

Abstract
A series of 2- and 3-OH Nile red dyes was prepared in order to generate water-soluble probes that could be used to probe lipid binding to proteins. Various substitutions in positions 2-/3-, 6-, and 7-shifted wavelengths while maintaining the environmental sensitivity of Nile red. In order to increase the solubility of the dyes in aqueous solutions, we attached butyric acid groups to the 2- or 3-OH position. In addition, phenothiazine dyes, which exhibited particularly long excitation properties, were synthesized and tested for the first time. All dyes showed Stoke’s shifts of 70–100 nm and changes in excitation and emission of over 100 nm, depending on the hydrophobicity of the environment. Binding studies with bovine serum albumin and the non-specific lipid transfer protein SCP2 revealed emission changes of more than 30 nm upon binding to the protein and a five-fold increase in emission intensity. Titration of the dye-loaded proteins with various lipids or drugs replaced the dye and thereby reversed the shift in wavelength intensity. This allowed us to estimate the lipid binding affinity of the investigated proteins. For SCP2, isothermal calorimetry (ITC) data verified the titration experiments. NMR titration experiments of SCP2 with Nile red 2-O-butyric acid (1a) revealed that the dye is bound within the lipid binding pocket and competes with lipid ligands for this binding site. These results give valuable insight into lipid and drug transport by proteins outside and inside cells.
Keywords: Nile red, Environmentally sensitive dyes, Binding, Isothermal calorimetry, Bovine serum albumin, SCP2
Graphical abstract
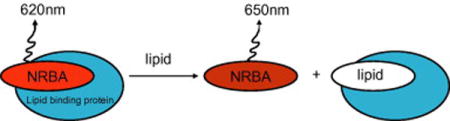
1. Introduction
The vast increase in bioinformatic information, in combination with solved protein structures, reveals great insight into protein domains, but does not permit determination of the biological function of the proteins. Among these domains, lipid-binding domains are of particular interest, because they are involved in many signal transduction events and the formation of protein complexes at membranes. Although many protein-binding domains have been used as fluorescently labeled reporters in living cells, it is still difficult to measure lipid–protein interaction in vitro. In addition, many proteins involved in drug transport, particularly albumins, feature domains that bind amphiphilic molecules like tryptophan or ibuprofen. Classical binding assays usually involve radioactively labeled drug candidates or lipids, allowing for estimates of protein–ligand interaction or investigations of fluorinated samples by NMR.1,2 Fluorescent methods simplify these tasks. In order to generate a fluorescent ligand that is as versatile as possible, we were looking for a lipophilic dye that is environmentally sensitive and would change its emission wavelength when switching from the aqueous environment to the relative hydrophobicity of a lipid-binding domain. One of the few dyes exhibiting these properties is Nile red, a dye traditionally used to stain lipid droplets in cells.3 Nile red is known to shift its emission wavelength over a large range of the visible spectra, giving shorter wavelength emission with increasing hydrophobicity of the environment.4 This property was used to study protein hydrophobicity in the past.5 However, Nile red has only a very limited solubility in water,4 potentially giving rise to unspecific binding or precipitation effects. We therefore decided to use more water-soluble hydroxy derivatives of Nile red that carry a charged extension. Hydroxy or O-alkyl groups in the 2- and 3-position shift the emission by 20 and 50 nm to longer wavelength, respectively. Such derivatives have previously been prepared by Briggs and co-workers as well as our own laboratory as fluorescent fatty acid derivatives.6–8 Here we describe the synthesis of novel phenoxazine and phenothiazine derivatives in the form of their butyric acid derivatives (Fig. 1). We report on the use of the new derivatives as probes to monitor lipid–albumin interaction. In addition, drugs like ibuprofen and natural ligands like tryptophan and lipids were investigated.
Figure 1.
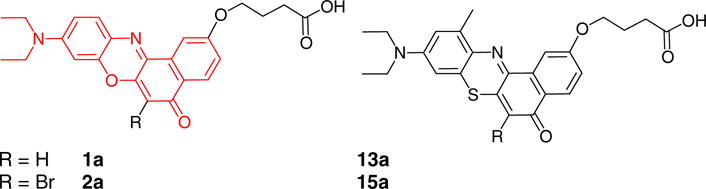
Structures of Nile red 2-O-butyric acid (1a, left) and phenothiazine 2-O-butyric acid (13a) and the respective 6-bromo derivatives 2a and 15a. The Nile red core is shown in red.
In order to demonstrate the accuracy and usefulness of the method, we investigated the non-specific lipid transfer protein SCP2 involved in organellar fatty acid metabolism.9 SCP2 is found in several intracellular locations, namely the peroxisome, cytosol, endoplasmic reticulum, and mitochondrion. Originally described as a sterol carrier protein, it was later determined that SCP2 in fact has a broad spectrum of lipid ligands, including fatty acids, fatty acyl-CoAs, phospholipids, sterols, and bile salts. The non-specific lipid binding activity of SCP2 is consistent with the numerous physiological functions ascribed to the protein, including cholesterol uptake and secretion, intracellular lipid trafficking, and activity in a variety of metabolic processes, for example fatty acid oxidation and esterification.9,10 Complex structures of SCP2 with both physiological and non-physiological lipids11,12 indicate some plasticity of the lipid binding pocket of SCP2.13.
In this study, we have used the mature peroxisomal form of human SCP2 (mSCP2) and high affinity acyl coenzyme A ligands to test the sensitivity of 1a as a probe for binding of physiological lipid ligands to an intracellular protein.13,14 It has previously been shown that SCP2 is functional as a long-chain fatty acyl CoA binding protein,15–17 with both saturated and unsaturated fatty acid chains of about 14–26 carbons being optimal ligands. Stearoyl and linoleoyl CoA contain C18:0 and C18:2Δ9,12 fatty acid chains, respectively. The competition assay using 1a, was verified by results from isothermal titration microcalorimetry (ITC), a direct measure of ligand binding. Furthermore, the interaction of 1a with mSCP2 was investigated by NMR to demonstrate that the binding sites of dye and lipid do in fact overlap.
2. Results
2.1. Synthesis
Environmentally sensitive dyes based on Nile red were prepared as functionalized derivatives to allow for the attachment of additional groups. The introduction of the butanoic acid moiety served to increase the solubility of the dyes in aqueous environment and to mimic the fatty acid character of common lipid components.
Compounds 7a and b were synthesized by reacting 5-diethylamino-2-nitrosophenol hydrochloride (5) and 1,6- or 1,7-dihydroxynaphthalene (6a and b), respectively, according to known procedures (Scheme 1).6 Alkylation of these compounds with trimethylsilylethyl protected bromobutyric acid was achieved at 80 °C. The protecting group of 8a and b was effectively removed with 12.5% trifluoroacetic acid in dichloromethane. The alkylation reaction was also performed with allyl protected bromobutyric acid (not shown). In this case, the protecting group was removed with tetrakistriphenylphosphine palladium. This route, however, was found to be lower yielding, particularly the deprotection step. In order to increase the excitation and emission wavelengths of the dyes, the protected butanoic acid derivatives 8a and b were brominated selectively at the 6-position using bromomethyl acetate in the presence of Huenig’s base to give compounds 9a and b. This method of bromination was much more selective and gave higher yields than the more standard reaction with bromine in chloroform. Deprotection as above gave the butanoic acid derivatives 2a and b.
Scheme 1.
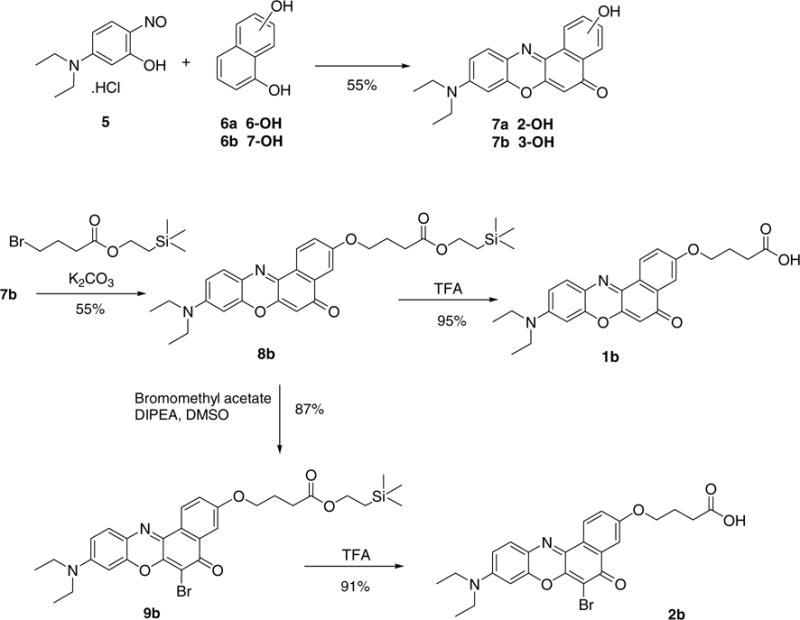
Synthesis of Nile red 3-O-butyric acid derivatives. The synthesis of 2-O-butyric acid derivatives worked accordingly.
The phenothiazine dyes were synthesized from 2-amino-5-diethylaminophenyl-thiosulfonic acid (10) which was prepared according to a previously reported procedure.18 Compound 10 was subsequently treated with an oxidizing agent, potassium dichromate, forming the semiquinonide ion in situ,19 which in turn was able to react with either 1,6- or 1,7-dihydroxynaphthalene (6a and b), respectively (Scheme 2). The resultant phenothiazine dyes (11a and b) were then alkylated at room temperature with trimethylsilylethanol protected bromobutyric acid. Bromination of the 6-position was undertaken as for the Nile red derivatives. Cleavage of the protecting group from 12a and b and 14a and b was effected with trifluoroacetic acid as described for the Nile red dyes above.
Scheme 2.
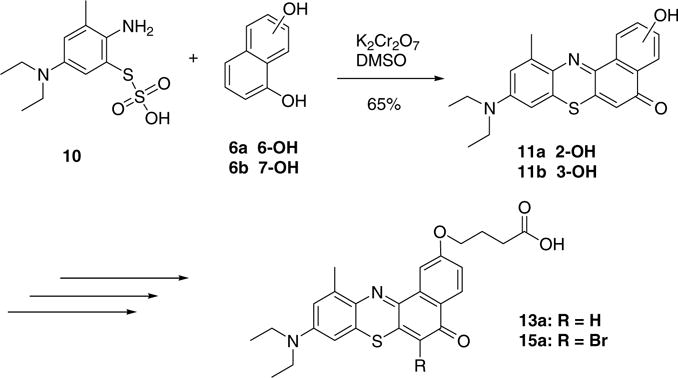
Synthesis of phenothiazine butyric acid derivatives.
2.2. Lipid binding assays
All Nile red dyes coupled to butyric acid exhibited good solubility (>100 μM) and exhibited a blue color in water and standard buffers. Phenothiazine dyes were less soluble by about a factor of 10. Upon addition of BSA the color changed to purple (Fig. 2), indicating a change in the dye’s environment. Titration of a 250 nM solution of dye with increasing concentrations of protein resulted in an emission shift from 657 to 620 nm for 1a and a five-fold increase in emission intensity with an EC50 of about 1.5 μM (Fig. 3A). All other dyes 2–6 gave similar shifts and fluorescence increases (Fig. 3B and Table S1 in the Supplement Material), but 1a provided the largest shift in the emission maxima. This shift was very similar when mSCP2 was added to a solution of 1a: free 1a gave a relatively weak maximum at ~657 nm, shifting to an approximately five-fold higher maximum at ~618 nm when mSCP2 was added. The EC50 was about 8 μM (Fig. 4A). As before, an isosbestic point could not be observed due to the strong emission increase at 618 nm.
Figure 2.

An aqueous solution of 1a before (left) and after addition of bovine serum albumin.
Figure 3.
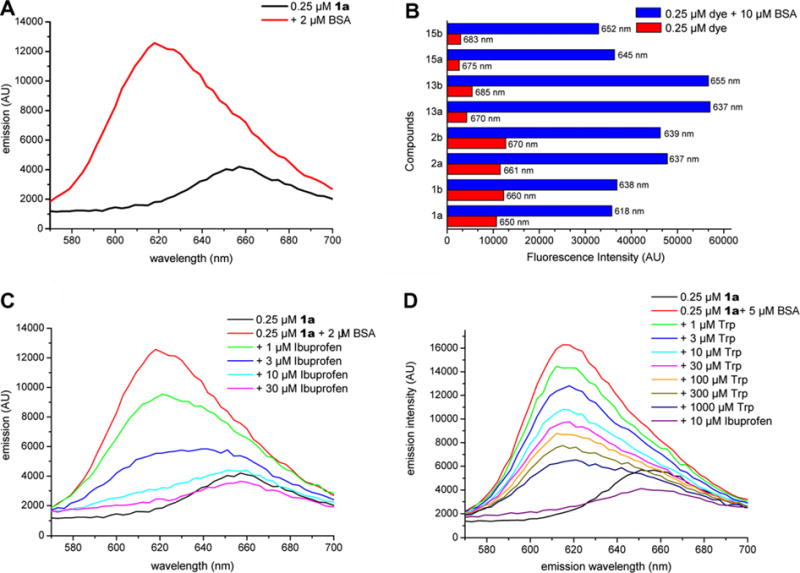
The curves shown in (A) reflect the color change shown in Figure 2 after addition of BSA. (B) Shift values and fluorescence increases of all tested dyes. (C) Titration of 1a loaded BSA with ibuprofen (Ibu). (D) A similar experiment with tryptophan (Trp) replaces only about half of the dye, even at very high concentrations. Subsequently, a small amount of ibuprofen releases all of the dye. Data are uncorrected for dilution effects due to the addition of ligands.
Figure 4.
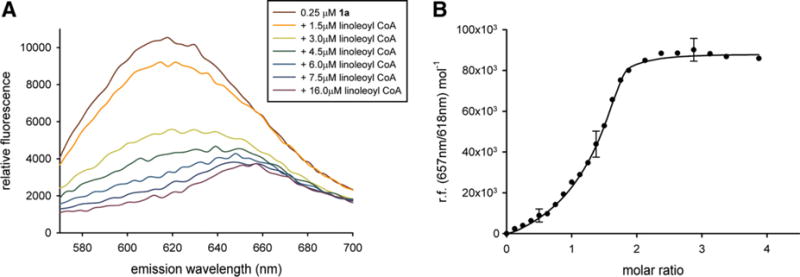
(A) Representative data showing the fluorescence response of mSCP2 preincubated with NRBA (1a) upon titration with the high-affinity physiological ligand, linoleoyl CoA. About 8 μM mSCP2 was incubated with 250 nM 1a and then titrated with linoleoyl CoA. The excitation wavelength was 540 nm. (B) Representative non-linear least squares fit to the fluorescence competition data in (A). The molar ratio describes relative amounts of linoleoyl CoA over mSCP2.
Dye-loaded BSA (2 μM protein, 250 nM dye) was then exposed to increasing concentrations of compound in order to replace the dye and thereby reverse the fluorescent shift. Titration with ibuprofen gave a complete reversal of the emission shift from 620 to 657 nm (Fig. 3C) with an apparent IC50 of about 3 μM. In contrast, similar experiments with tryptophan, one of the natural ligands of albumins, only reversed fluorescence by little more than 50%. This is in accordance with the fact that albumins have at least two lipid binding sites, of which only one binds tryptophan with high affinity.2 Figure 3D shows the dose-dependent replacement of the dye. Control experiments with lysozyme, a protein lacking a lipid-binding domain, did not result in fluorescence changes other than those from dilution effects when protein was added (data not shown).
In order to show that the method is applicable to intracellular proteins we turned to the well-characterized lipid transfer protein, mSCP2, which is known to bind fatty acyl-CoAs. Titration of 1a-preincubated mSCP2 with linoleoyl CoA showed a strong shift in the fluorescence maximum from 618 nm to 657 nm, suggesting release of 1a (Fig. 4A). Maximal free 1a was found at linoleoyl CoA concentrations roughly equimolar to mSCP2, indicating that linoleoyl CoA very efficiently competes with 1a for the lipid binding pocket of mSCP2.
A simple model was employed to fit the binding data (see Section 4), in which the rate of change in fluorescence ratio gives an estimate of the apparent dissociation constant (Kd) and the midpoint of the binding curve gives an estimate of stoichiometry. The data were best fit if two independent linoleoyl CoA binding sites were assumed in mSCP2 (Fig. 4B), as has previously been demonstrated for several long-chain fatty acid CoAs.20,21 Site 1 was estimated to have a higher occupancy (n ~ 0.73) and stronger binding properties (Kd ~ 339 nM) than Site 2 (n ~ 0.59, Kd ~ 947 nM), as shown in Table 1.
Table 1.
Summary of the binding parameters derived from fluorescence competition (1a) and ITC experiments
| n | Ka (M−1) | ΔH (kJ/mol) | TΔS (kJ/mol) | ΔG (kJ/mol) | Kd (nM) | |
|---|---|---|---|---|---|---|
| Site 1, ITC | 0.83 ± 0.05 | 1.127 × 107 ± 3.843 × 106 | −28.3 ± 0.5 | 13.3 | −41.6 | 89 ± 30 |
| Site 2, ITC | 0.74 ± 0.03 | 7.261 × 105 ± 1.009 × 105 | −17.7 ± 1.5 | 16.9 | −34.6 | 1377 ± 191 |
| Site 1, 1a | 0.73 ± 0.17 | 2.946 × 106 ± 1.101 × 106 | na | na | na | 339 ± 127 |
| Site 2, 1a | 0.59 ± 0.21 | 1.056 × 106 ± 6.074 × 105 | na | na | na | 947 ± 542 |
n, stoichiometry; Ka, per molar equilibrium association constant; ΔH, enthalpy change; TΔS, entropy change at 35 °C; ΔG, Gibbs free energy; Kd, molar equilibrium dissociation constant.
2.3. Calorimetric data
To verify these binding parameters as measured by competition with 1a, we employed a more direct binding assay, ITC. ITC has frequently been used to measure the equilibrium thermodynamics of lipid-protein interactions, for example fatty acid binding to FABP,22 although data quality may be limited by the poor solubility of lipid ligands, requiring them to be dissolved in organic solvents or prepared as salt derivatives at relatively high pH. These conditions can lead to very high ligand heats of dilution, which additionally may not be constant as a function of total lipid concentration.23 However, linoleoyl CoA was sufficiently soluble in aqueous solutions to circumvent the need for buffer/solvent mixing during the titration. As shown in Figure 5A (upper panel) while the endothermic ligand heat of dilution was not perfectly constant with changing concentration, it was relatively low compared to the exothermic heat of binding and could be subtracted for the binding data. ITC data were also best fit by assuming two independent binding sites (Fig. 5B, lower panel and Table 1). One relatively strong and one relatively weak binding site were again observed. Stoichiometries and dissociation constants were within a very similar range to those defined by the fluorescence assay, although Site 1 measured by ITC was found to have ~4-fold higher affinity than the corresponding site measured by fluorescence. This may be explained by the energy required to displace 1a in the competition assay or possibly by an energetic penalty due to unfavorable solvation of 1a. As determined by ITC, binding to both sites is enthalpically and entropically favorable.
Figure 5.
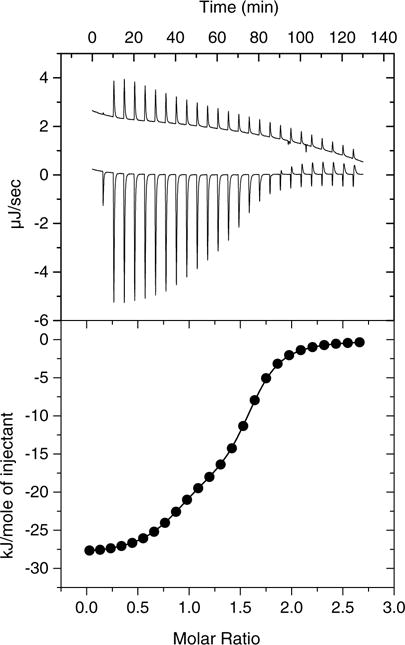
Representative ITC data and fit. Upper panel: Raw data for 50 μM mSCP2 titrated with linoleoyl CoA; the linoleoyl CoA heat of dilution (upper trace) is offset by 2 μJ/s for clarity. Lower panel: The integrated and background subtracted data showing a non-linear least squares fit to the ‘two sets of sites’ algorithm in MicroCal Origin 5.0. Please, note the similarity of this fit with the one depicted in Figure 4B. The molar ratio describes relative amounts of linoleoyl CoA over mSCP2.
Previous studies using fluorescence competition (for the intrinsically fluorescent parinaroyl CoA17) and competition coupled to FRET of pyrene dodecanoic acid and the single tryptophan in mSCP2 had determined Kd values of 2–4 nM and about 200–400 nM, respectively, for a variety of long-chain fatty acyl CoAs.13,15,24 In each of the assays described, a single lipid binding pocket was assumed. Broadly speaking, Kd values presented here are within the same range as those previously presented,13,15,24 given the different experimental specifications and ligands studied. Further, our data support the suggestion that mSCP2 has not one but two very long-chain fatty acyl CoA binding sites.13,20,21 Finally, the direct and high-fidelity binding assay, ITC, strongly supports the data obtained from our competition assay with 1a. The standard deviations from assays with 1a are rather higher than those obtained from ITC but can be considered tolerable for a relatively quick, easy, and more conservative (in terms of reagent consumption) assay.
2.4. Localization of dyes in the protein
In order to locate the binding of 1a to mSCP2, we mapped the binding site using NMR and compared it to the binding site of physiological lipid ligands. The lipid interaction of mSCP2 is analyzed by NMR using the spin-labeled paramagnetic fatty acid derivative 5-doxyl stearic acid (5DSA).14,25 The presence of 5DSA results in reduced intensities of amide protons at the inner side of the a-helices that frame the lipid binding pocket, defining this as the binding site for 5DSA. NMR titration experiments of mSCP2 with 1a reveal that the observed chemical shift perturbations in 1H, 15N-correlation spectra affect similar residues as those bleached by 5DSA (Fig. 6A–D). Similar chemical shift perturbations in mSCP2 are observed using physiological ligands, such as stearoyl CoA or linoleoyl CoA.13 Taken together, this demonstrates that 1a and physiological lipids utilize the same binding site and therefore can compete for binding. We note, that the region mapped by the NMR data would be compatible with a second lower affinity binding site. The overlapping binding sites are further corroborated by competitive binding of 1a with lipid ligands. We find in sequential NMR titration experiments that the interaction of a spin-labeled fatty acid can be competed by 1a with a higher affinity ligand. If the mSCP2–5DSA complex is incubated with a slight excess of 1a, reduction in the paramagnetic relaxation enhancement is almost completely lost (Supplementary Fig. S2). Moreover, identical NMR spectra are observed after addition of stearoyl CoA to a saturated 1a-mSCP2 complex resulting in the same spectrum as when mSCP2 was saturated solely with stearoyl CoA (Fig. 6E and F).
Figure 6.
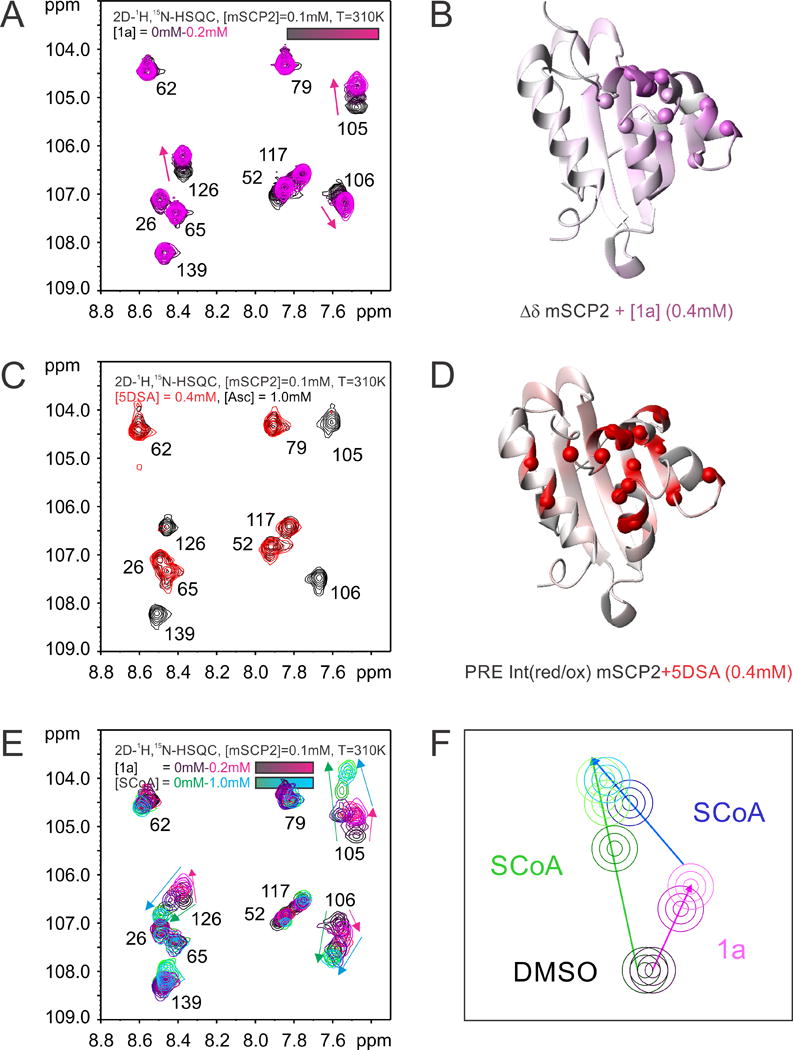
NMR titration experiments and ligand binding mapping of NRBA (1a) and 5DSA to mSCP2. (A) 1H, 15N-correlation spectra of mSCP upon titration with 1a. Protein and ligand concentrations are indicated. (B) The chemical shift perturbations are mapped at saturation with 1a onto the peptide backbone of mSCP2. Amide protons, which are affected by the addition of 1a are indicated as white to purple spheres with increasing chemical shift perturbation. (C) 1H,15N-correlation spectra of mSCP2 in the presence of the oxidized (red) and reduced (black, after addition of ascorbic acid) spin labeled lipid molecule (5-doxylstearic acid, 5DSA). With the oxidized 5DSA bound, peak intensities are reduced due to paramagnetic relaxation enhancement (PRE). (D) The PRE effects are mapped onto the structure of mSCP2. (E) Competitive binding of 1a and Stearoyl-CoA (SCoA) to mSCP2. Chemical shift changes observed upon addition of 1a and SCoA are colored in magenta and green-blue, respectively. (F) Schematic representation of the competition binding experiment.
3. Conclusions
Nile red and phenothiazine dyes in a water-soluble butyric acid form are readily accessible using standard chemical procedures. For the regioselective introduction of bromine in the 6-position, acetoxymethyl bromide was used for the first time. Due to the 30-nm blue shift in the emission (and excitation) wavelength and the large increase in fluorescence when entering a hydrophobic environment, Nile red and its derivatives are very suitable for quickly and cost-efficiently probing lipid binding sites in proteins. We addressed the question of whether competition assays using this class of dyes would provide data of sufficient quality to allow estimates of binding constants, applicable to drug screening and pharmacokinetics studies. Isothermal titration calorimetry was therefore employed to verify the results. The estimated Kd for the binding of linoleoyl CoA to mSCP2 was sufficiently similar by both methods to justify the use of the Nile red based assays in the future. Finally, to prove that NRBA is binding to the same site that is usually occupied by the fatty acid ligand or spin-labeled fatty acid derivatives, we investigated the binding of NRBA in mSCP2 by NMR. This revealed that 1a is a true competitor for this site. Since the latter is fairly hydrophobic, the large shift in fluorescence of 1a upon binding is easily explained. Future competition experiments with 1a or the longer wavelength derivatives may be a convenient method to determine the specificity of lipid binding pockets for their natural ligand(s).
4. Experimental
Mass spectra were recorded using a Jeol JMS 700 mass spectrometer. For chromatographic analysis a Waters Breeze HPLC system was used with a Kromosil RP18 column (10 μm, 250 × 4 mm). The purity of final compounds was assessed as being >95% by HPLC under two conditions. Method 1: 40 min isocratic (75/25 methanol/water containing 0.1% TFA, flow rate 1 ml/min) at room temperature, UV detection was taken at 280 nm. Method 2: 40 min isocratic (50/50 acetonitrile/water containing 0.1% TFA, flow rate 1 ml/min) at room temperature, UV detection was taken at 254 nm. Method 3: 40 min isocratic (60/40 acetonitrile/water containing 0.05% TFA, flow rate 1 ml/min) at room temperature, UV detection was taken at 254 nm. Columns were flushed after each run to check for more lipophilic by-products. 1H and 13C NMR spectra were obtained on a Bruker UltraShield™ Advance 400 (400 MHz, 1H; 100 MHz, 13C) spectrometer and are reported as parts per million downfield from Me4Si with the number of protons, multiplicities and coupling constants in hertz (Hz) indicated parenthetically.
Protein 2D 1H, 15N-correlation spectra were recorded at 310 K on a Bruker DRX600 spectrometer equipped with a cryogenic probe. Chemical shift perturbations (Δδ = [(Δδ1H)2 + (1/5Δδ15N)2]1/2, in parts per million) were monitored in 2D sensitivity enhanced 1H, 15N-HSQC experiments with 128 and 512 complex points in t1 and t2 using 16 scans per increment. Spin-label induced paramagnetic relaxation enhancements were analyzed from intensity changes in 1H,15N-HSQC experiments of mSCP2 recorded in the presence of the fatty acid derivative 5-doxyl stearic acid (5DSA) oxidized and after reduction with ascorbic acid.
Fluorescence spectra were recorded with a Quantamaster QM4/2000SE (Photon Technology International, The Netherlands) in 50 μl quartz cuvettes. Isothermal calorimetry was performed on a MicroCal VP-ITC (MicroCal, Milton Keynes, UK). All reagents and anhydrous solvents were purchased from Sigma–Aldrich, Germany, at the highest available purity. Linoleoyl CoA was purchased at 98% purity from Larodan Fine Chemicals (Malmö, Sweden). General solvents were purchased from Acros, Germany.
4.1. General procedure 1: Alkylation of dyes
The desired hydroxyphenoxazin or hydroxyphenothiazin derivative (1 eq), 4-bromobutyric acid 2-trimethylsilanyl-ethyl ester (1.5–2 eq), and potassium carbonate (3–5 eq) were dissolved in anhydrous DMF. The solutions were heated at 80 °C for 7 h. Water was then added and the solutions were extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residues were further purified by column chromatography [methanol/dichloromethane (1:99)] to afford the corresponding alkylated products.
4.2. General procedure 2: Deprotection of acid groups
Trimethylsilanyl-ethyl ester derivatives were dissolved in dichloromethane (40 ml) and cooled to 0 °C in an ice bath. Trifluoroacetic acid (5 ml) was added dropwise and the solutions subsequently stirred at RT for 18 h. The solvent was removed under reduced pressure (no heat) and the residue further dried under vacuum for 2 h. The residues were further purified by column chromatography to give the corresponding free butyric acid derivatives.
4.3. General procedure 3: Bromination of dyes
Hydroxyphenoxazin or hydroxyphenothiazin derivative (1 eq), respectively, was dissolved in anhydrous dimethylsulfoxide (5 ml). N-Ethyldiisopropylamine (2 eq) was added, followed by bromomethyl acetate (2 eq). The solutions were then stirred at RT for 1.5 h. After the addition of brine, the solutions were extracted three times with ethyl acetate. The combined organic fractions were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was further purified by column chromatography [methanol/dichloromethane (2:98)] to give the corresponding mono-brominated derivatives.
4.4. General procedure 4: Dye formation
Thiosulfuric acid S-(2-amino-5-diethylamino-3-methylphenyl) ester 10 (1 eq) and 1,6- or 1,7-dihydroxynaphthalene (1eq) were dissolved in anhydrous dimethylsulfoxide (10 ml) and stirred at RT for 45 min. Potassium dichromate (1 eq) was then added and the reaction mixture stirred for a further 20 min. Subsequently, a mixture of methanol (200 ml) and 1 N HCl (20 ml) was added and the solution was stirred for a further 1 h at RT. The excess methanol was then removed under reduced pressure and aq sodium bicarbonate was added to the residue in dimethylsulfoxide. The mixture was sealed and left at RT for 18 h. In some cases, the desired product precipitated and could be removed by filtration. If no precipitate formed, this mixture could be extracted with ethyl acetate and the organic layers were washed with brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to afford the desired product.
4.4.1. 4-(9-Diethylamino-5-oxo-5H-benzo[a]phenoxazin-2-yloxy)-butyric acid allyl ester (8a)
9-Diethylamino-2-hydroxy-benzo[a]phenoxazin-5-one 7a (1.50 g, 4.5 mmol), 4-bromobutyric acid allyl ester (1.40 g, 6.8 mmol), and potassium carbonate (3.60 g, 26.0 mmol) were added in anhydrous DMF (75 ml). The solution was heated at gentle reflux for 8 h. Brine was then added and the solution extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was further purified by column chromatography (dichloromethane) to give compound 8a (0.89 g, 43%). 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 8.6, 1H), 8.02 (d, J = 2.6, 1H), 7.58 (d, J = 9.1, 1H), 7.14 (dd, J = 8.6, 2.6; 1H), 6.64 (dd, J = 9.1, 2.6; 1H), 6.43 (d, J = 2.6, 1H), 6.28 (s, 1H), 5.95 (ddt, J = 17.3, 10.4, 5.8; 1H), 5.34 (dd, J = 17.3, 1.3; 1H), 5.25 (dd, J = 10.4,1.3;1H), 4.63 (d, J = 5.8, 2H), 4.22 (t, J = 5.9, 2H), 3.46 (q, J = 7.1, 4H), 2.64 (t, J = 7.3, 2H), 2.22 (qi, J = 6.7, 2H), 1.26 (t, J = 7.1, 6H); MS (FAB+) m/z: 461 (80%); HRMS (FAB+) m/z: Calcd 461.2076. Found: 461.2072.
4.4.2. 4-(9-Diethylamino-5-oxo-5H-benzo[a]phenoxazin-3-yloxy)-butyric acid 2-trimethylsilanyl-ethyl ester (8b)
9-Diethylamino-3-hydroxy-benzo[a]phenoxazin-5-one 7b (0.58 g, 1.7 mmol), 4-bromo-butyric acid 2-trimethylsilanyl-ethyl ester (0.77 g, 2.9 mmol), and potassium carbonate (1.0 g, 7.2 mmol) were reacted according to general procedure 1, affording compound 8b (0.50 g, 55%). 1H NMR (400 MHz, CDCl3) δ 8.58 (d, J = 8.8, 1H),7.73 (d, J = 2.7, 1H), 7.62 (d, J = 9.1, 1H), 7.28 (dd, J = 8.8,2.7;1H), 6.71 (dd, J = 9.1,2.4; 1H), 6.52 (d, J = 2.4, 1H),6.41 (s, 1H), 4.27–4.15 (m, 2H+2H),3.49 (q, J = 7.0, 4H), 2.55 (t, J = 7.3, 2H), 2.19 (qi, J = 6.7, 2H), 1.28 (t, J = 7.0, 6H), 1.02 (t, J = 8.7, 2H), 0.06 (s, 9H); MS (FAB+) m/z: 521 (60%); HRMS (FAB+) m/z: Calcd 521.2472. Found: 521.2488.
4.4.3. 4-(9-Diethylamino-5-oxo-5H-benzo[a]phenoxazin-2-yloxy)-butyric acid (1a)
Compound 8a (0.74 g, 1.6 mmol), diethylamine (0.35 g, 0.5 ml, 4.8 mmol), and tetrakis(triphenylphosphine)palladium (0.74 g, 0.64 mmol) were added together in anhydrous acetonitrile (50 ml). The solution was stirred at RT for 18 h. The solvent was removed under reduced pressure and the residue was further purified by column chromatography [methanol/dichloromethane/acetic acid (5:95:0.01)] to yield compound 1a (0.51 g, 76%). 1H NMR (400 MHz, DMSO-d6) δ 12.20 (v br s, 1H), 8.04 (d, J = 8.6, 1H), 7.94 (d, J = 1.8, 1H), 7.64 (d, J = 9.1, 1H), 7.26 (dd, J = 8.6, 1.8; 1H), 6.82 (dd, J = 9.1, 2.3, 1H), 6.66 (d, J = 2.3, 1H), 6.20 (s, 1H), 4.19 (t, J = 5.8, 2H), 3.51 (q, J = 6.9, 4H), 2.44 (t, J = 7.2, 2H), 2.03 (qi, J = 6.5, 2H), 1.17 (t, J = 6.9, 6H); MS (FAB+) m/z: 421 (20%); HRMS (FAB+) m/z: Calcd 421.1763. Found: 421.1780; Degree of purity: HPLC method 1, retention time of 7.8 min, 97.4%; method 2, retention time of 8.7 min, 95.2%.
4.4.4. 4-(9-Diethylamino-5-oxo-5H-benzo[a]phenoxazin-3-yloxy)-butyric acid (1b)
Compound 8b (0.28 g, 0.5 mmol) was treated with trifluoroacetic acid as described in general procedure 2, and purified by column chromatography [methanol/dichloromethane/acetic acid (15:85:0.01)] to give compound 1b (0.22 g, 95%). 1H NMR (400 MHz, DMSO-d6) δ 8.49 (d, J = 9.0, 1H), 7.62 (d, J = 9.4, 1H), 7.54 (d, J = 2.8, 1H), 7.39 (dd, J = 9.0, 2.8; 1H), 6.84 (dd, J = 9.4, 2.7; 1H), 6.68 (d, J = 2.7, 1H), 6.31 (s, 1H), 4.15 (t, J = 6.4, 2H), 3.50 (q, J = 7.1, 4H), 2.40 (t, J = 7.2, 2H), 2.00 (qi, J = 7.0, 2H), 1.17 (t, J = 7.1, 6H); MS (FAB+) m/z: 421 (60%); HRMS (FAB+) m/z: Calcd 421.1763. Found: 421.1770; Degree of purity: HPLC method 1, retention time of 9.2 min, 95.1%; method 2, retention time of 10.7 min, 95.3%.
4.4.5. 4-(6-Bromo-9-diethylamino-5-oxo-5H-benzo[a]phenoxazin-2-yloxy)-butyric acid allyl ester (9a)
Compound 8a (0.08 g, 0.17 mmol) was dissolved in anhydrous dimethylsulfoxide (5 ml) and reacted with N-ethyldiisopropylamine (0.05 g, 0.4 mmol) and bromomethylacetate (0.05 g, 0.4 mmol) according to general procedure 3 to give compound 9a (0.07 g, 78%). 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 8.8, 1H), 8.03 (d, J = 2.6, 1H), 7.62 (d, J = 9.3, 1H), 7.15 (dd, J = 8.8, 2.6;1H), 6.72 (dd, J = 9.3,2.5; 1H), 6.58 (d, J = 2.5, 1H), 5.95 (ddt, J = 17.2,10.4, 5.6; 1H), 5.35 (dd, J = 17.2, 1.3; 1H), 5.26 (dd, J = 10.4, 1.3; 1H),4.64 (d, J = 5.6, 2H), 4.25 (t, J = 6.1, 2H), 3.51 (q, J = 7.0, 4H), 2.65 (t, J = 7.2, 2H), 2.24 (qi, J = 6.4, 2H), 1.30 (t, J = 7.0, 6H); MS (FAB+) m/z: 541 (100%), 539 (100%).
4.4.6. 4-(6-Bromo-9-diethylamino-5-oxo-5H-benzo[a]phenoxazin-3-yloxy)-butyric acid 2-trimethylsilanyl-ethyl ester (9b)
Compound 8b (0.08 g, 0.15 mmol) was dissolved in dimethylsulfoxide (5 ml), and reacted with N-ethyldiisopropylamine (0.04 g, 0.3 mmol) and bromomethylacetate (0.05 g, 0.3 mmol) according to general procedure 3, to give compound 9b (0.08 g, 87%). 1H NMR (400 MHz, CDCl3) δ 8.62 (d, J = 8.8, 1H), 7.82 (d, J = 2.6, 1H), 7.71 (d, J = 9.1, 1H), 7.31 (dd, J = 8.8, 2.6; 1H), 6.86 (dd, J = 9.1, 2.2; 1H), 6.76 (d, J = 2.2, 1H), 4.26−4.18 (m, 2H + 2H), 3.53 (q, J = 7.2, 4H), 2.57 (t, J = 7.3, 2H), 2.20 (qi, J = 6.6, 2H), 1.32 (t, J = 7.2, 6H), 1.02 (t, J = 8.6, 2H), 0.07 (s, 9H); MS (FAB+) m/z: 601 (40%), 599 (40%); HRMS (FAB+) m/z: Calcd 599.1577. Found 599.1504.
4.4.7. 4-(6-Bromo-9-diethylamino-5-oxo-5H-benzo[a]phenoxazin-2-yloxy)-butyric acid (2a)
Compound 9a (0.07 g, 0.14 mmol), diethylamine (0.04 g, 0.5 mmol) and tetrakis(triphenylphosphine)palladium (0.05 g, 0.04 mmol) were added together in anhydrous dimethylformamide (10 ml). The solution was stirred at RT for 18 h. The solvent was removed under reduced pressure and the residue was further purified by column chromatography [methanol/dichloromethane/acetic acid (6:94:0.01)] to give compound 2a (0.04 g, 51%). 1H NMR (400 MHz, DMSO-d6) δ 12.14 (v br s, 1H), 8.11 (d, J = 8.6, 1H), 7.95 (d, J = 2.4, 1H), 7.70 (d, J = 9.0, 1H), 7.28 (dd, J = 8.6, 2.4; 1H), 6.91 (dd, J = 9.0, 2.2; 1H), 6.70 (d, J = 2.2, 1H), 4.24−4.17 (m, 2H), 3.55 (q, J = 6.9, 4H), 2.46 (t, J = 7.2, 2H), 2.03 (qi, J = 6.6, 2H), 1.19 (t, J = 6.9, 6H); MS (FAB+) m/z: 501 (25%), 499 (25%); HRMS (FAB+) m/z: Calcd 499.0868. Found: 499.0911; Degree of purity: HPLC method 1, retention time of 11.8 min, 96.4%; method 2, retention time of 16.9 min, 95.2%.
4.4.8. 4-(6-Bromo-9-diethylamino-5-oxo-5H-benzo[a]phenoxazin-3-yloxy)-butyric acid (2b)
Compound 9b (0.08 g, 0.13 mmol) was treated with trifluoroacetic acid as described in general procedure 2. The resultant residue was purified by column chromatography [methanol/dichloromethane/acetic acid (7:93:0.01)] to give compound 2b (0.06 g, 91%). 1H NMR (400 MHz, DMSO-d6) δ 12.21 (v br s, 1H), 8.49 (d, J = 9.0, 1H), 7.67 (d, J = 9.2, 1H), 7.60 (d, J = 2.5, 1H), 7.42 (dd, J = 9.0, 2.5; 1H), 6.93 (dd, J = 9.2, 2.5; 1H), 6.71 (d, J = 2.5, 1H), 4.16 (t, J = 6.4, 2H), 3.54 (q, J = 6.9, 4H), 2.44 (t, J = 7.2, 2H), 2.02 (qi, J = 6.8, 2H), 1.19 (t, J = 6.9, 6H); MS (FAB+) m/z: 499 (25%), 501 (25%); HRMS (FAB+) m/z: Calcd 499.0868. Found: 499.0849; Degree of purity: HPLC method 1, retention time of 14.7 min, 95.1%; method 2, retention time of 21.7 min, 95.3%.
4.4.9. 9-Diethylamino-2-hydroxy-11-methyl-benzo[a]phenothiazin-5-one (11a)
Compound 10 (0.50 g, 1.7 mmol) and 1,6 dihydroxynaphthalene (0.27 g, 1.7 mmol) were reacted with potassium dichromate (0.50 g, 1.7 mmol) as described in general procedure 4. The mixture was extracted with ethyl acetate and the organic layers further washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford compound 11a (0.42 g, 68 %). 1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.06 (d, J = 2.4, 1H), 7.96 (d, J = 8.6, 1H), 7.08 (dd, J = 8.6, 2.4; 1H), 6.84 (d, J = 2.5, 1H), 6.74 (d, J = 2.5, 1H), 6.64 (s, 1H), 3.49 (q, J = 7.0, 4H), 2.66 (s, 3H), 1.16 (t, J = 7.0, 6H); MS (FAB+) m/z: 365 (70%); HRMS (FAB+) m/z: Calcd 365.1324. Found: 365.1309.
4.4.10. 9-Diethylamino-3-hydroxy-11-methyl-benzo[a]phenothiazin-5-one (11b)
Compound 10 (0.50 g, 1.7 mmol) and 1,7 dihydroxynaphthalene (0.28 g, 1.7 mmol) were reacted with potassium dichromate (0.51 g, 1.7 mmol) as described in general procedure 4. The resultant precipitate was removed by filtration, washed with water and dried under high vacuum to yield compound 11b (0.46 g, 73 %). 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 8.59 (d, J = 8.8, 1H), 7.44 (d, J = 2.7, 1H), 7.22 (dd, J = 8.8, 2.7; 1H), 6.85 (d, J = 2.6, 1H), 6.75 (s, 1H), 6.74 (d, J = 2.6, 1H), 3.47 (q, J = 7.0, 4H), 2.64 (s, 3H), 1.15 (t, J = 7.0, 6H); MS (FAB+) m/z: 365 (20%); HRMS (FAB+) m/z: Calcd 365.1324. Found: 365.1306.
4.4.11. 4-(9-Diethylamino-11-methyl-5-oxo-5H-benzo[a]phenothiazin-2-yloxy)-butyric acid 2-trimethylsilanyl-ethyl ester (12a)
Compound 11a (0.39 g, 1.1 mmol), 4-bromo-butyric acid 2-trimethylsilanyl-ethyl ester (0.45 g, 1.7 mmol), and potassium carbonate (0.44 g, 3.2 mmol) were reacted according to general procedure 1 to give compound 12a (0.30 g, 51%). 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 8.7, 1H), 8.17 (d, J = 2.4, 1H), 7.15 (dd, J = 8.7, 2.4; 1H), 6.72 (s, 1H), 6.63 (s, 1H), 6.39 (d, J = 2.0, 1H), 4.25−4.17 (m, 4H), 3.44 (q, J = 7.2, 4H), 2.69 (s, 3H), 2.59 (t, J = 7.3, 2H), 2.22 (qi, J = 6.4, 2H), 1.26 (t, J = 7.2, 6H), 1.01 (t, J = 8.5, 2H), 0.05 (s, 9H); MS (FAB+) m/z: 551 (90%); HRMS (FAB+) m/z: Calcd 551.2400. Found: 551.2364.
4.4.12. 4-(9-Diethylamino-11-methyl-5-oxo-5H-benzo[a]phenothiazin-3-yloxy)-butyric acid 2-trimethylsilanyl-ethyl ester (12b)
Compound 11b (0.29 g, 0.8 mmol), 4-bromo-butyric acid 2-trimethylsilanyl-ethyl ester (0.40 g, 1.5 mmol) and potassium carbonate (0.33 g, 2.3 mmol) were reacted according to general procedure 1 to give compound 12b (0.22 g, 52%). 1H NMR (400 MHz, Acetone-d6) δ 8.69 (d, J = 8.8, 1H), 7.61 (d, J = 2.5, 1H), 7.31 (dd, J = 8.8, 2.5; 1H), 6.84 (d, J = 2.8, 1H), 6.67 (d, J = 2.8, 1H), 6.65 (s, 1H), 4.24−4.16 (m, 4H), 3.54 (q, J = 6.9, 4H), 2.67 (s, 3H), 2.57 (t, J = 7.2, 2H), 2.17 (qi, J = 6.8, 2H), 1.25 (t, J = 6.9, 6H), 1.02 (t, J = 8.3, 2H), 0.07 (s, 9H); MS (FAB+) m/z: 551 (100%); HRMS (FAB+) m/z: Calcd 551.2400. Found: 551.2368.
4.4.13. 4-(9-Diethylamino-11-methyl-5-oxo-5H-benzo[a]phenothiazin-2-yloxy)-butyric acid (13a)
Compound 12a (0.25 g, 0.45 mmol) was treated with trifluoroacetic acid as described in general procedure 2. The residue was further purified by column chromatography [methanol/dichloromethane/acetic acid (15:85:0.01)], giving compound 13a (0.18 g, 90%). 1H NMR (400 MHz, DMSO-d6) δ 12.18 (v br s, 1H), 8.11 (d, J = 2.5, 1H), 8.04 (d, J = 8.6, 1H), 7.26 (dd, J = 8.6,2.5; 1H), 6.87 (d, J = 2.1, 1H), 6.77 (d, J = 2.1, 1H), 6.69 (s, 1H), 4.20 (t, J = 6.2, 2H), 3.49 (q, J = 6.9, 4H), 2.66 (s, 3H), 2.45 (t, J = 7.5, 2H), 2.04 (qi, J = 6.9, 2H), 1.16 (t, J = 6.9, 6H); MS (FAB+) m/z: 451 (100%); HRMS (FAB+) m/z: Calcd 451.1691. Found: 451.1665; Degree of purity: HPLC method 1, retention time of 6.7 min, 95.9%; method 2, retention time of 8.8 min, 95.1%.
4.4.14. 4-(9-Diethylamino-11-methyl-5-oxo-5H-benzo[a]phenothiazin-3-yloxy)-butyric acid (13b)
Compound 12b (0.22 g, 0.40 mmol) was treated with trifluoroacetic acid as described in general procedure 2. The residue was purified by column chromatography [methanol/dichloromethane/acetic acid (10:90:0.01)] to give compound 13b (0.10 g, 56%). 1H NMR (400 MHz, DMSO-d6) δ 8.63 (d, J = 8.8, 1H), 7.51 (d, J = 2.6, 1H), 7.36 (dd, J = 8.8, 2.6; 1H), 6.87 (d, J = 2.4, 1H), 6.78 (s, 1H), 6.76 (d, J = 2.4, 1H), 4.13 (t, J = 6.4, 2H), 3.48 (q, J = 7.0, 4H), 2.65 (s, 3H), 2.42 (t, J = 7.3, 2H), 2.01 (qi, J = 6.8, 2H), 1.16 (t, J = 7.0, 6H); MS (FAB+) m/z: 451 (50%); HRMS (FAB+) m/z: Calcd 451.1691. Found: 451.1680; Degree of purity: HPLC method 1, retention time of 7.1 min, 96.0%; method 2, retention time of 7.9 min, 95.5%.
4.4.15. 4-(6-Bromo-9-diethylamino-11-methyl-5-oxo-5H-benzo[a]phenothiazin-2-yloxy)-butyric acid 2-trimethylsilanyl-ethyl ester (14a)
Compound 12a (0.04 g, 0.07 mmol) was dissolved in dimethylsulfoxide (5 ml) and reacted with N-ethyldiisopropylamine (0.02 g, 0.15 mmol) and bromomethylacetate (0.02 g, 0.15 mmol) according to general procedure 3 to give compound 14a (0.03 g, 68%). 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 8.6, 1H), 8.19 (d, J = 2.5, 1H), 7.14 (dd, J = 8.6, 2.5; 1H), 6.70 (d, J = 2.4, 1H), 6.54 (d, J = 2.4, 1H), 4.30−4.16 (m, 4H), 3.47 (q, J = 7.0, 4H), 2.72 (s, 3H), 2.60 (t, J = 7.2, 2H), 2.23 (qi, J = 6.4, 2H), 1.28 (t, J = 7.0, 6H), 1.02 (t, J = 8.5, 2H), 0.06 (s, 9H); MS (FAB+) m/z: 629 (40%), 631 (40%); HRMS (FAB+) m/z: Calcd 629.1505. Found 629.1505.
4.4.16. 4-(6-Bromo-9-diethylamino-11-methyl-5-oxo-5H-benzo[a]phenothiazin-3-yloxy)-butyric acid 2-trimethylsilanyl-ethylester (14b)
Crude compound 12b (0.16 g, 0.30 mmol) was dissolved in dimethylsulfoxide (5 ml) and reacted with N-ethyldiisopropylamine (0.09 g, 0.60 mmol) and bromomethylacetate (0.08 g, 0.60 mmol) according to general procedure 3 to give compound 14b (0.06 g, 33%). 1H NMR (400 MHz, Acetone-d6) δ 8.60 (d, J = 8.8, 1H), 7.60 (d, J = 2.8, 1H), 7.27 (dd, J = 8.8, 2.8; 1H), 6.84 (d, J = 2.0, 1H), 6.76 (d, J = 2.0, 1H), 4.26−4.17 (m, 4H), 3.55 (q, J = 7.1, 4H), 2.63 (s, 3H), 2.58 (t, J = 7.4, 2H), 2.17 (qi, J = 6.8, 2H), 1.27 (t, J = 7.1, 6H), 1.03 (t, J = 8.3, 2H), 0.08 (s, 9H); MS (FAB+) m/z: 629 (40%), 631 (40%); HRMS (FAB+) m/z: Calcd 629.1505. Found 629.1442.
4.4.17. 4-(6-Bromo-9-diethylamino-11-methyl-5-oxo-5H-benzo[a]phenothiazin-2-yloxy)-butyric acid (15a)
Compound 14a (0.03 g, 0.05 mmol) was treated with trifluoroacetic acid as described in general procedure 2. The residue was purified by column chromatography [dichloromethane–methanol/dichloromethane/acetic acid (7:93:0.01)] to give compound 15a (0.01 g, 53%). 1H NMR (400 MHz, DMSO-d6) δ 12.17 (v br s, 1H), 8.13 (d, J = 2.6, 1H), 8.11 (d, J = 8.8, 1H), 7.27 (dd, J = 8.8, 2.6; 1H), 7.02 (d, J = 2.4, 1H), 6.96 (d, J = 2.4, 1H), 4.21 (t, J = 6.3, 2H), 3.53 (q, J = 6.9, 4H), 2.68 (s, 3H), ±2.45 (hidden t, 2H), 2.05 (qi, J = 6.7, 2H), 1.18 (t, J = 6.9, 6H); MS (FAB+) m/z: 529 (90%), 531 (90%); HRMS (FAB+) m/z: Calcd 529.0796. Found: 529.0793; Degree of purity: HPLC method 1, retention time of 22.3 min, 97.8%; method 3, retention time of 14.1 min, 97.8%.
4.4.18. 4-(6-Bromo-9-diethylamino-11-methyl-5-oxo-5H-benzo[a]phenothiazin-3-yloxy)-butyric acid (15b)
Compound 14b (0.06 g, 0.09 mmol) was treated with trifluoroacetic acid as described in general procedure 2. The residue was purified by column chromatography [methanol/dichloromethane/acetic acid (8:92:0.01)] to give compound 15a (0.05 g, 98%). 1H NMR (400 MHz, DMSO-d6) δ 12.23 (v br s, 1H), 8.59 (d, J = 9.1, 1H), 7.53 (d, J = 2.8, 1H), 7.35 (dd, J = 9.1, 2.8; 1H), 6.94 (d, J = 2.6, 1H), 6.90 (d, J = 2.6, 1H), 4.13 (t, J = 6.5, 2H), 3.49 (q, J = 7.0, 4H), 2.64 (s, 3H), 2.45 (t, J = 7.4, 2H), 2.02 (qi, J = 7.2, 2H), 1.17 (t, J = 7.0, 6H); MS (FAB+) m/z: 529 (70%), 531 (70%); HRMS (FAB+) m/z: Calcd 529.0796. Found: 529.0793; Degree of purity: HPLC method 1, retention time of 28.1 min, 95.2%; method 3, retention time of 16.6 min, 95.9%.
5. mSCP2 protein preparation
mSCP2 expression, isotope labeling, and purification has been described before.13,14 In brief, the mature form of human sterol carrier protein 2 (mSCP2, residues 21–143) was expressed from a modified pET24d vector (G. Stier, EMBL-Heidelberg) in Escherichia coli BL21(DE3). Expression incorporated an N-terminal His6-GST fusion, cleavable with tobacco etch virus (His6-TEV) protease. Cultures were grown in LB medium with 1% (w/v) glucose or M9 minimal medium supplemented with 15NH4Cl and induced mid-log-phase with 1.0 mM IPTG for 3 h at 37 °C. Cell pellets were resuspended, lysed by sonication in the presence of protease inhibitors, and the lysate was cleared by centrifugation. The lysate was loaded onto a glutathione Sepharose 4B resin (Pharmacia) and eluted with 20 mM reduced glutathione. TEV protease was added until fusions were completely cleaved and the mixture was applied to Ni-NTA agarose (Qiagen).
6. Binding assay procedures
Assays were performed with a Quantamaster QM4/2000SE (Photon Technology International) spectrofluorimeter in a 50 μl quartz cuvette. A 10 mM solution of dye in DMSO was diluted with HEPES buffer (pH 7.4) to give a 10 μM stock solution. The final concentration for the experiment was determined to be 250 nM. The dye was excited at 540 nm. Addition of protein was measured by full emission scans from 570 to 750 nm with 3 nm step intervals and 0.1-s integration time. Titrations with BSA showed that fluorescence at 620 nm peaked at a concentration of 3 μM. We therefore used submaximal BSA concentrations (2 μM) for standard competition experiments. The change in fluorescence was instantaneous on the timescale of the measurement. Repetition of scans after several minutes showed no significant difference to the previous scan. Competitors for the binding of 1a were added in a dose-dependent manner in the lowest possible volume. Dose-response curves (see Fig. S1 in the Supplementary material), but not titration curves, were corrected for the dilution effect when competitors were added. In cases where site-selective inhibitors were added (tryptophan), a supramaximal dose of ibuprofen was added at the end of the experiments to fully replace dye from all relevant binding sites. All experiments were performed at least in triplicate with very small variations being observed between single experiments.
For experiments with mSCP2, an 8 μM protein solution in 100 mM phosphate buffer, pH 7.0, was preincubated with 250 nM 1a for 30 min. The mixture was titrated with aliquots of linoleoyl CoA from a 1 mM stock solution in 100 mM potassium phosphate, pH 7.0. Data were expressed in relative fluorescence (657 nm/618 nm) per mole mSCP2 as a function of molar ratio (linoleoyl CoA: mSCP2) and normalized for the small dilutions resulting from addition of linoleoyl CoA. Thus, data could be fitted in MicroCal Origin 5.0 using the non-linear least squares fit ‘two sets of sites’ model for ease of comparison with ITC data.
7. Isothermal titration calorimetry ITC
mSCP2 and linoleoyl CoA were co-dialyzed against 100 mM potassium phosphate pH 7.0 using Spectra/Por dialyzers with a 500 Da nominal molecular weight cut-off (Carl Roth & Co., Germany). Dialysates were degassed and the concentration measured by A280 nm. ITC measurements were conducted on a MicroCal VP-ITC (Milton Keynes, UK) using mSCP2 at 50 μM as sample and linoleoyl CoA at 1 mM as titration ligand. Experiments were conducted at 35 °C. Titrations used 24 linoleoyl CoA injections, each of 7.5 μl, thus giving a final molar excess of approximately 2.5-fold over mSCP2. Ligand heats of dilution were subtracted and data fitted using the non-linear least squares fit ‘two sets of sites’ model in MicroCal Origin 5.0.
8. NMR titrations
15N isotope labeled samples of mSCP2 were exchanged into 15mM potassium phosphate buffer (pH 6.0) by gel filtration on a 16/60 Superdex 75 column (Pharmacia). 2D 1H, 15N-correlation spectra were acquired with protein concentrations of 0.1 mM at 310 K with 128 and 512 complex points in t1 and t2 using 16 scans per increment. Annotation of backbone amide chemical shifts was based on previous work.25
Supplementary Material
Acknowledgments
The authors thank Marc Niebuhr (Stanford) and Santosh Panjikar (Hamburg) for helpful discussions. The work was supported by the EU sixth framework Grants LSHG-CT-2003-503259 to C.S.
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc. 2007.10.080.
References and notes
- 1.Cao BL, Endsley S, Andersen NH. Bioorg Med Chem. 2003;11:69. doi: 10.1016/s0968-0896(02)00324-3. [DOI] [PubMed] [Google Scholar]
- 2.Gerig JT, Klinkenborg JC. J Am Chem Soc. 1980;102:4267. [Google Scholar]
- 3.Klinkner AM, Bugelski PJ, Waites CR, Louden C, Hart TK, Kerns WD. J Histochem Cytochem. 1997;45:743. doi: 10.1177/002215549704500513. [DOI] [PubMed] [Google Scholar]
- 4.Sackett DL, Wolff J. Anal Biochem. 1987;167:228. doi: 10.1016/0003-2697(87)90157-6. [DOI] [PubMed] [Google Scholar]
- 5.Ruvinov SB, Yang XJ, Parris KD, Banik U, Ahmed SA, Miles EW, Sackett DL. J Biol Chem. 1995;270:6357. doi: 10.1074/jbc.270.11.6357. [DOI] [PubMed] [Google Scholar]
- 6.Briggs MSJ, Bruce I, Miller JN, Moody CJ, Simmonds AC, Swann EJ. Chem Soc Perkin Trans. 1997;1:1051. [Google Scholar]
- 7.Wichmann O, Schultz C. Chem Commun. 2001;2500 doi: 10.1039/b107670c. [DOI] [PubMed] [Google Scholar]
- 8.Dinkel C, Wichmann O, Schultz C. Tetrahedron Lett. 2003;44:1153. [Google Scholar]
- 9.Gallegos AM, Atshaves BP, Storey SM, Starodub O, Petrescu AD, Huang H, McIntosh AL, Martin GG, Chao H, Kier AB, Schroeder F. Prog Lipid Res. 2001;40:498. doi: 10.1016/s0163-7827(01)00015-7. [DOI] [PubMed] [Google Scholar]
- 10.Stolowich NJ, Petrescu AD, Huang H, Martin GG, Scott AI, Schroeder F. Cell Mol Life Sci. 2002;59:193. doi: 10.1007/s00018-002-8416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haapalainen AM, van Aalten DM, Merilainen G, Jalonen JE, Pirila P, Wierenga RK, Hiltunen JK, Glumoff T. J Mol Biol. 2001;313:1127. doi: 10.1006/jmbi.2001.5084. [DOI] [PubMed] [Google Scholar]
- 12.Dyer DH, Lovell S, Thoden JB, Holden HM, Rayment I, Lan Q. J Biol Chem. 2003;278:39085. doi: 10.1074/jbc.M306214200. [DOI] [PubMed] [Google Scholar]
- 13.Filipp FV, Sattler M. Biochemistry. 2007;46:7980. doi: 10.1021/bi6025616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanley WA, Filipp FV, Kursula P, Schuller N, Erdmann R, Schliebs W, Sattler M, Wilmanns M. Mol Cell. 2006;24:653. doi: 10.1016/j.molcel.2006.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frolov A, Cho TH, Billheimer JT, Schroeder F. J Biol Chem. 1996;271:31878. doi: 10.1074/jbc.271.50.31878. [DOI] [PubMed] [Google Scholar]
- 16.Wirtz KW, Wouters FS, Bastiaens PH, Wanders RJ, Seedorf U, Jovin TM. Biochem Soc Trans. 1998;26:374. doi: 10.1042/bst0260374. [DOI] [PubMed] [Google Scholar]
- 17.Dansen TB, Westerman J, Wouters FS, Wanders RJ, van Hoek A, Gadella TW, Jr, Wirtz KW. Biochem J. 1999;339:193. [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner SJ, Skripchenko A, Robinette D, Foley JW, Cincotta L. Photochem Photobiol. 1998;67:343. [PubMed] [Google Scholar]
- 19.Kubo Y, Kuwana M, Yoshida K. Chem Express. 1988;3:663. [Google Scholar]
- 20.Stolowich N, Frolov A, Petrescu AD, Scott AI, Billheimer JT, Schroeder F. J Biol Chem. 1999;274:35425. doi: 10.1074/jbc.274.50.35425. [DOI] [PubMed] [Google Scholar]
- 21.Stanley WA, Versluis C, Schultz C, Heck AJR, Wilmanns M. Arch Biochem Biophys. 2007;461:50. doi: 10.1016/j.abb.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 22.Wolfrum C, Borchers T, Sacchettini JC, Spener F. Biochemistry. 2000;39:1469. doi: 10.1021/bi991638u. [DOI] [PubMed] [Google Scholar]
- 23.Kurian E, Kirk WR, Prendergast FG. Biochemistry. 1996;35:3865. doi: 10.1021/bi952589y. [DOI] [PubMed] [Google Scholar]
- 24.Seedorf U, Raabe M, Ellinghaus P, Kannenberg F, Fobker M, Engel T, Denis S, Wouters F, Wirtz KW, Wanders RJ, Maeda N, Assmann G. Genes Dev. 1998;12:1189. doi: 10.1101/gad.12.8.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia FL, Szyperski T, Dyer JH, Choinowski T, Seedorf U, Hauser H, Wuthrich K. J Mol Biol. 2000;295:595. doi: 10.1006/jmbi.1999.3355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.