

Abstract
Objective
Co-secretion of cortisol and aldosterone can be observed in adrenal adenomas. The aim of the present study was to investigate the molecular characteristics of a co-existing aldosterone- and a cortisol-producing adenoma in the same patient.
Design and Methods
Two different adenomas within the same adrenal from a forty nine year-old female patient with primary aldosteronism and Cushing syndrome were studied. Multiple formalin-fixed paraffin-embedded tumor blocks were used for the analysis. Immunohistochemistry (IHC) was performed using a specific antibody against aldosterone synthase (CYP11B2). DNA and RNA were isolated separately from CYP11B2 positive and negative tumor regions based on CYP11B2 IHC results.
Results
CYP11B2 IHC clearly demonstrated that three pieces from one adenoma were positive for CYP11B2 and the remaining three from the other adenoma were negative for CYP11B2. In quantitative real-time RT-PCR, CYP11B2 mRNA was upregulated in CYP11B2 positive tumor specimens (219-fold vs. CYP11B2 negative tumor specimens). Targeted next generation sequencing detected novel KCNJ5 gene mutations (p.T148I/T149S, present in the same reads) and a PRKACA gene hotspot mutation (p.L206R) in the CYP11B2 positive and negative tumors, respectively. Sanger sequencing of DNA from each tumor specimen (CYP11B2 positive tumor, n=3; CYP11B2 negative tumor, n=3) showed concordant results with targeted next generation sequencing.
Conclusion
Our findings illustrate the co-existence of two different adrenocortical adenomas causing the concurrent diagnosis of primary aldosteronism and Cushing syndrome in the same patient. Molecular analysis was able to demonstrate that the two diseases resulted from independent somatic mutations seen in double adrenocortical adenomas.
Key terms: primary aldosteronism, Cushing syndrome, somatic mutation, CYP11B2, next generation sequencing
Introduction
Primary aldosteronism (PA) is a common form of secondary hypertension caused by autonomous adrenal aldosterone production. PA is clinically characterized by hypertension and hypokalemia. Aldosterone-producing adenoma (APA) and idiopathic hyperaldosteronism are the major subtypes of PA. Adrenal Cushing syndrome (CS) is caused by glucocorticoid excess due to a cortisol-producing adenoma (CPA), resulting in hypertension, obesity, type 2 diabetes, and vertebral osteoporosis. As a result of cortisol excess, CS patients present with unique clinical features such as easy bruising, skin atrophy, facial plethora, striae, and dorsocervical fat pad. Over the last decades, an increasing number of patients with subclinical Cushing syndrome (SCS) have been identified. SCS is often characterized by mild hypercortisolism without specific signs or symptoms for CS. In patients with PA, concurrent autonomous cortisol production has been increasingly recognized (1). The majority of the reported cases have had SCS, whereas overt CS has been rarely found.
Next generation sequencing (NGS) has identified several somatic and germline mutations underlying sporadic APA and/or familial PA, such as mutations in an inwardly rectifying potassium channel (KCNJ5) (2), the P-type ATPase gene family (ATP1A1 and ATP2B) (3), and an L-type voltage-gated calcium channel (CACNA1D) (4, 5), and T-type voltage-gated calcium channel (CACNA1H) (6). These mutations alter the pathway involved in regulation of aldosterone production, such as potassium sensing, membrane potential regulation, intracellular Ca2+ levels, and steroidogenesis. More recently, recurrent activating mutations in PRKACA, encoding the catalytic subunit α of protein kinase A (PKA) have been identified in CPA (7). PRKACA gene mutations are found in 35-65% of the patients with CS (8). Herein, we report a case of PA and CS due to the co-existence of two adenomas harboring novel KCNJ5 somatic mutations and a PRKACA somatic mutation.
Patient and Methods
Case Report
The patient was a forty nine year-old African American woman referred for further evaluation of endocrine hypertension. Her past medical history was positive for a pregnancy-associated deep venous thrombosis and her family history was negative for any adrenal or endocrine diseases. She had a five year history of high blood pressure and three years of hypokalemia necessitating replacement doses of 30 to 90 mmol/day of potassium. Over the course of the recent year she had a weight gain of 27 kg (weight at presentation 154 kg). She noted some facial fullness and rounding and had recently been diagnosed with prediabetes. She complained of depression, easy bruising and muscle weakness, particularly climbing stairs. On examination she was found to be obese with facial rounding and increased prominence of the supraclavicular fat pads. Blood pressure was 166/97 mmHg. There were some pale striae on the abdomen, no skin atrophy, but some acanthosis of the neck. Laboratory evaluation revealed an aldosterone of 45 ng/dL (1248 pmol/L) [4-31 ng/dL], a renin of 0.4 ng/mL/hr (0.11 ng/L·s) [1-7 ng/mL/hr] and a 24 hr urine cortisol of 47.3 μg/24hr (130.5 nmol/d) [20-90 μg/24hr] with a low morning ACTH of < 9 pg/mL (1.98 pmol/L) [9-52 pg/mL]. Computed tomography (CT) showed two adrenocortical adenomas in the right adrenal (2.7 cm and 3.0 cm). The posterior mass measured 4 Hounsfield Unit (HU) on unenhanced CT scan (lipid rich adenoma), and the anterior mass measured 20 HU, with a percentage enhancement washout value of 63% (lipid poor adenoma) (9) (Figure 1A-C). NP59 scan showed suppressed uptake in the contralateral gland. Both lesions were identified in the pathological specimen and the diagnosis was that of an adrenal adenoma in both masses. Following surgery the patient felt significant mood improvement and blood pressure and potassium levels normalized. She lost 6 kg over the course of 3 months following surgery. She was diagnosed with adrenal insufficiency following surgery with stimulation to maximum of 1.4 μg/dl (38.6 nmol/L) [> 18 μg/dL] cortisol and did require glucocorticoid replacement therapy.
Figure 1. Imaging and histopatohogical findings of adrenal tumors.
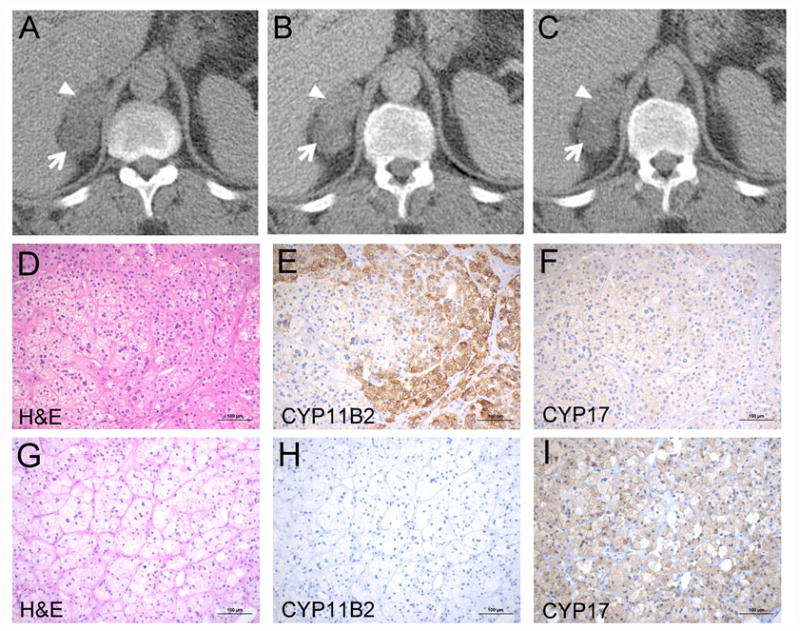
A-C. Abdominal CT showing adrenocortical adenomas in the right adrenal. A. Unenhanced CT scan. The Hounsfield Unit (HU) of the anterior tumor (arrowhead) and the posterior tumor (arrow) were 20 HU and 4 HU, respectively. B. Enhanced CT at 100 sec. The HU of the anterior tumor (arrowhead) and posterior tumor (arrow) were 47 HU and 23 HU, respectively. C. Delayed CT at 15 min. The HU of the anterior tumor (arrowhead) and the posterior tumor (arrow) were 30 HU and 14 HU, respectively. CT, computed tomography. D-I. Histopathological findings of the anterior tumor (D-F) and the posterior tumor (G-I). The anterior tumor showed positive staining for CYP11B2 and the posterior tumor was negative for CYP11B2 (E and H). The posterior tumor had higher expression of CYP17 compared to the anterior tumor (F and I). Scale bars, 100 μm. D and G, H&E; E and H, CYP11B2 IHC; F and I, CYP17 IHC. H&E, Hematoxylin and eosin staining.
Materials
Multiple formalin-fixed paraffin-embedded (FFPE) tumor blocks were used for the analysis. This study was approved by Institutional Review Board of the University of Michigan (HUM00042749).
Immunohistochemistry
Immunohistochemistry was performed using antibodies against aldosterone synthase (CYP11B2) (mouse monoclonal; 1:1500, Millipore, Billerica, MA, USA, #MABS1251) (10) and cytochrome P450 17A1 (CYP17) (mouse monoclonal; 1: 500, kindly provided by Dr. Richard Parker, University of Alabama at Birmingham) as described previously (11).
DNA/RNA isolation
For each sample, six unstained FFPE slides were used for genomic DNA (gDNA) and RNA isolation. gDNA and RNA of CYP11B2 positive and CYP11B2 negative regions were isolated separately based on CYP11B2 IHC using the AllPrep DNA/RNA FFPE kit (QIAGEN, Valencia, CA, USA) as described previously (11).
Quantitative real-time RT-PCR
Total RNA was reverse transcribed using the High Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA, USA). Quantitative real-time RT-PCR (RT-qPCR) was performed in the ABI StepOnePlus Real-Time PCR systems (Applied Biosystems). CYP11B2 primer/probe mixtures were prepared as described previously (12). Primer/probe mixtures for the amplification of β-actin (ACTB, Hs01060665_g1) were purchased from Applied Biosystems. ACTB transcript was used for normalization of sample loading. Relative quantification was determined using comparative threshold cycle method (13).
Targeted next generation sequencing
For mutation analysis of CYP11B2 positive tumor specimen 1 (B2T1) and CYP11B2 negative tumor specimen 1 (T1) samples, 20 ng of isolated gDNA was used for barcoded library generation by multiplexed PCR using a custom Ion AmpliSeq Panel and the Ion AmpliSeq Library kit 2.0 (Life Technologies, Foster City, CA, USA) according to the manufacturer's instructions. The custom Ion AmpliSeq Panel was designed to target genes previously shown to be mutated in APA or other adrenal hyperplasias/neoplasms. The panel contains 499 independent primer pairs targeting the entire coding regions of genes with reported germline or somatic mutations in PA (KCNJ5, ATP1A1, ATP2B3, CACNA1D, and CACNA1H), genes shown to harbor germline or somatic variants associated with adrenal hyperplasia (phosphodiesterase 11A [PDE11A], phosphodiesterase 8B [PDE8B], protein kinase, cAMP-dependent, regulatory, type 1, α [PRKAR1A], PRAKACA, and armadillo repeat containing 5 [ARMC5]), and oncogene hotspots in guanine nucleotide-binding protein subunit α (GNAS) and catenin, β1, 88kDa (CTNNB1). Template preparation and NGS of multiplexed templates were performed as described previously (11) using Ion 318 Chip v2 on the Ion Torrent Personal Genome Machine (PGM) Sequencer (Life Technologies). Data analysis methods for somatic variant identification are described in Supplementary methods.
Direct sequencing of PCR products and subsequent analysis
Bidirectional Sanger sequencing analyses of KCNJ5 and PRKACA gene for DNA samples isolated from each tumor piece were performed using primers as follows: KCNJ5 forward 5′-GGACCATGTTGGCGACCAAGAGTG-3′, reverse 5′-GACAAACATGCACCCCACCATGAAG-3′; PRKACA forward 5′-GGTGACAGACTTCGGTTTCGC-3′, reverse 5′- CCTTGTTGTAGCCCTGGAGCA-3′ (14). For each PCR reaction, 20 ng of template gDNA was used. Sanger sequencing of the purified products was undertaken on the Applied Biosystems 3730 XL sequencer. The PCR product from CYP11B2 positive specimen 3 (B2T3) DNA sample was inserted into pCR2.1-TOPO vector using a TOPO TA cloning kit (Invitrogen, Carlsbad, CA, USA) for subsequent analysis. After chemical transformation using TOP10 competent cells, several carbenicillin resistant clones were picked and sequenced.
Results
CYP11B2 IHC clearly demonstrated that the anterior adenoma showed positive staining for CYP11B2 (B2T1, B2T2, B2T3) and the posterior adenoma was negative for CYP11B2 (T1, T2, T3). CYP11B2 positive tumor specimens exhibited positive but lower expression of CYP17 compared to the CYP11B2 negative tumor specimens (Figure 1D-I). In RT-qPCR, mean of CYP11B2 mRNA expression in three specimens from the CYP11B2 positive adenoma was 219-fold higher than that in CYP11B2 negative adenoma, confirming accurate sample collection. Targeted NGS identified novel KCNJ5 mutations (c.443C>T and c.445A>T, p.T148I/T149S) in a specimen from the anterior adenoma (B2T1) and a PRKACA hotspot mutation (c.617T>G, p.L206R) in a specimen from the posterior adenoma (T1) (Table 1). Read level analysis in Integrative Genomics Viewer demonstrated that the KCNJ5 mutations were present on the same reads (in cis) as shown in Supplemental Figure 1. We further investigated mutational heterogeneity through assessing multiple tumor specimens. As shown in Figure 2, KCNJ5 mutations were identified in all CYP11B2 positive anterior adenoma specimens by Sanger sequencing, but not in CYP11B2 negative tumor specimens from the posterior adenoma. On the other hand, PRKACA mutations were identified only in CYP11B2 negative tumor specimens from the posterior adenoma. Subsequent analysis of PCR product from B2T3 DNA confirmed the NGS identified KCNJ5 mutations (Figure 2B).
Table 1. Somatic mutations identified by targeted next generation sequencing.
| Sample | Gene | Reference allele | Variant allele | Amino acid change | FAO | FDP | Variant allele frequency (FAO/FDP; %) | Variant allele frequency in matched tumor specimen (%) | Reference sequence |
|---|---|---|---|---|---|---|---|---|---|
| B2T1 | KCNJ5 | C | T | T148I | 691 | 1999 | 35 | 0 | NM_000890 |
| B2T1 | KCNJ5 | A | T | T149S | 702 | 1997 | 35 | 0 | NM_000890 |
| T1 | PRKACA | A | C | L206R | 553 | 1996 | 28 | 0 | NM_002730 |
All high-confidence somatic nonsynonymous variants identified in CYP11B2 positive tumor specimen 1 (B2T1) and CYP11B2 negative tumor specimen 1 (T1) are shown. The variant allele frequency in the matched tumor specimen, i.e. T1 for B2T1 and B2T1 for T1, respectively, is shown for comparison. FAO, flow-corrected variant allele-containing read; FDP, flow-corrected read depth.
Figure 2. CYP11B2 immunohistochemistry and Sanger sequencing results of each tumor piece.
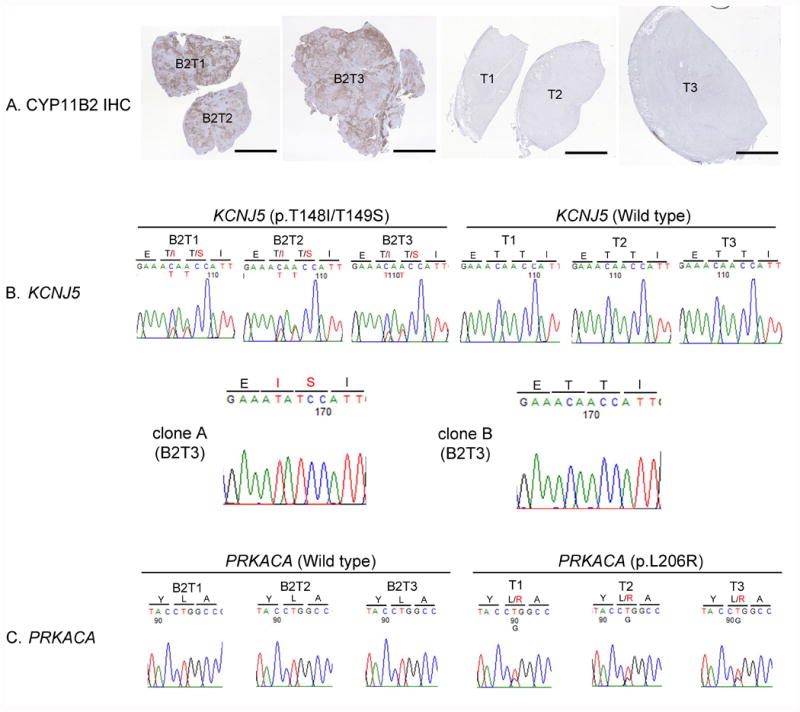
A. Low magnification scan of tumor pieces with CYP11B2 IHC (CYP11B2; brown). Scale bars, 5 mm. B. Results of Sanger sequencing of KCNJ5 gene (upper panel) and subsequent analysis of PCR product (lower panel). KCNJ5 gene mutations (p.T148I/T149S) were detected in B2T1, B2T2, and B2T3 but not in T1, T2, and T3. Subsequent analysis of PCR product (B2T3) showed both mutations were present in the same allele (clone A) and the other allele was wild type (clone B). C. Results of Sanger sequencing of PRKACA gene. PRKACA gene mutations (p.L206R) were identified in T1, T2, and T3 but not in B2T1, B2T2, and B2T3. IHC, immunohistochemistry; B2T1, CYP11B2 positive tumor specimen 1; B2T2, CYP11B2 positive tumor specimen 2; B2T3, CYP11B2 positive tumor specimen 3, T1, CYP11B2 negative tumor specimen 1; T2, CYP11B2 negative tumor specimen 2, T3, CYP11B2 negative tumor specimen 3.
Discussion
Adrenal tumors are common neoplasms and the prevalence increases with age. Most are benign cortical adenomas and some are associated with endocrine disorders such as PA and CS. Although adrenal tumors with concomitant autonomous aldosterone and cortisol secretion have been increasingly recognized, their molecular characteristics remain to be elucidated.
The present study demonstrates that the adrenal tumors in our patient have different characteristics in terms of steroidogenic enzyme expression and mutation status. KCNJ5 encodes the G-protein-activated inwardly-rectifying potassium channel 4 (GIRK4). Although the KCNJ5 gene mutations observed in this study have not been reported previously, two somatic mutations involving T148 and T149 located near the selectivity filter of the GIRK4 channel pore, have been identified in APA (p.T148_T149insT, p.T148_T149insR) and in vitro analyses demonstrated that both mutations caused membrane depolarization, raised intracellular Ca2+, and increased aldosterone production (15, 16), supporting a pathological potential of the KCNJ5 mutations identified in this case (p.T148I/T149S). Additionally, considering higher conservation of T149 residue among orthologues and paralogues compared to T148 (2), the mutation altering the amino acid in position 149 likely has a more important pathophysiological role than the change in position 148. PRKACA encodes the catalytic subunit of cyclic AMP-dependent PKA. The p.L206R mutation is located in a highly conserved core of the domain responsible for the interaction between the regulatory and catalytic subunits of PKA (7). Functional experiments revealed that the mutation caused constitutive PKA activation, providing a molecular explanation for the development of CPA (7, 8). The mutation analysis suggests that the CYP11B2 positive tumor was the main source of autonomous aldosterone production and the CYP11B2 negative and CYP17 positive tumor was the main source of autonomous cortisol production in this case. It remains speculative, whether the co-occurrence of two adenomas in a single patient might be caused by a common underlying genetic predisposition or simply a chance event. In most cases the pathomechanism of hypercortisolism accompanying primary aldosteronism is believed to be co-secretion of cortisol and aldosterone by the same adenoma. Considering the rare co-appearance of APA and CPA in a single adrenal gland, the adenomas likely have occurred independently through two independent genetic events rather than having a common progenitor of the two adenomas. There remains a possible involvement of a susceptibility gene to tumor development. However, more patients and families would be necessary to attempt to define this susceptibility locus.
In the present study, we demonstrate a case of PA and CS due to two different adrenocortical adenomas. This is certainly a quite rare case. However, it is important to notice any hypercortisolism in patients undergoing adrenal surgery in order to consider the necessity of perioperative hydrocortisone supplementation. This study also has implications for the pathologic distinction of nodular hyperplasia and double adenoma in endocrine neoplasia. Our finding of distinct mutations in discrete masses provides compelling evidence for a double adenoma model that also applies to other endocrine glands.
Supplementary Material
Acknowledgments
We thank Dr. Richard Parker (University of Alabama at Birmingham) for the generous gift of CYP17 antibody and Tina Fields for excellent immunohistochemical support.
Funding: This work was supported by grants from the NIDDK (DK106618) to W.E.R. and S.A.T.. T.E. is sponsored by American Heart Association 14SDG17990000. K.N. is supported by American Heart Association 14POST20020003. S.A.T. is supported by the A. Alfred Taubman Medical Research Institute and a National Cancer Institute Grant CA46592 (to the Michigan Cancer Center Core). This research is also supported (in part) by the National Institutes of Health through the University of Michigan's Cancer Center Support Grant (5 P30 CA46592).
Footnotes
Disclosure: S.A.T has received honoraria from Thermo Fisher Scientific and had a separate sponsored research agreement with Thermo Fisher Scientific. None of the study described herein was supported by Thermo Fisher Scientific and they had no role in the data collection, interpretation, or analysis, and did not participate in the study design or the decision to submit for publication. The remaining authors have nothing to disclose.
References
- 1.Spath M, Korovkin S, Antke C, Anlauf M, Willenberg HS. Aldosterone- and cortisol-co-secreting adrenal tumors: the lost subtype of primary aldosteronism. European journal of endocrinology / European Federation of Endocrine Societies. 2011;164:447–455. doi: 10.1530/EJE-10-1070. [DOI] [PubMed] [Google Scholar]
- 2.Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331:768–772. doi: 10.1126/science.1198785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, Penton D, Schack VR, Amar L, Fischer E, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nature genetics. 2013;45:440–444. doi: 10.1038/ng.2550. 444e441-442. [DOI] [PubMed] [Google Scholar]
- 4.Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, Fonseca AL, Korah R, Starker LF, Kunstman JW, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nature genetics. 2013;45:1050–1054. doi: 10.1038/ng.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azizan EAB, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, Maniero C, Garg S, Bochukova EG, Zhao W, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nature genetics. 2013;45:1055–1060. doi: 10.1038/ng.2716. [DOI] [PubMed] [Google Scholar]
- 6.Scholl UI, Stolting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, Prasad ML, Goh G, Carling T, Juhlin CC, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. eLife. 2015;4:e06315. doi: 10.7554/eLife.06315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beuschlein F, Fassnacht M, Assie G, Calebiro D, Stratakis CA, Osswald A, Ronchi CL, Wieland T, Sbiera S, Faucz FR, et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing's syndrome. The New England journal of medicine. 2014;370:1019–1028. doi: 10.1056/NEJMoa1310359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calebiro D, Di Dalmazi G, Bathon K, Ronchi CL, Beuschlein F. cAMP signaling in cortisol-producing adrenal adenoma. European journal of endocrinology / European Federation of Endocrine Societies. 2015;173:M99–106. doi: 10.1530/EJE-15-0353. [DOI] [PubMed] [Google Scholar]
- 9.Caoili EM, Korobkin M, Francis IR, Cohan RH, Platt JF, Dunnick NR, Raghupathi KI. Adrenal masses: characterization with combined unenhanced and delayed enhanced CT. Radiology. 2002;222:629–633. doi: 10.1148/radiol.2223010766. [DOI] [PubMed] [Google Scholar]
- 10.Gomez-Sanchez CE, Qi X, Velarde-Miranda C, Plonczynski MW, Parker CR, Rainey W, Satoh F, Maekawa T, Nakamura Y, Sasano H, et al. Development of monoclonal antibodies against human CYP11B1 and CYP11B2. Molecular and cellular endocrinology. 2014;383:111–117. doi: 10.1016/j.mce.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nanba K, Chen AX, Omata K, Vinco M, Giordano TJ, Else T, Hammer GD, Tomlins SA, Rainey WE. Molecular Heterogeneity in Aldosterone-Producing Adenomas. The Journal of clinical endocrinology and metabolism. 2016 doi: 10.1210/jc.2015-3239. jc20153239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pezzi V, Mathis JM, Rainey WE, Carr BR. Profiling transcript levels for steroidogenic enzymes in fetal tissues. The Journal of steroid biochemistry and molecular biology. 2003;87:181–189. doi: 10.1016/j.jsbmb.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 14.Larkin SJ, Ferrau F, Karavitaki N, Hernandez-Ramirez LC, Ansorge O, Grossman AB, Korbonits M. Sequence analysis of the catalytic subunit of PKA in somatotroph adenomas. European journal of endocrinology / European Federation of Endocrine Societies. 2014;171:705–710. doi: 10.1530/EJE-14-0545. [DOI] [PubMed] [Google Scholar]
- 15.Kuppusamy M, Caroccia B, Stindl J, Bandulik S, Lenzini L, Gioco F, Fishman V, Zanotti G, Gomez-Sanchez C, Bader M, et al. A novel KCNJ5-insT149 somatic mutation close to, but outside, the selectivity filter causes resistant hypertension by loss of selectivity for potassium. The Journal of clinical endocrinology and metabolism. 2014;99:E1765–1773. doi: 10.1210/jc.2014-1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng FF, Zhu LM, Nie AF, Li XY, Lin JR, Zhang K, Chen J, Zhou WL, Shen ZJ, Zhu YC, et al. Clinical characteristics of somatic mutations in Chinese patients with aldosterone-producing adenoma. Hypertension. 2015;65:622–628. doi: 10.1161/HYPERTENSIONAHA.114.03346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.