertinent to the outcomes of I/R. This idea of compartmentalization for the control of ROS is both logical and supported by a large body of evidence (reviewed in [58-60]). In normal physiology, the cell highly regulates the amount of ROS, particularly through superoxide dismutase (SOD) which degrades ROS to H2O2, which can be further degraded to water and molecular oxygen. The mitochondrial matrix contains manganese-SOD (Mn-SOD or SOD-2) while the cytoplasm and the intermembrane space in mitochondria contain copper/zinc-SOD (Cu/Zn-SOD or SOD-1) [52,61]. Insults like I/R ignite overwhelming quantities of ROS that overpower these safeguards and leads to senescence, autophagy and apoptotic pathways associated with cardiovascular diseases [1].
In the present study, we observed that not only is it the subcellular compartment key to ROS impact, but the mitochondrial ETC complex generating that ROS is crucial as well. By studying isolated mitochondria’s ability to sequester calcium before opening the mPTP, we found that the impact of ROS produced by activated complexes I and III on the opening of the mPTP is contradictory (Figure 4). The mPTP opening is known to be associated with an increase in ROS and its opening is a trademark event of mitophagy following I/R injury [20]. Our data indicate that the treatment of fresh and healthy mitochondria with succinate buffer (5 mM) (for complex II) delays the opening of the mPTP as the calcium retention capacity (CRC) increases, while the stimulation of complex I (glutamate/malate buffer, 5 mM) actually reduces mitochondrial CRC (Figure 4A, 4B). For each of these substrates, addition of the other substrate either aids or impedes the CRC effect. As shown in Figure 4, when glutamate/malate buffer (5 mM) is supplemented with either succinate or G3P (3 mM), this serves to improve the mitochondria’s CRC; on the contrary, addition of glutamate/malate (3 mM) to succinate buffer (5 mM) solution slightly reduces CRC and, as can be deduced from our hypothesis, addition of 3 mM G3P (for complex III) to succinate buffer (5 mM) increases CRC. As discussed earlier, G3P by itself is a poor cardiac ROS substrate, and this was also shown by its low CRC values when it was used as substrate buffer (5 mM) (Figure 4C); however, it was improved significantly in the presence of the natural substrates, glutamate/malate (3 mM) and succinate (3 mM).
Following our hypothesis that the ETC source of ROS is critical in cardioprotection, we studied the CRC effects of increased ROS production when complexes I and III were sequentially blocked by use of rotenone and antimycin A as blockers. Rotenone inhibits oxidation of the iron-sulfur clusters in complex I while also blocking RET from complex II [36,62] (Figure 1). Antimycin A blocks the Qi site of complex III resulting in increased ROS production at the complex’s Q0 site, with this ROS being expelled into the intermembrane space from the Q0 site [43,51,63,64]. Application of antimycin A, a drug with no biological equivalent, to mitochondria results in the collapse of membrane potential, opening of the mPTP [62,65] and hence, the inability to absorb Ca2+ as we also found (Figure 5). Furthermore, addition of rotenone to activated complex I, which increases ROS at this site also depleted mitochondrial CRC when using glutamate/malate; but rotenone’s blocking of RET improved CRC when complex II was activated probably due to more “good” ROS coming from complex III. We found that limiting ROS from resting state complex I by applying rotenone and stimulating complex II using succinate, served to enhance the cardio-positive effects of the ROS from complex III by increasing CRC (Figure 5D). The fact that the stimulation of the complexes II and III (while inhibiting influence of complex I) increases the mitochondrial CRC indicate that ROS produced under these conditions are cardioprotective since it is well established that the inhibition of mPTP opening protects the heart during pre- and post-conditioning, and controlled reperfusion [37].
The differential effects of ROS from complexes I and III in isolated mitochondria suggested opposite roles and we sought to determine if these roles extended to intact myocardium. For this, we compared cardiac functional recovery as well as myocardial infarct size in isolated hearts perfused with KH buffer versus KH with supplementation of 3 mM of each substrate. Although our previous experiments had been in isolated mitochondria with substrates easily accessing the inner membrane of mitochondria, all three substrates are found in the cytosol and can be transported across cell membranes from exogenous sources [66,69].
The rate-pressure product (RPP), or cardiovascular product, used in hemodynamic stress studies in cardiac physiology, is used to predict myocardial oxygen consumption, or quite simply, myocardial workload [70]. As the product of heart rate and systolic blood pressure, RPP can be used to monitor the functional impact of I/R injury on the heart [18,71,72] and in our studies, the RPP was used to determine the level of functional recovery following I/R. In this study, we found that G3P showed the greatest recovery with glutamate/malate showing the least, further evidence for the opposing complexes’ ROS. The left ventricular developed (LVDP) was similar in all groups and the improvement of the RPP with G3P infusion was mostly due to the increase in the heart rate (HR) (Figure 6A-C), suggesting that G3P may also directly modulate pace maker activity.
Myocardial infarct size was measured using histological staining and planimetry and the results showed significantly different outcomes following I/R (Figure 7). Following the trends, we observed in mitochondrial CRC, applying the substrates to whole tissue further cemented our proposition that complex III ROS are cardio-protective and complex I ROS are cardio-deleterious: G3P resulted in reduced infarct size compared to glutamate/malate. As KH buffer already contains a mitochondrial stimulant and Krebs cycle driver in the form of glucose, we did not expect dramatically pronounced differences between the samples. This was because glucose was stimulating complexes I and II, as well as supporting an active tricarboxylic acid cycle, which is necessary for the heart to survive ex vivo. Nevertheless, we did observe significant differences between glutamate/malate and G3P treatment, enough to support the notion of opposed impacts of ROS. With regards to succinate, we also did observe smaller infarct sizes compared to glutamate/malate but not to the same extent as those for G3P (Figure 7). As succinate/complex II feeds electrons to both complexes I and III, this intermediate result was not surprising. Furthermore, Chouchani and colleagues have shown that in vivo, the accumulation of succinate during ischemia is the main driver for ROS generation by complex I via RET during reperfusion and that a reduction of this RET is adequate enough to mitigate I/R injury in murine models [73].
Impact on cardioprotection and antioxidant therapy
Based on our results, a fundamental question that arises is how exactly cardiomyocytes, or specifically, mitochondria recognize or delineate between “good” versus “bad” ROS in times of stress? As ROS in normal conditions and controlled quantities features as a signaling conduit [10], it may be possible that under such circumstances neither complex I nor complex III ROS are good or bad. In pathophysiological conditions resulting from I/R or exogenous inhibitors, we have shown that ROS from complexes I and III are not the same, but the mechanism or even the sites of action of these ROS leading to cardiac protection or damage are the subject of future studies. Drose et al. have suggested a mechanistic model to explain ‘signaling ROS’ by studying diazoxide effects on mitochondrial ROS generation [63]. They proposed that diazoxide, a complex II blocker, could stimulate the production of ‘signalling ROS’ at the Q0 site of complex III by inhibition of the succinate-ubiquinone oxidoreductase (complex II) thereby partially oxidizing the Q-pool. They hypothesized that following stress, ‘signaling ROS’ activates mitochondrial signaling pathways that during reoxygenation eventually reduce the production of ‘deleterious’ ROS by NADH-ubiquinone oxidoreductase (complex I) via RET and that in addition, diazoxide can directly reduce oxidative damage evoked by RET at complex I by inhibition of complex II. However, studies in H9c2 myoblasts by other investigators have suggested complex I being a source of signaling ROS for muscle differentiation [74]. In our study, our data agree with the Drose group in suggesting that the source of this ‘signaling’ ROS in heart mitochondria to be complex III, while also highlighting the deleterious effects of ROS from complex I. We extend this premise into the impact that both ETC sources have on cardiac tissue recovery following ischemic insult. Despite this model offering some insights into ROS classification, much is still needed to fully elucidate how cells respond to ROS from different ETC sites. Multiple factors need consideration when defining such pathways including the varying needs and responses of mitochondria in different organs [25,50] and how, for example, in the brain, glutamate is both an excitatory neurotransmitter and a cause of neurotoxicity [75]. Understanding these mechanisms will be vital to designing targeted therapeutic agents for cardioprotection as current models of coenzyme Q10 treatment [6] or applying indiscriminant antioxidants as one-size fits all panaceas have proven to be somewhat fruitless as both good and bad ROS are destroyed [3].
In conclusion, we demonstrated the opposite impact of ROS generated by stimulation of complexes I, II and III after ischemia-reperfusion and also application of pro-apoptotic drugs in fresh mitochondria. We propose an extension to the concept that the quantity of ROS is important by showing that the mitochondrial ETC source complex of that ROS is also pivotal. We went on to demonstrate the positive impact of the ROS from mitochondrial complex III in cardioprotection via the inhibition of the mPTP opening, and following I/R, reduction of myocardial infarct size and improvements in cardiac functional recovery, while showing the cardio-deleterious effects of ROS from complex I.
Acknowledgements
Supported by Bopassa’s AHA fellowship 09POST2190008, Voelcker fund (Bopassa).
Disclosure of conflict of interest
None.
References
- 1.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res. 2015;116:531–549. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinhubl SR. Why have antioxidants failed in clinical trials? Am J Cardiol. 2008;101:14D–19D. doi: 10.1016/j.amjcard.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005;25:29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- 4.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. 2007;297:842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 5.Miller ER 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 6.Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion. 2007;7(Suppl):S154–167. doi: 10.1016/j.mito.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Chandel NS, Tuveson DA. The promise and perils of antioxidants for cancer patients. N Engl J Med. 2014;371:177–178. doi: 10.1056/NEJMcibr1405701. [DOI] [PubMed] [Google Scholar]
- 8.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 9.Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 10.Bartosz G. Reactive oxygen species: destroyers or messengers? Biochem Pharmacol. 2009;77:1303–1315. doi: 10.1016/j.bcp.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Das DK, Maulik N, Sato M, Ray PS. Reactive oxygen species function as second messenger during ischemic preconditioning of heart. Mol Cell Biochem. 1999;196:59–67. [PubMed] [Google Scholar]
- 12.Maslov LN, Naryzhnaya NV, Podoksenov YK, Sementsov AS, Gorbunov AS. [Signaling Mechanism of Cardioprotective Effect of Reactive Oxygen Species] . Ross Fiziol Zh Im I M Sechenova. 2015;101:377–385. [PubMed] [Google Scholar]
- 13.Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta. 2007;1767:1007–1031. doi: 10.1016/j.bbabio.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou T, Chuang CC, Zuo L. Molecular Characterization of Reactive Oxygen Species in Myocardial Ischemia-Reperfusion Injury. Biomed Res Int. 2015;2015:864946. doi: 10.1155/2015/864946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun HY, Wang NP, Kerendi F, Halkos M, Kin H, Guyton RA, Vinten-Johansen J, Zhao ZQ. Hypoxic postconditioning reduces cardiomyocyte loss by inhibiting ROS generation and intracellular Ca2+ overload. Am J Physiol Heart Circ Physiol. 2005;288:H1900–1908. doi: 10.1152/ajpheart.01244.2003. [DOI] [PubMed] [Google Scholar]
- 16.Kin H, Zhao ZQ, Sun HY, Wang NP, Corvera JS, Halkos ME, Kerendi F, Guyton RA, Vinten-Johansen J. Postconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res. 2004;62:74–85. doi: 10.1016/j.cardiores.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 17.Penna C, Rastaldo R, Mancardi D, Raimondo S, Cappello S, Gattullo D, Losano G, Pagliaro P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res Cardiol. 2006;101:180–189. doi: 10.1007/s00395-006-0584-5. [DOI] [PubMed] [Google Scholar]
- 18.Feng Y, Bopassa JC. Oxygen surrounding the heart during ischemic conservation determines the myocardial injury during reperfusion. Am J Cardiovasc Dis. 2015;5:127–139. [PMC free article] [PubMed] [Google Scholar]
- 19.Rahman S, Li J, Bopassa JC, Umar S, Iorga A, Partownavid P, Eghbali M. Phosphorylation of GSK-3beta mediates intralipid-induced cardioprotection against ischemia/reperfusion injury. Anesthesiology. 2011;115:242–253. doi: 10.1097/ALN.0b013e318223b8b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bopassa JC, Eghbali M, Toro L, Stefani E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2010;298:H16–23. doi: 10.1152/ajpheart.00588.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bienert GP, Chaumont F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim Biophys Acta. 2014;1840:1596–1604. doi: 10.1016/j.bbagen.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 22.Loschen G, Azzi A, Richter C, Flohe L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974;42:68–72. doi: 10.1016/0014-5793(74)80281-4. [DOI] [PubMed] [Google Scholar]
- 23.Bopassa JC, Ferrera R, Gateau-Roesch O, Couture-Lepetit E, Ovize M. PI 3-kinase regulates the mitochondrial transition pore in controlled reperfusion and postconditioning. Cardiovasc Res. 2006;69:178–185. doi: 10.1016/j.cardiores.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 24.Ferrera R, Bopassa JC, Angoulvant D, Ovize M. Post-conditioning protects from cardioplegia and cold ischemia via inhibition of mitochondrial permeability transition pore. J Heart Lung Transplant. 2007;26:604–609. doi: 10.1016/j.healun.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 25.Vial G, Dubouchaud H, Couturier K, Cottet-Rousselle C, Taleux N, Athias A, Galinier A, Casteilla L, Leverve XM. Effects of a high-fat diet on energy metabolism and ROS production in rat liver. J Hepatol. 2011;54:348–356. doi: 10.1016/j.jhep.2010.06.044. [DOI] [PubMed] [Google Scholar]
- 26.Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A, Van Remmen H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1159–1168. doi: 10.1152/ajpregu.00767.2006. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 28.Tahara EB, Navarete FD, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med. 2009;46:1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 29.Wolvetang EJ, Johnson KL, Krauer K, Ralph SJ, Linnane AW. Mitochondrial respiratory chain inhibitors induce apoptosis. FEBS Lett. 1994;339:40–44. doi: 10.1016/0014-5793(94)80380-3. [DOI] [PubMed] [Google Scholar]
- 30.Argaud L, Gateau-Roesch O, Muntean D, Chalabreysse L, Loufouat J, Robert D, Ovize M. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J Mol Cell Cardiol. 2005;38:367–374. doi: 10.1016/j.yjmcc.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Kabir ME, Singh H, Lu R, Olde B, Leeb-Lundberg LM, Bopassa JC. G Protein-Coupled Estrogen Receptor 1 Mediates Acute Estrogen-Induced Cardioprotection via MEK/ERK/GSK-3beta Pathway after Ischemia/Reperfusion. PLoS One. 2015;10:e0135988. doi: 10.1371/journal.pone.0135988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemasters JJ. The mitochondrial permeability transition and the calcium, oxygen and pH paradoxes: one paradox after another. Cardiovasc Res. 1999;44:470–473. doi: 10.1016/s0008-6363(99)00368-5. [DOI] [PubMed] [Google Scholar]
- 33.Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res. 2009;81:449–456. doi: 10.1093/cvr/cvn280. [DOI] [PubMed] [Google Scholar]
- 34.Rolo AP, Palmeira CM. Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicol Appl Pharmacol. 2006;212:167–178. doi: 10.1016/j.taap.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114:524–537. doi: 10.1161/CIRCRESAHA.114.300559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bopassa JC. Protection of the ischemic myocardium during the reperfusion: between hope and reality. Am J Cardiovasc Dis. 2012;2:223–236. [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294:C460–466. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- 39.Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chance B, Williams GR, Hollunger G. Inhibition of electron and energy transfer in mitochondria. I. Effects of Amytal, thiopental, rotenone, progesterone, and methylene glycol. J Biol Chem. 1963;238:418–431. [PubMed] [Google Scholar]
- 41.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH: ubiquinone oxidoreductase (complex I) J Biol Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 42.Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 2012;287:27255–27264. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 44.Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. doi: 10.1042/bse0470053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H2023–2031. doi: 10.1152/ajpheart.01283.2006. [DOI] [PubMed] [Google Scholar]
- 46.Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem. 2003;278:5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- 47.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 48.Lee HL, Chen CL, Yeh ST, Zweier JL, Chen YR. Biphasic modulation of the mitochondrial electron transport chain in myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2012;302:H1410–1422. doi: 10.1152/ajpheart.00731.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rottenberg H, Covian R, Trumpower BL. Membrane potential greatly enhances superoxide generation by the cytochrome bc1 complex reconstituted into phospholipid vesicles. J Biol Chem. 2009;284:19203–19210. doi: 10.1074/jbc.M109.017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kwong LK, Sohal RS. Substrate and site specificity of hydrogen peroxide generation in mouse mitochondria. Arch Biochem Biophys. 1998;350:118–126. doi: 10.1006/abbi.1997.0489. [DOI] [PubMed] [Google Scholar]
- 51.Mracek T, Pecinova A, Vrbacky M, Drahota Z, Houstek J. High efficiency of ROS production by glycerophosphate dehydrogenase in mammalian mitochondria. Arch Biochem Biophys. 2009;481:30–36. doi: 10.1016/j.abb.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 52.Kalogeris T, Bao Y, Korthuis RJ. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014;2:702–714. doi: 10.1016/j.redox.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Victor VM, Rocha M. Targeting antioxidants to mitochondria: a potential new therapeutic strategy for cardiovascular diseases. Curr Pharm Des. 2007;13:845–863. doi: 10.2174/138161207780363077. [DOI] [PubMed] [Google Scholar]
- 54.Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension. 2003;42:1075–1081. doi: 10.1161/01.HYP.0000100443.09293.4F. [DOI] [PubMed] [Google Scholar]
- 55.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther. 2006;319:1405–1412. doi: 10.1124/jpet.106.110262. [DOI] [PubMed] [Google Scholar]
- 56.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr. 1996;28:131–138. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- 58.Jones DP, Go YM. Redox compartmentalization and cellular stress. Diabetes Obes Metab. 2010;12(Suppl 2):116–125. doi: 10.1111/j.1463-1326.2010.01266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cardoso AR, Chausse B, da Cunha FM, Luevano-Martinez LA, Marazzi TB, Pessoa PS, Queliconi BB, Kowaltowski AJ. Mitochondrial compartmentalization of redox processes. Free Radic Biol Med. 2012;52:2201–2208. doi: 10.1016/j.freeradbiomed.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 60.Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu Rev Pharmacol Toxicol. 2006;46:215–234. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- 61.Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 62.Drose S. Differential effects of complex II on mitochondrial ROS production and their relation to cardioprotective pre- and postconditioning. Biochim Biophys Acta. 2013;1827:578–587. doi: 10.1016/j.bbabio.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 63.Drose S, Hanley PJ, Brandt U. Ambivalent effects of diazoxide on mitochondrial ROS production at respiratory chain complexes I and III. Biochim Biophys Acta. 2009;1790:558–565. doi: 10.1016/j.bbagen.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 64.Drose S, Brandt U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J Biol Chem. 2008;283:21649–21654. doi: 10.1074/jbc.M803236200. [DOI] [PubMed] [Google Scholar]
- 65.Bernardi P, Broekemeier KM, Pfeiffer DR. Recent progress on regulation of the mitochondrial permeability transition pore; a cyclosporin-sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr. 1994;26:509–517. doi: 10.1007/BF00762735. [DOI] [PubMed] [Google Scholar]
- 66.Inoue K, Zhuang L, Ganapathy V. Human Na+ -coupled citrate transporter: primary structure, genomic organization, and transport function. Biochem Biophys Res Commun. 2002;299:465–471. doi: 10.1016/s0006-291x(02)02669-4. [DOI] [PubMed] [Google Scholar]
- 67.Nakayama T, Kawakami H, Tanaka K, Nakamura S. Expression of three glutamate transporter subtype mRNAs in human brain regions and peripheral tissues. Brain Res Mol Brain Res. 1996;36:189–192. doi: 10.1016/0169-328x(95)00297-6. [DOI] [PubMed] [Google Scholar]
- 68.Hara-Chikuma M, Verkman AS. Physiological roles of glycerol-transporting aquaporins: the aquaglyceroporins. Cell Mol Life Sci. 2006;63:1386–1392. doi: 10.1007/s00018-006-6028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bartoloni L, Wattenhofer M, Kudoh J, Berry A, Shibuya K, Kawasaki K, Wang J, Asakawa S, Talior I, Bonne-Tamir B, Rossier C, Michaud J, McCabe ER, Minoshima S, Shimizu N, Scott HS, Antonarakis SE. Cloning and characterization of a putative human glycerol 3-phosphate permease gene (SLC37A1 or G3PP) on 21q22.3: mutation analysis in two candidate phenotypes, DFNB10 and a glycerol kinase deficiency. Genomics. 2000;70:190–200. doi: 10.1006/geno.2000.6395. [DOI] [PubMed] [Google Scholar]
- 70.Fletcher GF, Ades PA, Kligfield P, Arena R, Balady GJ, Bittner VA, Coke LA, Fleg JL, Forman DE, Gerber TC, Gulati M, Madan K, Rhodes J, Thompson PD, Williams MA American Heart Association Exercise, Cardiac Rehabilitation, and Prevention Committee of the Council on Clinical Cardiology, Council on Nutrition, Physical Activity and Metabolism, Council on Cardiovascular and Stroke Nursing, and Council on Epidemiology and Prevention. Exercise standards for testing and training: a scientific statement from the American Heart Association. Circulation. 2013;128:873–934. doi: 10.1161/CIR.0b013e31829b5b44. [DOI] [PubMed] [Google Scholar]
- 71.Pasdois P, Parker JE, Griffiths EJ, Halestrap AP. Hexokinase II and reperfusion injury: TAT-HK2 peptide impairs vascular function in Langendorff-perfused rat hearts. Circ Res. 2013;112:e3–7. doi: 10.1161/CIRCRESAHA.112.274233. [DOI] [PubMed] [Google Scholar]
- 72.Cadete VJ, Arcand SA, Chaharyn BM, Doroszko A, Sawicka J, Mousseau DD, Sawicki G. Matrix metalloproteinase-2 is activated during ischemia/reperfusion in a model of myocardial infarction. Can J Cardiol. 2013;29:1495–1503. doi: 10.1016/j.cjca.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 73.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa AS, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee S, Tak E, Lee J, Rashid MA, Murphy MP, Ha J, Kim SS. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res. 2011;21:817–834. doi: 10.1038/cr.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Atlante A, Calissano P, Bobba A, Giannattasio S, Marra E, Passarella S. Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett. 2001;497:1–5. doi: 10.1016/s0014-5793(01)02437-1. [DOI] [PubMed] [Google Scholar]