

Abstract
TRAF2- and NCK-interacting kinase (TNIK) represents one of the crucial targets for Wnt-activated colorectal cancer. In this study, we curated two datasets and conducted a comprehensive modeling study to explore novel TNIK inhibitors with desirable biopharmaceutical properties. With Dataset I, we derived Comparative Molecular Similarity Indices Analysis (CoMSIA) and variable-selection k-nearest neighbor models, from which 3D-molecular fields and 2D-descriptors critical for the TNIK inhibitor activity were revealed. Based on Dataset II, predictive CoMSIA-SIMCA (Soft Independent Modelling by Class Analogy) models were obtained and employed to screen 1,448 FDA-approved small molecule drugs. Upon experimental evaluations, we discovered that mebendazole, an approved anthelmintic drug, could selectively inhibit TNIK kinase activity with a dissociation constant Kd = ~1 μM. The subsequent CoMSIA and kNN analyses indicated that mebendazole bears the favorable molecular features that are needed to bind and inhibit TNIK.
Loss of function of the adenomatous polyposis coli (APC), a Wnt signaling pathway inhibitor, and activation mutation of β-catenin are the two major forces driving transformations in colorectal cancers1,2. However, to date few druggable targets involved in the Wnt pathway have been identified. TRAF2 and NCK-interacting kinase (TNIK) was recently characterized as an essential activator of TCF4/β-catenin transcriptional programme. It is recruited to the promoters of the Wnt target genes and directly phosphorylates TCF43,4. This kinase also regulates cytoskeleton rearrangements and stress responses through the Rap2A and c-Jun N-terminal kinase (JNK) pathway, respectively5,6. Knockdown or mutation of the TNIK kinase domain downregulates the canonical Wnt pathway and JNK pathway, and thus triggers the apoptosis7. Since the kinase activity is essential for activation of the β-catenin pathway, TNIK is an attractive therapeutic target against colorectal cancer that obtains aberrant Wnt signaling.
While numerous clinical-relevant kinase inhibitors have been approved8,9, development of inhibitors targeting TNIK is still in the very early stage. Recently, Yamada et al. patented a series of thiazole-4-carboxamide derivatives, including 48 compounds which are able to inhibit TNIK at sub-micromolar concentration (defined as Dataset I)10. Similarly, Davis et al. profiled the binding affinities of 72 known, chemically diverse kinase inhibitors against 442 kinases, and found a number of compounds exhibited significant binding to TNIK, with dissociation constant ranging from 4.7 nM to 8.5 μM (defined as Dataset II)11. However, the biopharmaceutical properties (bioavailability, pharmacokinetics, etc.) of the Dataset I compounds need to be further optimized, and most of the kinase inhibitors in Dataset II are notorious for their known side effects12.
Herein, we conducted 2D- and 3D-QSAR studies based on these two datasets and attempted to identify novel TNIK inhibitors with preferred biopharmaceutical properties. With Dataset I, we derived Comparative Molecular Field Analysis (CoMFA), Comparative Molecular Similarity Indices Analysis (CoMSIA), and variable-selection k-nearest neighbor (kNN) models, from which the 3D-molecular fields and 2D-descriptors critical for TNIK inhibitory activity were discovered. Based on Dataset II, a CoMSIA-SIMCA (Soft Independent Modeling by Class Analogy13) classification model was obtained and used to screen 1,448 currently marketed drugs. Upon experimental validation using the KINOMEscan platform14 (Ambit Biosciences; http://www.kinomescan.com), we found that an anthelmintic drug, mebendazole, could selectively inhibited 91.8% of the TNIK signal at 10 μM, with a dissociation constant (Kd) of ~1 μM. Mebendazole binds to the ATP-binding pocket of TNIK in a similar fashion as dasatinib, and both of our CoMSIA and kNN models demonstrated that the compound possesses the required molecular pharmacophores and properties to bind TNIK. This study represents a unique ligand-based framework for drug repurposing against a specific protein target critical for colorectal cancer treatment.
Results
CoMFA and CoMSIA modeling
As all compounds in Dataset I are thiazole-4-carboxamide derivatives (Fig. 1), it provides a reliable structural basis to perform ligand alignment for quantitative CoMFA and CoMSIA studies. Using the Sphere Exclusion (SE) algorithm15, we divided this dataset into a training set (38 compounds) and a test set (10 compounds). The statistics of the CoMFA and CoMSIA models are shown in Table 1. Compared to the CoMFA model, the CoMSIA model achieved a slightly better leave-one-out cross-validation q2 (0.685), along with comparable 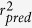 (0.774) and standard error of prediction (0.273). Intriguingly, the hydrophobic term, rather than the steric or electrostatic term, has the largest contribution to the IC50 prediction (Table 1).
(0.774) and standard error of prediction (0.273). Intriguingly, the hydrophobic term, rather than the steric or electrostatic term, has the largest contribution to the IC50 prediction (Table 1).
Figure 1. Chemical structures of the thiazole-4-carboxamide derivatives (dataset I).

The values in the parentheses are pIC50.
Table 1. Summary of CoMFA and CoMSIA models.
| Statistics | CoMFA | CoMSIA |
|---|---|---|
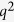 a a
|
0.622 | 0.685 |
| SEPb | 0.586 | 0.534 |
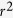 c c
|
0.987 | 0.992 |
| SEEd | 0.108 | 0.084 |
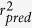 e e
|
0.779 | 0.774 |
| SEPpredf | 0.263 | 0.273 |
| Componentsg | 10 | 10 |
| Fh | 216.204 | 353.298 |
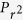 i i
|
0.000 | 0.000 |
| Fraction | ||
| Steric | 0.573 | 0.215 |
| Electrostatic | 0.427 | 0.330 |
| Hydrophobic | NAj | 0.455 |
aLOO cross-validated correlation coefficient (training set).
bLOO cross-validated standard error of prediction (training set).
cNon-cross-validated correlation coefficient (training set).
dStandard error of estimate (training set).
eCorrelation coefficient for the test set.
fStandard error of prediction for the test set.
gOptimal number of components.
hF-test value.
iProbability of obtaining the F value by chance.
jHydrophobic contribution not available in CoMFA.
This CoMSIA model also offers structural insights for lead optimization of this thiazole-4-carboxamide series. For example, extension of the ring I on the para-position is sterically favorable, as indicated by the S2 region in Fig. 2A. In contrast, modifications on the ortho- or meta- position, especially the meta-position, are sterically prohibited (S3 in Fig. 2A). One may notice that the S3 region is located at the carboxamide side of ring I, and compounds such as 5, 11 and 12, may flip ring I to avoid the sterically unfavorable S3 region when binding to TNIK. However, compound 17, which adopts bulky substituents on both sides of ring I, has no means to circumvent the S3 region, and thus it obtains the highest IC50. Furthermore, CoMSIA model suggested that the orth-/meta-position of ring I favors single electropositive and hydrophilic group (e.g., amide in compound 103), and disfavors hydrophobic or electronegative group (e.g., Cl or Br as in compound 11 and 12), as indicated by E3, H2 and H3 regions (Fig. 2B,C).
Figure 2. CoMSIA model derived from dataset I.

The most active inhibitor, A84 (in sticks), is used as an example to illustrate the CoMSIA fields (in grids). CoMSIA fields (A) Yellow – sterically unfavorable region; Green – sterically favorable region; (B) Blue – electronegative unfavorable (or electropositive favorable) region; Red – electronegative favorable (or electropositive unfavorable) region; (C) Cyan – hydrophobicity unfavorable region; Black – hydrophobicity favorable region. (D) Overlapping the CoMSIA fields to TNIK kinase domain (in lines). Yellow dashed lines indicated the hydrogen bonds with the hinge.
Further analysis of the S2 and H1 regions showed that a linear, three-heavy-atom para modification of ring I contributes positively to the IC50 (Fig. 2A,C). The 4th heavy atom favors an electronegative one, such as oxygen (E1 region in Fig. 2B). For example, compared with compound A11, the addition of a hydroxyl group to the 4th heavy atom (compound A37) lowers IC50. For the distal modifications of ring I, the sterically unfavorable S1 region restricts the maximum length of the substituent. To achieve the best TNIK inhibition, the estimated ideal length should range from 5.0 Å to 7.0 Å (Fig. 2A). However, it is worth noting that the contributions of S1, E1 and H1 regions are usually below 0.3 pIC50 units, significantly less than other regions (Supplementary Table 2 for comparisons).
On the other side of the core, we observed that a small electronegative substituent on the para-position is energetically favorable on ring II, as indicated by S4 and E5 in Fig. 2A,B. This finding is consistent to IC50 values that follow the ascendant trend: 4-pyridine (X3 = N) < 3-pyridine or 5-pyrimidine (X1 or/and X3 = N) < Phe (R2 = H) < Phe-4-OMe (R2 = OMe) (Supplementary Table 2). Accordingly, the existence of electronegative-favorable E5 region could result in the electropositive-favorable E5’ region.
Molecular docking confirmed CoMSIA model
As we analyzed, no compound in Dataset I bears the pharmacophore of the allosteric inhibitors of TNIK (type 2 kinase inhibitor16) (data not shown). Indeed, this chemical series most likely functions as type 1 kinase inhibitors16, which bind the ATP-binding pocket without flanking to the allosteric site. The molecular docking confirmed this hypothesis, because the thiazole-4-carboxamide core was consistently docked to the adenine site in three difference receptor models, including TNIKclose, TNIKopen and TNIKDFG-out. Also the ligand binding mode is in agreement with the aforementioned CoMSIA model. Figure 2D demonstrated the predicted binding mode for compound A84 in TNIKclose. Based on the Traxler model which breaks the ATP-binding pocket into five subcomponents17, the adenine site is occupied by thiazole-4-carboxamide forming the hydrogen bonds to the hinge (E106 and C108 in Fig. 2D). Ring I is located at the hydrophobic pocket I, while ring II is buried in the hydrophobic pocket II and close to the gatekeeper residue (M105).
In particular, the meta-carbon of the ring I (corresponding to the E3 and H2 regions) is only 4.4 Å away from the carboxylatic oxygen of D115 (Fig. 2D). Moreover, the sterically-unfavorable S3 and hydrophobicity-unfavorable H3 regions are located in the hydrophobic pocket I. These observations buttressed the CoMSIA model that a small, electropositive and hydrophilic, meta- substituent of ring I is structurally preferred. Surprisingly, the electropositively favorable E2 region forms a polar contact with the carbonyl group of G109 (Fig. 2D). Finally, due to the bulky gatekeeper residue M105, the small electronegative modification(s) of ring II is apparently more energetically favorable than the large hydrogen-rich ones. This is consistent with the above analysis of the S4 and E5 regions (Fig. 2A,B).
Ligand-based kNN modeling
The best six kNN models are shown in Table 2. All of these models had less than five descriptors, and both leave-one-out cross-validated training set q2 and testing set r2 were over 0.74, indicating the high predictive abilities of the resultant models. Of note, the kNN model #1, which contains 27 compounds in the training set and 21 in the test set, was able to accurately predict the pIC50 values with q2 = 0.81 and r2 = 0.78 using only three descriptors. The right plot in Fig. 3 demonstrated that our variable selection method particularly determined four types of descriptors that contribute mostly to the kNN models: KierA2 (second alpha modified shape index), diameter/radius, GCUT_SLogP/BCUT_SLogP, and PEOE_VSA_HYD (total hydrophobic van der Waals surface area).
Table 2. Summary of kNN models.
| Models | Data splitting (training/testing) | Neighborsa |
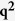 b b
|
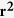 c c
|
Descriptors |
|---|---|---|---|---|---|
| 1 | 27/21 | 2 | 0.81 | 0.78 | KierA2, GCUT_SLogP_0, radius |
| 2 | 28/20 | 2 | 0.82 | 0.74 | KierA2, BCUT_SLogP_3, diameter |
| 3 | 33/15 | 2 | 0.75 | 0.83 | PEOE_VSA_HYD, vsa_acc, radius |
| 4 | 35/13 | 2 | 0.77 | 0.86 | PEOE_VSA_HYD, SMR_VSA6, PEOE_PC+, radius |
| 5 | 36/12 | 2 | 0.74 | 0.89 | KierA2, b_ar, radius |
| 6 | 37/11 | 4 | 0.76 | 0.93 | KierA2, GCUT_SLogP_0, radius |
aOptimal number of nearest neighbors.
bLOO cross-validated correlation coefficient for the training set.
cCorrelation coefficient for the test set.
Figure 3. Predicted pIC50 values versus actual pIC50 values for CoMSIA model (left) and kNN model (right).

The predicted pIC50 for both training sets are predicted by leave-one-out cross-validation.
Intriguingly, we discovered that there are inverse correlations between pIC50 with KierA2 and PEOE_VSA_HYD for molecules with a radius below 7 (Supplementary Fig. 1). The correlation coefficients for pIC50 with KierA2 and PEOE_VSA_HYD are 0.78 and 0.55, respectively. KierA2 represents the overall shape of the molecule. Generally, a linear compound has alower KierA2 than a complex one. PEOE_VSA_HYD describes the total hydrophobic van der Waals surface area, in which the partial charges are computed with the partial equalization of orbital electronegatives. These two inverse correlations implied that the core scaffold of TNIK inhibitors (e.g., radius < 7) should be linear and hydrophilic in order to maximize the efficiency of design. This is consistent to the observation that the steric bulky or hydrophobic unfavorable regions, such as S3, S4, H2 and H4, are located at the hydrophobic pockets I and II (Fig. 2D).
Accurate categorization of TNIK-binding kinase inhibitors by CoMSIA-SIMCA
While the 2D- and 3D-QSAR of Dataset I have provided essential structural guidance for computer-aided design of TNIK inhibitors, additional studies of Dataset II, which covers much more chemical spaces, can expand the applicability domain of our models. To this end, we conducted SIMCA13 analysis with Dataset II. SIMCA is a widely-used classification algorithm that develops predictive models using principal component analysis (PCA) based on the precategorized training set. Due to the structural diversity of Dataset II, we filtered out all type 2 kinase inhibitors and allosteric inhibitors, leading to 54 type 1 kinase inhibitors (Supplementary Table 1). These compounds were then grouped into four categories. According to their respective pKd values, we intentionally name them as Category IV: pKd < 5; Category V: 5 ≤ pKd < 6; Category VI: 6 ≤ pKd < 7; Category VII: pKd ≥ 7. We also divided the dataset into a training set (48 compounds) and a test set (6 compounds) for model building (Supplementary Table 5). The CoMSIA fields were computed based on the receptor-guided alignment using rigorously-designed molecular dockings with respect to the available structural data (see Materials and Methods for details).
As indicated by the confusion matrix (Table 3), we derived a robust CoMSIA-SIMCA model in which 91.7% of kinase inhibitors were accurately classified (cross-validated) for the training set, while only four compounds were misclassified after five-group cross-validation. The predictive ability of this model was further validated on the test set, in which only one compound (AZD-2171) was misclassified. It is worth noting that three (out of five) misclassified compounds, including SB-203580 (pKd = 6.086), TAE-684 (pKd = 5.921) and AZD-2171 (pKd = 6.092), have pKd values close to the classification margin. As expected, these three compounds were classified to the adjacent categories: SB-203580 - Category V, TAE-684 - Category VI and AZD-2171 - Category V. Furthermore, the distances between categories followed the trend of binding affinities (Table 4), indicating that the SIMCA model can semi-quantitatively reflect the strength of TNIK binding. The details of the categorization results are available in Supplementary Table 5. To exclude the possibility of overfitting, we also conducted Y-randomization as we described previously18,19,20,21. It failed to accurately predict the activity (data not shown), demonstrating the robustness of our models.
Table 3. CoMSIA-SIMCA analysis for the training set upon five-group cross-validation.
| Actual/Predicted | IV | V | VI | VII | Total |
|---|---|---|---|---|---|
| IV | 21 | 1 | 0 | 0 | 22 |
| V | 0 | 8 | 1 | 0 | 9 |
| VI | 0 | 1 | 9 | 0 | 10 |
| VII | 0 | 0 | 1 | 6 | 7 |
Category IV: pKd < 5; Category V: 5 ≤ pKd < 6; Category VI: 6 ≤ pKd < 7; Category VII: pKd ≥ 7.
Table 4. Distance between categories obtained from CoMSIA-SIMCA model.
| ActualCat4 | ActualCat5 | ActualCat6 | ActualCat7 | |
|---|---|---|---|---|
| ProjectedCat4 | 201.395 | 270.533 | 320.406 | 381.345 |
| ProjectedCat5 | 262.338 | 165.989 | 241.476 | 292.651 |
| ProjectedCat6 | 305.968 | 231.206 | 181.780 | 244.579 |
| ProjectedCat7 | 377.213 | 277.938 | 239.415 | 170.452 |
Identification of mebendazole as TNIK inhibitor
Herein, we have derived predictive CoMSIA-SIMCA models that can be utilized to efficiently screen a drug-like database to discover novel TNIK inhibitors. The structural insights provided by our modeling can further improve the virtual screening accuracy. To this end, we screened 1,448 US Food and Drug Administration (FDA)-approved small-molecule drugs, aiming to identify potential TNIK inhibitor from the marketed drugs that have desired absorption, distribution, metabolism, and elimination (ADME) properties and already passed the strict safety/toxicity investigations. Not surprisingly, the CoMSIA-SIMCA model identified three kinase inhibitors, sunitinib, gefitinib and dasatinib, which could block TNIK activities. Such predictions are in agreement with some experimental reports: Sunitinib was classified in Category VII (actual Kd = 25 nM), whereas gefitinib was classified in Category V (actual Kd = 6.9 μM) (Table 5).
Table 5. The experimental results from KINOMEscan scanELECT.
| Compound | Kinase | % Ctrl |
Kd (μM) | |
|---|---|---|---|---|
| 0.1 μM | 10 μM | |||
| Sunitinib | TNIK | 37 | 0 | 0.025 |
| Dasatinib | TNIK | 82 | 11 | 2.0 |
| Gefitinib | TNIK | 100 | 34 | 6.9 |
| Lapatinib | TNIK | 90 | 93 | >10 |
| Flavopiridol | TNIK | 92 | 33 | >10 |
| Mebendazole | TNIK | 97 | 8.2 | ND |
| ABL2 | 100 | 33 | ND | |
| MEK1 | 100 | 38 | ND | |
| EGFR | 100 | 83 | ND | |
| ACK1 | 100 | 98 | ND | |
| PDPK1 | 100 | 100 | ND | |
| PIK3CA | 100 | 100 | ND | |
The values were reported as percentage of control, where lower value indicates stronger binding. ND – not determined.
For the first time, mebendazole (MBZ), which was originally designed as an anthelmintic drug22, was identified to be a TNIK inhibitor. The CoMSIA-SIMCA model classified MBZ as a potent agent (Category V - Kd ranging from 1 μM to 10 μM). Our follow-up molecular docking indicates that MBZ binds to the adenine site of TNIK. The 3-N of benzimidazole forms hydrogen bonding interactions with the C108 amide, whereas the carbamate group interacts with C108 via its carbonyl group. This hydrogen bonding pattern of MBZ binding to TNIK is similar to that of dasatinib (Fig. 4). When referred to the CoMSIA fields shown in Fig. 2A–D, both dasatinib and MBZ circumvent the unfavorable bulky S3 region and minimize the hydrophobicity in the H2 region. However, MBZ is even less bulky around the hydrophobic pocket II (corresponding to S4 region) than dasatinib. Conforming to our kNN modeling, MBZ has the desired molecular properties as a TNIK inhibitor: Its radius was 7, whereas the KierA2 (5.24) and PEOE_VSA_HYD (213.47) were significantly below the averages of KierA2 (8.03) and PEOE_VSA_HYD (297.95) obtained from Dataset I.
Figure 4. The binding modes of dasatinib (white) and Mebendazole (yellow) in TNIK kinase domain.

The blue ribbons represents TNIK kinase domain, and the hinge residues and D115 side chain are highlighted with sticks. The magenta dashed lines represent the hydrogen bonds between Mebendazole and hinge. Chemical structures of Dasatinib and Mebendazole are also shown.
Experimental validation of MBZ as a TNIK inhibitor
We performed experimental studies of MBZ using the KINOMEscan platform to evaluate its binding to TNIK and selectivity among different kinases. Our result shows that MBZ inhibits 91.8% of TNIK activity at 10 μM. Compared with other inhibitors, such as dasatinib (89% inhibition at 10 μM) and gefitinib (66% inhibition at 10 μM), the Kd of MBZ binding to TNIK is likely to be ~1.0 μM (Table 5). In contrast, MBZ does not exhibit significant inhibitory effect, defined as over 70% inhibition of the control, on ABL2, EGFR, MEK1, PDPK1, PIK3CA, and ACK1 at 10 μM. Since TNIK acts as an activator of the Wnt/β-catenin/TCF4 pathway3,4, this finding is consistent to some previous study showing that MBZ exhibits a potent cytotoxicity against β-catenin-active colon and non-small-cell lung cancer cell lines23.
Discussion
In the present study, we conducted comprehensive modeling studies of TNIK inhibitors and developed CoMFA/CoMSIA, kNN, and CoMSIA-SIMCA models, which were then rationally applied to screen and identify an approved drug, mebendazole, as a potent TINIK inhibitor for cancer therapy. In particular, CoMSIA and kNN model provided valuable structural insights that the TNIK inhibitors usually favor the linear and hydrophilic moieties rather than the complex and hydrophobic groups. Meanwhile, CoMSIA-SIMCA classification model provided a platform to conduct efficient primary screening of potential TNIK inhibitors. As part of the recent polypharmacology and drug repurposing efforts24,25 such comprehensive studies and the resultant models are ready to be used for compound repositioning against other targets.
Mebendazole (MBZ), as a marketed drug, is suited for further clinical study considering its promising safety profile22. Several studies have suggested MBZ as a potent antitumor agent. For instance, MBZ was demonstrated significant inhibition against the growth and metastasis of the adrenocortical carcinoma, both in vitro and in vivo26. In addition, studies have shown that MBZ could cause a mitotic arrest and a time-dependent apoptotic response in various cancercell lines27,28. However, the mechanism of its antitumor property remains elusive. The most well-known target of MBZ is tubulin, in which MBZ directly binds tubulin and impairs the tubulin polymerization27,29.
Herein, we identified a novel mechanism of action for MBZ which targets an oncogenic protein, TNIK, in a clinical-relevant signaling pathway, particular in colorectal cancer. TNIK phosphorylates S154 of TCF4, and its catalytic activity has proved to be essential for the colorectal cancer growth3. According to our experimental testing results, the Kd for MBZ against TNIK kinase domain is around 1 μM, which is of the same order of magnitude as the reported Kd value to mammalian brain tubulin (0.5 μM)30. In addition, mebendazole is a FDA-approved drug and can be used at a significantly higher dosage (up to 200 mg/kg with daily use31) without severe side effects. In addition, mebendazole may exhibit a synergic anti-tumor effect with other anti-cancer drugs through disrupting cytoskeleton because TNIK kinase activity has proven essential to regulate the F-actin fiber formation32. Such advantages render the possibility of quickly translating the discovery into clinical setting for cancer treatment in the near future.
Materials and Methods
Dataset
We collected a series of potent TNIK inhibitors from a previous study, including 48 patented TNIK inhibitors with known IC50 values (Dataset I)10, and 72 kinase inhibitors with known dissociation constant values (Kd) (Dataset II)11. All of the patented compounds along with the inhibitory IC50 values are available in Fig. 1. The chemical structures and the Kd values were retrieved from the ChEMBL database (ID: 1908790). The kinase inhibitors in Dataset II were classified into type 1, type 2 and allosteric inhibitors based on the available literatures and protein-ligand complex structures (Supplementary Table 1). –log(IC50) and –log(Kd) values were computed as the dependent variable for all analyses. Since Dataset I and II cover different chemical space, they are curated herein to achieve different goals and thus we kept them separate during model building.
3D-QSAR by CoMFA and CoMSIA
3D-molecular alignment were prepared using SYBYL 8.1 (Tripos, Ltd, US) and Molecular Operating Environment (MOE 2010.10). For dataset I, molecular alignments were performed using substructure overlap thiazole-4-carboxamide core with respect to the docking poses (see molecular docking for details). The resulting alignment was refined by flexible alignment using MOE. The alignment is available from the authors upon request. CoMFA and CoMSIA were performed by SYBYL 8.1 using the default parameter (Tripos standard field, 2 Å grid spacing, dielectric distance 1/r2, 30 kcal/mol cutoff) probed by an sp3 carbon with a charge of +1. CoMFA steric energy (Lennard-Jones) and electrostatic (Coulomb) energy were calculated. CoMSIA steric, electrostatic and hydrophobic energies were calculated with the attenuation factor = 0.3. Pullman charges were used for electrostatic field calculations. 3D-QSAR was performed by partial least squares (PLS) analyses. Dataset I was divided into a training set (38 compounds) and a test set (10 compounds) based on the Sphere Exclusion (SE) algorithm15. The details of data splitting are available in Supplementary Table 3. Leave-one-out cross-validation was used for the training set. The predictive ability the non-cross-validated models were validated using the test set. The statistics of the resulted CoMFA and CoMSIA models are available in Table 1.
2D-QSAR by variable-selected kNN
2D-QSAR was performed on dataset I. A total of 186 2D-descriptors were calculated with MOE 2010.10. The resulting descriptors were normalized to [0, 1], and the descriptors with zero standard deviation were removed. As described previously, dataset splitting was performed using an SE8 program implemented with sphere exclusion algorithm15. The details of data splitting are available in Supplementary Table 4. The resulting 50 training and test sets which consider the diversity of descriptors were used to build the predictive k-nearest neighbor (kNN) models. The implementation of this variable-selection kNN algorithm has been published elsewhere33. Concisely, the simulated annealing algorithm was employed to explore the entire descriptor spaces and optimize the descriptor subset based upon the leave-one-out (LOO) cross-validation q2. The models whose q2s satisfied the cutoff were validated on their respective test sets (non-cross-validated). Only the kNN models satisfying the criteria that “q2 > 0.7 and r2 > 0.7 and number of descriptors <10” were kept for further analysis.
Molecular docking
High-resolution crystal structure of TNIK in complex with Wee1 kinase inhibitor was obtained from Protein Data Bank (PDB ID: 2X7F). Due to the structural defects in the ATP binding site, we aligned all available five chains in 2X7F, and chain B was used as receptor because its ATP-binding site had the least number of missing residues (e.g., E29, T35, K41, K54, M56, E69, K155, E163). The missing ζ-amine of K54 and K155 were manually added, and these two ζ-amine moieties were subjected to energy minimization optimization. Considering the dynamics of the TNIK kinase domain, we established three different models, TNIKopen, TNIKclose and TNIKDFG-out, to represent the lobe-open, lobe-close and DFG-out conformations. TNIKclose used the original coordinates in 2X7F. Based on TNIKclose, we built TNIKopen by modeling the TNIK lobe (residue 29–41) with respect to a template in which lobe is in open conformation (PDB ID: 3NIZ, residue 27–39). Similarly, TNIKDFG-out was built by modeling the TNIK activation loop (residue 170–173) with respect to the sorafenib-bound template (PDB ID: 3HEG34, residue 167–170). GOLD 5.1 (CCDC)35 was used for molecular docking. Based on the expert knowledges on kinase and the crystal structure of p38α in complex with an isothiazole-4-carboxamdide derivative, CP-547632 (PDB ID: 3L8S)36, we constrained the hydrogen bonds with the carbonyl group of M106 localized at the hinge. All of the docking parameters were the same as previously described37, except that ChemScore-Kinase scoring function was used because its ability to recognize the activated CH groups (e.g., the CH group next to the nitrogen in a pyridine) for H-bonds. In addition, the soft potentials on Y36 were employed for TNIKclose to account for the flexibility of the tip of the lobe.
CoMSIA-SIMCA
A total of 54 type 1 kinase inhibitors were selected from Dataset II for CoMSIA-SIMCA modeling (Supplementary Table 1). The selected molecules were aligned by molecular docking to TNIKopen and TNIKclose. For each molecule, only the top-ranking pose that was similar to the one crystallized in the reference structures (root mean square deviation <3.0 Å and same H-bond pattern to the hinge) is kept for CoMSIA-SIMCA modeling. The CoMSIA fields (steric, electrostatic, hydrophobic, H-bond acceptor, H-bond donor) were computed using the parameters described above. These type 1 inhibitors were classified into four groups based on their respective pKd values (Table 3), and the dataset was divided into a training set (48 compounds) and a test set (6 compounds). The SIMCA model derived from the training set was validated by five-group cross-validation. The details of data splitting and predictions are available in Supplementary Table 5.
Experimental validation
We collected 1,448 FDA-approved small-molecule drugs from the DrugBank38. These molecules were docked to TNIKopen and TNIKclose using the same procedure described above, and the binding affinities to TNIK were predicted by CoMSIA-SIMCA model. Ten hits (Amodiaquine, Gefitinib, Sunitinib, Mebendazole, Dasatinib, Lapatinib, Imatinib, Sorafenib, Flavoxate, and Flavopiridol) that were predicted to be Category V, VI or VII were selected for experimental validation using KINOMEscan (DiscovRx, San Diego, CA) against seven kinases (TNIK, Activated CDC42 kinase 1 (ACK1), phosphatidylinositol–4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), 3-phosphoinositide dependent protein kinase-1 (PDPK1), Abelson tyrosine-protein kinase 2 (ABL2), Epidermal growth factor receptor (EGFR), Mitogen-activated protein kinase kinase (MEK)). Briefly, this KINOMEscan platform quantifies the amount of DNA-tagged kinase that is unable to bind the immobilized reference ligand after adding the testing ligand by qPCR. The selected compounds were tested at 0.1 μM and 10 μM. The results of KINOMEscan testing were reported as the percentage of the control (% Ctrl) where lower values suggest stronger hits: % Ctrl = (test compound signal − positive control signal)/(negative control signal − positive control signal).
Additional Information
How to cite this article: Tan, Z. et al. Comprehesnive Modeling and Discovery of Mebendazole as a Novel TRAF2- and NCK-interacting Kinase Inhibitor. Sci. Rep. 6, 33534; doi: 10.1038/srep33534 (2016).
Supplementary Material
Acknowledgments
We thank OpenEye and ChemAxon for the free academic licenses of their software packages. Special thanks to the support from Research Information Systems and Technology Services of M.D. Anderson Cancer Center along with their HPC resources as well as Texas Advanced Computing Center (TACC). We also thank Allison Chang for proof-reading the manuscript. S.Z. is an Endowed Faculty Scholar of MD Anderson Cancer Center. He is also partially supported by CPRIT DP150086, NSF CHE-1411859, NIH/NIGMS GM070737, and the University Cancer Foundation via the Institutional Research Grant program.
Footnotes
Author Contributions S.Z. is responsible for experimental design, project planning, and manuscript writing. Z.T. and L.C. performed all the experiments and wrote the manuscript. All authors read the manuscript and contributed to scientific discussion.
References
- Scholer-Dahirel A. et al. Maintenance of adenomatous polyposis coli (APC)-mutant colorectal cancer is dependent on Wnt/beta-catenin signaling. Proceedings of the National Academy of Sciences of the United States of America 108, 17135–17140, 10.1073/pnas.1104182108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus A. & Birchmeier W. Wnt signalling and its impact on development and cancer. Nature reviews. Cancer 8, 387–398, 10.1038/nrc2389 (2008). [DOI] [PubMed] [Google Scholar]
- Shitashige M. et al. Traf2- and Nck-interacting kinase is essential for Wnt signaling and colorectal cancer growth. Cancer Res 70, 5024–5033, 10.1158/0008-5472.CAN-10-0306 (2010). [DOI] [PubMed] [Google Scholar]
- Mahmoudi T. et al. The kinase TNIK is an essential activator of Wnt target genes. EMBO J 28, 3329–3340, 10.1038/emboj.2009.285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taira K. et al. The Traf2- and Nck-interacting kinase as a putative effector of Rap2 to regulate actin cytoskeleton. The Journal of biological chemistry 279, 49488–49496, 10.1074/jbc.M406370200 (2004). [DOI] [PubMed] [Google Scholar]
- Hussain N. K., Hsin H., Huganir R. L. & Sheng M. MINK and TNIK differentially act on Rap2-mediated signal transduction to regulate neuronal structure and AMPA receptor function. The Journal of neuroscience: the official journal of the Society for Neuroscience 30, 14786–14794, 10.1523/JNEUROSCI.4124-10.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui J., Yang B., Wu J. & Zhou X. Enormous influence of TNIK knockdown on intracellular signals and cell survival. Hum Cell 24, 121–126, 10.1007/s13577-011-0023-2 (2011). [DOI] [PubMed] [Google Scholar]
- Chen L. et al. From laptop to benchtop to bedside: structure-based drug design on protein targets. Curr Pharm Des 18, 1217–1239, CPD-EPUB-20120203-005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Yang P. L. & Gray N. S. Targeting cancer with small molecule kinase inhibitors. Nature reviews. Cancer 9, 28–39, 10.1038/nrc2559 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T., Shitashige M., Yokota K., Sawa M. & Moriyama H. TNIK inhibitor and the use (2010). [Google Scholar]
- Davis M. I. et al. Comprehensive analysis of kinase inhibitor selectivity. Nature biotechnology 29, 1046–1051, 10.1038/nbt.1990 (2011). [DOI] [PubMed] [Google Scholar]
- Hartmann J. T., Haap M., Kopp H. G. & Lipp H. P. Tyrosine kinase inhibitors - a review on pharmacology, metabolism and side effects. Current drug metabolism 10, 470–481 (2009). [DOI] [PubMed] [Google Scholar]
- Wold S. & SjÖStrÖM M. In Chemometrics: Theory and Application Vol. 52 ACS Symposium Series Ch. 12 , 243–282 (American Chemical Society, 1977). [Google Scholar]
- Fabian M. A. et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nature biotechnology 23, 329–336, 10.1038/nbt1068 (2005). [DOI] [PubMed] [Google Scholar]
- Golbraikh A. et al. Rational selection of training and test sets for the development of validated QSAR models. Journal of computer-aided molecular design 17, 241–253 (2003). [DOI] [PubMed] [Google Scholar]
- Liu Y. & Gray N. S. Rational design of inhibitors that bind to inactive kinase conformations. Nature chemical biology 2, 358–364, 10.1038/nchembio799 (2006). [DOI] [PubMed] [Google Scholar]
- Traxler P. & Furet P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol Ther 82, 195–206 (1999). [DOI] [PubMed] [Google Scholar]
- Du-Cuny L. et al. Computational modeling of novel inhibitors targeting the Akt pleckstrin homology domain. Bioorganic & medicinal chemistry 17, 6983–6992, 10.1016/j.bmc.2009.08.022 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Golbraikh A., Oloff S., Kohn H. & Tropsha A. A novel automated lazy learning QSAR (ALL-QSAR) approach: method development, applications, and virtual screening of chemical databases using validated ALL-QSAR models. J Chem Inf Model 46, 1984–1995, 10.1021/ci060132x (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. et al. Antitumor agents 252. Application of validated QSAR models to database mining: discovery of novel tylophorine derivatives as potential anticancer agents. Journal of computer-aided molecular design 21, 97–112, 10.1007/s10822-007-9102-6 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du-Cuny L., Chen L. & Zhang S. A critical assessment of combined ligand- and structure-based approaches to HERG channel blocker modeling. J Chem Inf Model 51, 2948–2960, 10.1021/ci200271d (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter S., Jensen B., Buttenschoen K., Kratzer W. & Kern P. Benzimidazoles in the treatment of alveolar echinococcosis: a comparative study and review of the literature. The Journal of antimicrobial chemotherapy 46, 451–456 (2000). [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay T., Sasaki J., Ramesh R. & Roth J. A. Mebendazole elicits a potent antitumor effect on human cancer cell lines both in vitro and in vivo. Clin Cancer Res 8, 2963–2969 (2002). [PubMed] [Google Scholar]
- Tan Z., Chaudhai R. & Zhang S. Polypharmacology in Drug Development: A Minireview of Current Technologies. ChemMedChem 11, 1211–1218, 10.1002/cmdc.201600067 (2016). [DOI] [PubMed] [Google Scholar]
- Reddy A. S., Tan Z. & Zhang S. Curation and analysis of multitargeting agents for polypharmacological modeling. J Chem Inf Model 54, 2536–2543, 10.1021/ci500092j (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martarelli D., Pompei P., Baldi C. & Mazzoni G. Mebendazole inhibits growth of human adrenocortical carcinoma cell lines implanted in nude mice. Cancer Chemother Pharmacol 61, 809–817, 10.1007/s00280-007-0538-0 (2008). [DOI] [PubMed] [Google Scholar]
- Sasaki J. et al. The anthelmintic drug mebendazole induces mitotic arrest and apoptosis by depolymerizing tubulin in non-small cell lung cancer cells. Mol Cancer Ther 1, 1201–1209 (2002). [PubMed] [Google Scholar]
- Doudican N., Rodriguez A., Osman I. & Orlow S. J. Mebendazole induces apoptosis via Bcl-2 inactivation in chemoresistant melanoma cells. Mol Cancer Res 6, 1308–1315, 10.1158/1541-7786.MCR-07-2159 (2008). [DOI] [PubMed] [Google Scholar]
- Bai R. Y., Staedtke V., Aprhys C. M., Gallia G. L. & Riggins G. J. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro-oncology 13, 974–982, 10.1093/neuonc/nor077 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell G. J., Gill J. H. & Lacey E. Binding of [3H]benzimidazole carbamates to mammalian brain tubulin and the mechanism of selective toxicity of the benzimidazole anthelmintics. Biochemical pharmacology 43, 1095–1100 (1992). [DOI] [PubMed] [Google Scholar]
- Bryceson A. D., Woestenborghs R., Michiels M. & van den Bossche H. Bioavailability and tolerability of mebendazole in patients with inoperable hydatid disease. Transactions of the Royal Society of Tropical Medicine and Hygiene 76, 563–564 (1982). [DOI] [PubMed] [Google Scholar]
- Fu C. A. et al. TNIK, a novel member of the germinal center kinase family that activates the c-Jun N-terminal kinase pathway and regulates the cytoskeleton. The Journal of biological chemistry 274, 30729–30737 (1999). [DOI] [PubMed] [Google Scholar]
- Zheng W. & Tropsha A. Novel variable selection quantitative structure--property relationship approach based on the k-nearest-neighbor principle. J Chem Inf Comput Sci 40, 185–194 (2000). [DOI] [PubMed] [Google Scholar]
- Namboodiri H. V. et al. Analysis of imatinib and sorafenib binding to p38alpha compared with c-Abl and b-Raf provides structural insights for understanding the selectivity of inhibitors targeting the DFG-out form of protein kinases. Biochemistry 49, 3611–3618, 10.1021/bi100070r (2010). [DOI] [PubMed] [Google Scholar]
- Verdonk M. L., Cole J. C., Hartshorn M. J., Murray C. W. & Taylor R. D. Improved protein–ligand docking using GOLD. Proteins: Structure, Function, and Bioinformatics 52, 609–623, 10.1002/prot.10465 (2003). [DOI] [PubMed] [Google Scholar]
- Simard J. R. et al. Fluorophore labeling of the glycine-rich loop as a method of identifying inhibitors that bind to active and inactive kinase conformations. J Am Chem Soc 132, 4152–4160, 10.1021/ja908083e (2010). [DOI] [PubMed] [Google Scholar]
- Chen L., Calin G. A. & Zhang S. Novel insights of structure-based modeling for RNA-targeted drug discovery. J Chem Inf Model 52, 2741–2753, 10.1021/ci300320t (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox C. et al. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic acids research 39, D1035–D1041, 10.1093/nar/gkq1126 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.