

Abstract
Rare bone diseases, generally defined as monogenic traits with either autosomal recessive or dominant patterns of inheritance, have provided a rich database of genes and associated pathways over the past 2–3 decades. The molecular genetic dissection of these bone diseases has yielded some major surprises in terms of the causal genes and/or involved pathways. The discovery of genes/pathways involved in diseases such as osteopetrosis, osteosclerosis, osteogenesis imperfecta and many other rare bone diseases have all accelerated our understanding of complex traits. Importantly these discoveries have provided either direct validation for a specific gene embedded in a group of genes within an interval identified through a complex trait genome-wide association study (GWAS) or based upon the pathway associated with a monogenic trait gene, provided a means to prioritize a large number of genes for functional validation studies. In some instances GWAS studies have yielded candidate genes that fall within linkage intervals associated with monogenic traits and resulted in the identification of causal mutations in those rare diseases. Driving all of this discovery is a complement of technologies such as genome sequencing, bioinformatics and advanced statistical analysis methods that have accelerated genetic dissection and greatly reduced the cost. Thus, rare bone disorders in partnership with GWAS have brought us to the brink of a new era of personalized genomic medicine in which the prevention and management of complex diseases will be driven by the molecular understanding of each individuals contributing genetic risks for disease.
Introduction
Complex genetic traits are defined as those phenotypes controlled by multiple genes, environmental factors and gene–environment interactions. In the decades leading up to the Human Genome Project, the identification of genes underlying bone traits was confined largely to the study of monogenic disorders of bone in which a particular trait segregated in a large family or several smaller families. In addition, a cadre of candidate gene association studies have been reported in the literature for complex traits. Not surprisingly these candidate gene association studies with few exceptions produced findings of both positive and negative associations depending upon the ethnicity of the population under study, the phenotype chosen and the sample size. In contrast, the identification of causal mutations in rare bone disorders has provided a wealth of knowledge regarding bone biology and provided a number of candidate genes and pathways whose components can be associated with complex traits such as osteoporosis and fracture. Just as important, as candidate genes are proposed that reside within chromosomal regions identified through genome-wide association studies (GWAS), these rare bone disorders can provide a critical validation of a role for those GWAS candidate loci in bone regulation.
The Human Genome Project established a variety of tools that have greatly accelerated gene discovery in complex bone traits such as bone mineral density (BMD), fracture and many other phenotypes that have often been associated with diseases such as osteoporosis. Also, large consortiums were established that began to provide adequate power in terms of subjects enrolled in the studies. In recent years, high throughput and relatively low cost DNA sequencing technologies, high density marker and physical maps, large genetic databases, and advances in statistical approaches, as well as multiple approaches using animal models to identify or more important biologically validate candidate genes, has greatly facilitated the identification of genes underlying complex bone traits. However, guidance for the study of complex bone traits from studies of rare, monogenic bone diseases still remains an important contributor to our molecular genetic dissection of complex diseases. In fact, many of the same technologies that have accelerated the genetic dissection of complex bone diseases have also facilitated the discovery of genes underlying rare diseases of bone.
Two recent reviews on the genetics of bone mass have been published that the reader is referred to for additional details beyond the scope of this perspective. Boudin et al.1 have published an excellent review of a number of genes that are central to the control of bone mass and their associated monogenic bone disorders and detailed descriptions of their human bone phenotypes. Karasik et al.,2 summarized the current state of GWAS for bone traits and listed a total of 132 single-nucleotide polymorphisms (SNPs) with a genome level of statistical significance that have been identified for BMD, bone ultrasound and fracture. These SNPs are associated with ∼144 neighboring candidate genes. Thirteen of these candidate genes have causal mutations identified in monogenic traits and have been found in GWAS based association studies as highly significant loci. Among these genes are those associated with Wnt/β-catenin signaling (LRP5,3,4,5,6,7,8,9,10 LRP46,8,11,12,13,14 and SOST6,8,15,16,17,18,19), osteoclast differentiation or function (TNFSF11,6,7,8,10,19,20,21 TNFRSF11A,6,8,10,22,23 TNFRSF11B6,8,9,10,19,23,24,25,26,27 and CLCN726,28,29,30) and osteoblast differentiation or function (RUNX,8,31,32,33,34,35 SP76,8,36,37 and COL1A138,39), and other signaling pathways important in bone cell regulation (JAG1,40,41,42 ESR16,7,9,27,39,43 and PTHLH34,44). The relationship between the key bone cells expressing these genes and their bone cell targets are illustrated in Figure 1.
Figure 1.
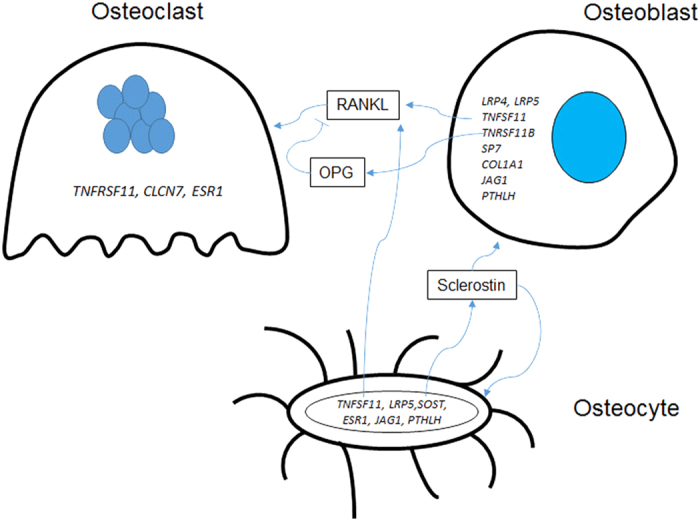
Major genes that are causal for rare bone disorders and have genome level significant single nucleotide polymorphisms for bone mass, bone ultrasound or fracture as identified by GWAS. Genes are associated with the bone cell in which the gene is expressed, although many have a site of action that affects a different bone cell. For example, the SOST gene is expressed in osteocytes, but its protein product sclerostin appears to act on any cell expressing the LRP4/5/6 co-receptors that regulate Wnt/β-catenin signaling, such as the osteoblast and the osteocyte. RANKL, the product of the TNFSF11 gene is expressed in both osteoblasts and osteocytes and regulates osteoclast differentiation by binding to its receptor, RANK (product of the TNFRSF11 gene). OPG, the product of the TNFRSF11B gene, is a decoy receptor for RANKL and blocks RANKL binding to RANK.
In this review, I will discuss some of the key ‘historical' gene discoveries that have greatly informed our biology of bone and some of the more recent gene identifications that continue to direct and validate our studies of complex traits. The focus of my discussions will be on three broad categories of monogenic bone syndromes/disorders; namely osteopetrosis, hyperostosis and osteosclerosis, and osteogenesis imperfecta. Presented in Table 1 is a brief description of the skeletal and important clinical features associated with these rare bone disease that in the past have served to differentiate them from each other. However, as our molecular/genetic dissection of these conditions has evolved, it is interesting to note that the ‘traditional' clinical categorization of many of these syndromes/disorders has been or needs to be reconsidered based upon our knowledge of the underlying genetic basis of the trait.
Table 1. Skeletal features of monogenic bone syndromes/disorders.
| Syndrome/disorder | General skeletal and other important clinical features | Defective bone cell (s) | OMIM reference IDa |
|---|---|---|---|
| Osteopetrosis | |||
| Autosomal dominant osteopetrosis type II (ADO II) (Albers-Schönberg Disease) | Endplate thickening of vertebrae, Increased cortical thickening, Compromised marrow space, Diffuse increased bone density but associated with multiple fractures, ‘Marbled' or bone-within-bone in the pelvis | Osteoclasts | 166600 |
| Autosomal recessive osteopetrosis | |||
| Type 1 (severe neonatal or infantile form) | Osteomyelitis, Uniform increase in bone density, Increased fracture incidence, thick dense skull, sandwich appearance of vertebrae, Coxa vara in the hip | Osteoclasts | 259700 |
| Type 2 | Hyperostosis of the skull, osteosclerosis of the skeleton, increased number of multiple fractures, tendency for osteomyelitis particularly in the jaw, mandible prognathism | Osteoclasts | 259710 |
| Type 3 | Osteosclerosis of the skeleton, short stature, renal tubular acidosis and elevated serum acid phosphatase | Osteoclasts | 259730 |
| Type 4 | Severe osteopetrosis and associated multiple fractures | Osteoclasts | 611490 |
| Type 5 | Severe osteopetrosis, in utero fractures, hydrocephaly and stillborn cases reported | Osteoclasts | 259720 |
| Type 6 | ‘Erlenmeyer flask' deformity of distal femur | Osteoclasts | 611497 |
| Type 7 | Large areas of cartilage retention and trabecular structures | Osteoclasts | 612301 |
| Type 8 | Macrocephaly of skull, open fontanels, dense bones with reduced marrow cavity, anemia | Osteoclasts | 615085 |
| Pycnodysostosis | Dense skull with open anterior fontanelle, aplastic clavicle, delayed tooth eruption, scoliosis of the spine | Osteoclasts | 265800 |
| Hyperostosis and osteosclerosis | |||
| Osteoporosis pseudoglioma syndrome | Low bone density (childhood osteoporosis), early onset blindness, kyphosis | Osteoblast/osteocyte | 259770 |
| High bone mass (HBM) | Generalized increased in bone density across skeleton with thick cortices | Osteoblast/osteocyte | 601884 |
| Van Buchem's | Cranial hyperostosis and generalized increased in bone density throughout skeleton, hearing loss | Osteoblast/osteocyte | 239100 |
| Sclerosteosis | Increased bone density with asymmetric/prominent mandible, cranial hyperostosis, sclerotic vertebral endplates, high cortical density of long bones | Osteoblast/osteocyte | 269500 |
| Endosteal hyperostosis | Increased bone density, skull particularly affected with elongated mandible, mild sclerosis of spine and long bones with thickened cortices | Osteoblast/osteocyte | 144750 |
| Robinow syndrome | Macrocephaly, frontal bossing and other facial features common. Short limbs and small hands also common. | Osteoblast | 180700 |
| Al-Awadi-Rass Rothchild syndrome | Asymmetric, long face. Hemivertebrae with shortened forearms and aplastic fibula and tibia | 276820 | |
| Fuhrmann syndrome | Bowing of forearm and femur, missing patella, short stature, congenital hip dislocation | 228930 | |
| Autosomal dominant osteopetrosis type I (ADO I) | Increased bone density, generalized skeletal sclerosis particularly pronounced in the cranial vault. No ‘Rugger-Jersey spine' (vertebral end-plate thickening) observed in ADO II | Osteoblast/osteoclast | 607634 |
| Camurati-Engelmann or progressive diaphyseal dysplasia | Sclerosis of skull and mandible. Progressive diaphyseal widening and 'Erlenmeyer' appearance, long bone marrow cavity narrowing | Osteoblast | 131300 |
| Raine syndrome | Neonatal generalized sclerosis, short stature, usually results in death within first few weeks after birth. Severe craniofacial anomalies common | Osteoblast | 259775 |
| Craniofacialmetaphyseal dysplasia | Sclerotic skull base and cranial vault with normal spine and pelvis. Limb metaphyses are widened and ‘Erlenmeyer' appearance in childhood and club-shaped appearance in adulthood of distal femur | Osteoblast | 123000 |
| Occulodentodigital dysplasia | Skull and vertebrae hyperostosis with broad tubular long bones. Dental issues and cleft lip/palate, microcephaly, various eye problems | Osteoblast | 164200 |
| Osteogenesis imperfectaa | |||
| Type I | Blue sclerae throughout life, mild osteopenia, multiple fractures during childhood to puberty | Osteoblast | 166200 |
| Type II | Multiple fractures present at birth. Soft calvaria. Perinatal lethal | Osteoblast | 166200 |
| Type III | Blue sclerae at birth but becomes normal as patient ages. Multiple fractures at birth. Sciolosis/kyphosis of spine, hypomineralized cranium. Long bone deformity at birth and bowing of limbs due to fractures. | Osteoblast | 259420 |
| Type IV | Mild to moderate skeletal deformities with varying degrees of fractures, scoliosis/kyphosis and biconcave and flattened vertebrae with limb bowing due to fractures. | Osteoblast | 166220 |
Summary of key skeletal and/or clinical features of rare bone syndromes/disorders discussed in this review. The Online Mendelian Inheritance in Man (OMIM) database (http://omim.org) Accession number is listed for further detailed clinical and genetic information related to these diseases. The major bone cell type involved in each disease is also indicated.
aOnly the forms of osteogenesis imperfecta that involve either the Col1A1 or Col1A2 genes are listed. For a complete description of all osteogenesis imperfecta types refer to the recent review by Forlino and Marini.102
Osteopetrosis
Historically speaking the period from 1900–1990 provided a wealth of detailed clinical descriptions of rare diseases of bone. The decade of the 1990s and up to the present date, because of our enhanced abilities to genetically dissect monogenic traits, has yielded a detailed molecular understanding of these rare bone diseases and identified critical genes/proteins/pathways that in some cases confirmed, but in many instances identified, new components of bone mass regulation that clinical studies could not provide.
Osteopetrosis was first described in 1904 by Albers-Schönberg,45 which generally occurs in adulthood and is less severe that the autosomal recessive (AR) forms. Radiographically it is distinguished by pronounced endplate thickening of the vertebrae and the ‘bone within bone' appearance of the iliac wings. Collectively the autosomal dominant (AD) and AR forms of osteopetrosis represent a class of osteoclast defect diseases. Traditionally, each of the different types of osteopetrosis has been distinguished by clinical descriptions that relied heavily on radiographic features, severity of disease and age of onset, although oftentimes differences between disorders are very subtle. Our current ability to determine causal mutations has provided a means to more finely categorize these diseases and also provides valuable directions for clinical management of the disorders.
The gene for Albers-Schönberg disease or AD osteopetrosis type II (ADO II), as it is now classified, was shown by linkage studies of several families to reside on chromosome 16p13.3 and each these families carried mutations in the chloride channel 7 (CLCN7) gene.46 At the same time, Kornak et al.28 reported a patient with infantile malignant osteopetrosis that was a compound heterozygote with a nonsense mutation (Q555X) in exon 18 of one allele and a missense mutation (R762Q) in exon 24 of the other allele of the CLCN7 gene. This suggests that CLCN7 can underlie different forms of osteopetrosis in a genotype–phenotype relationship that is not clearly understood. CLCN7 is an integral component of the ruffled membrane of osteoclasts involved in the transport of Cl− ions into the resorption compartment on bone surfaces. Most cases of ADO II are due to heterozygous mutations in CLCN7, whereas a few cases of homozygous or compound heterozygous mutations have been attributed to AR osteopetrosis.47
AR forms of osteopetrosis (ARO) were first shown by clinical studies to be due to a deficiency in carbonic anhydrase II (CA II)48 and subsequently a point mutation in the CA2 gene49 was identified in a Belgian family in 1991. We now identify eight different forms of ARO, each associated with different genes/mutations (see Review by Boudin et al.1) that encode several different proteins involved in osteoclast differentiation or function. Some of the major genes that have been identified are TCIRG1 (the osteoclast specific vacuolar proton pump V-type ATPase a3 subunit)50,51,52 and two members of the tumor necrosis factor (TNF) receptor superfamily; TNFSF11 (encoding RANKL)20 and TNFRSF11A (encoding RANK).22 A closely related disease, Pycnodysostosis, is caused by mutations in CTSK (Cathepsin-K).53,54 All of these studies of ADO and ARO have illustrated the critical role of the osteoclast and control of its differentiation and function in bone mass regulation. Somewhat surprisingly of all of the genes causal for various forms of osteopetrosis, GWAS studies to date have only identified TNFSF11 and TNFRSF11A as candidate genes potentially underlying normal variation in BMD (at the lumbar spine).2
Hyperostosis and osteosclerosis
Hyperostosis and osteosclerosis have generally been considered one large group of sclerosing bone diseases. These syndromes manifest clinically with generalized changes in bone density (decreased or increased) throughout the skeleton, but with varying degrees of involvement at specific skeletal sites and in many cases other tissue involvement that have served to distinguish them in the past. However as a result of our emerging molecular/genetic characterization we need to begin incorporating knowledge of the genes/pathways involved in the classification of these syndromes as clinical classifications do not fully discriminate the subtle differences that we now appreciate.
One of the most significant pathways to be identified as a result of the genetic dissection of rare bone diseases is the Wnt/β-catenin signaling pathway that was identified within the past 20 years as critical for many aspects of bone biology and particularly bone mass regulation. In 1996, Warman and colleagues55 described the localization of the loci responsible for Osteoporosis Pseudoglioma Syndrome (OPPG) (Online Mendelian Inheritance in Man (OMIM) #259770), a rare human disease characterized by juvenile onset osteoporosis and progressive blindness, to human chromosome 11q12-13. The next year, Johnson and colleagues56 localized the loci responsible for an AD high-bone mass (HBM) (OMIM #601884) trait to the same 11q12–13 chromosomal region and suggested that perhaps these two opposite phenotypes resulted from allelic variants in the same gene. Follow-up work to identify the gene in both of these traits led to the discovery of inactivating mutations in the low-density lipoprotein-related protein 5 (LRP5) by Warman and colleagues published in November of 200157 and a G171V missense mutation in LRP5 as causal for the HBM kindred phenotype by Johnson and colleagues that was published in January of 2002.58 A few months later Boyden and colleagues59 published the identification of the same LRP5 G171V mutation in another, extreme high bone mass family that was unrelated to the Johnson and colleagues kindred. Critical to the discoveries of the LRP5 mutations in these monogenic disorders were studies by other groups demonstrating the role of LRP5 and its close homolog, LRP6, in the Wnt/β-catenin signaling pathway.60,61,62,63 The Wnt/β-catenin pathway was known to be an oncogenic pathway since the studies by Nusse and colleagues in the 1980s demonstrating that the mouse mammary oncogene int-1 was identical to the Drosophila segment polarity gene Wingless (Wg).64 Thus, the role of this pathway in bone mass accrual and regulation of adult mass came as a complete surprise. The convergence of these studies identifying LPR5 and its associated Wnt/β-catenin signaling pathway, served as a catalyst for the thousands of studies that followed over the last 15 years into this pathway's role in bone. However, although common allelic variants and SNPs of LRP5 have consistently correlated with different bone phenotype in many association studies and in several GWAS analysis, the best estimates indicate that they explain only 2–3% of the variation in bone mass in any given population.
Also in 2001–2002 there were a number of papers published that identified SOST gene mutations as causal for Van Buchem's disease (OMIM #239100) and Sclerosteosis (OMIM #269500).65,66,67,68 The importance of SOST was not fully recognized until 2005 when sclerostin, the product of the SOST gene, was shown to inhibit Wnt/β-catenin signaling by binding to LRP5/LRP6,69,70 although one study suggested sclerostin was a BMP antagonist.71 This second convergence which brought SOST and LRP5 together created a broader landscape of regulation of bone mass by the Wnt/β-catenin signaling pathway and brought into focus the potential role of several other regulators of the pathway such as the Dkk and sFRP families of proteins. LRP5 mutations have now been described in several bone related phenotypes.72,73,74
LRP4 is another member of the Wnt pathway family of proteins important in bone mass regulation. It was first identified in genome-wide studies6,9,19 and in both mice75,76 and humans11,12 and shown to regulate bone mass. LRP4 functions as a binding or docking protein for sclerostin.11,12,75 Thus, while LRP4 was first identified as a candidate gene for bone mass from GWAS studies, the identification of human mutations with the rare bone disorders provided important confirmatory evidence for its role in human bone mass regulation.
Recently, from a GWAS study of the Icelandic population77 a rare nonsense variant in LGR4 (leucine-rich-repeat-containing G-protein-coupled receptor 4) was identified as being strongly associated with low BMD and osteoporotic fracture. LGR4 binds the R-spondin family of proteins that are known to potentiate Wnt/β-catenin signaling (see review78). Zhu et al.79 have shown that LGR4 is a key receptor for R-spondin 2. What these LRP4 and LGR4 findings clearly illustrate is that the recognized importance of the Wnt/β-catenin pathway in regulating bone mass has enabled investigators to scrutinize regions that reach genome level significance in GWAS studies and focus on genes whose proteins regulate or interact with the pathway.
Several other diseases that have a bone associated phenotype related to the Wnt pathway family of genes have been described. WNT5A mutations have been reported in patients with autosomal dominant Robinow Syndrome (OMIM #180700).80 WNT7A appears to be important in limb development as evidence by reports relating to Al-Awadi-Rass Rothchild syndrome (OMIM #276820) and Fuhrmann syndrome (OMIM #228930).81,82 Several other members of the Wnt/β-catenin signaling pathway in bone have been identified as causal for rare bone disorders and this list is likely to grow as the genetic dissection of rare traits becomes more commonplace in the clinical characterization of these diseases. Conversely, GWAS studies have also identified WNT16 polymorphisms with bone mass and osteoporotic fracture,83,84 but no rare syndrome has yet been identified due to mutations in this gene.
Interestingly, the ADO I form presents with a more generalized sclerosis (not the bone-within-bone typical of ADO II), which is especially pronounced in the cranial vault, and has been shown to be due to a number of different mutations in LRP5.72,85 The fact that a bone formation gene/pathway, which principally acts in the osteoblast lineage, calls into question whether ADO I is correctly associated with osteopetrosis? Studies in mice and bone cell lines86,87,88,89,90,91,92,93 have shown that Wnt/β-catenin signaling in osteoblasts/osteocytes controls the expression regulators of osteoclastogenesis (for example, osteoprotegerin and RANKL). Thus while ADO I has the appearance of an osteopetrotic condition,85 the primary defect is acting at the osteoblast/osteocyte cell level, which begs the question of how best to classify the disease. Given the genetic basis of ADO I, perhaps it should be more appropriately categorized as an osteosclerotic phenotype as suggested by de Vernejoul.47
Other sclerosing bone diseases that have had causal genes identified include Camurati-Engelmann disease (OMIM #131300), which has been shown to be due to mutations in TGFB1 (transforming growth factor β-1 gene),94 Raine Syndrome (OMIM #259775) that is caused by mutations in FAM20C,95 and craniofacialmetaphyseal dysplasia (OMIM #123000) that has been shown to be due to mutations in ANKH96,97 and interestingly to a rare mutation in GJA1.98 Mutations in GJA1 have been more commonly reported in association with occulodentodigital dysplasia (OMIM #164200).99 Several other sclerosing diseases have been described and excellent reviews have been published in the literature that details these syndromes.1,47,100,101
At present, other than the Wnt/β-catenin pathway-associated genes, GWAS has yet to identify variants of these genes as underlying normal variation in bone traits. This highlights one of the limitations of GWAS studies, namely that common allelic variants are generally covered by GWAS and not rare variants that contribute to monogenic diseases. However, as the speed of genome sequencing increases and costs decrease, the potential exists for performing complete genome sequencing of all individuals within a population. Such was the case for the above mentioned Icelandic population study,77 which identified LGR4 as being associated with low BMD and osteoporotic fracture in that population.
Osteogenesis imperfecta
Perhaps one of the best examples of how rare bone disorders has propelled our understanding of bone biology is illustrated by the studies from several groups who have provided a detailed molecular/genetic dissection of various forms of osteogenesis imperfecta. Several excellent reviews have been published and the reader is referred to one of the more recent reviews by Forlino and Marini102 for detailed information. What has evolved is a detailed understanding of the molecular events necessary for collagen synthesis (COL1A1 and COL1A2), post-translational processing/modification (CRTAP, LEPRRE1, PPIB and TMEM38B), folding and crosslinking (SERPINH1, FKBP10 and PLOD2) and many of the key genes/proteins involved in this pathway.102 Interestingly, of the studies to date, for these collagen associated genes only the COL1A1 gene has been shown to reach a genome wide level of significance in GWAS studies.2 What this perhaps suggests is that the numerous players involved are so finely tuned to their associated roles in collagen production that variants in these genes all lead to disease, rather than more subtle variations in bone density or fracture susceptibility as in the case of osteoporosis.
Recently, there has been a report of mutations in WNT1 have been shown to cause bone fragility in one family specifically diagnosed with osteogenesis imperfecta.103,104 Many other non-collagen genes105 have been implicated in osteogenesis imperfecta including CREB3L1106 and SP7,36 which have roles in osteoblast differentiation. To account for this rapidly growing list of new genes associated with osteogenesis imperfecta, Forlino and Marini have proposed a functional metabolic classification scheme that incorporates both clinical and genetic information to subtype the disease.102
Conclusions
In this review I have focused on a few of the rare bone diseases that have contributed some of the greatest impact on our body of molecular/genetic knowledge with respect to bone biology. A complete listing of all rare diseases of bone is not possible in this short review. A simple search of the OMIM (http://www.ncbi.nlm.nih.gov/omim) database using terms such as bone dysplasia, osteopetrosis, osteosclerosis, osteogenesis imperfecta, Paget's Disease of Bone yielded a list of over 700 disorders in the catalog, and easily over one-third of which still have yet to have a causal gene identified. The advances made in recent years with exomic and whole genome sequencing, for example, now make it possible to identify the casual mutation in a single patient and it seems likely that the causal genes/mutations for these currently unknown disorders will be identified within the next decade.
However, once this catalog is created we still will have much work to do in the area functional studies aimed at understanding how common allelic variants in genes mutated in rare bone disease contribute to bone mass traits. For example, the types of studies that are needed include defining whether these common allelic variants contribute to changes in gene expression, mRNA stability, or induce subtle changes in protein conformation that alter protein function or interactions with other regulatory proteins (for example, inhibitors or activators). Another critical aspect that needs to be understood in the cases of many noncoding (exonic) allelic variants is if/whether they contribute to altered epigenetic regulation of the gene.
Given that our catalog of loci from GWAS studies now includes hundreds of potential genes, all with small effect sizes, and the relative contributions of any given gene to variation in a given bone trait in any one patient is likely to be highly variable; individual genetic profiling is ultimately going to be required to provide personalized diagnosis. The ability to rapidly sequence genomes at relatively low cost means that we are at the threshold of this reality. How these polymorphisms alter function remains a big challenge and this knowledge is essential. Once we can equate genetic changes to functional consequences, we will be one step closer to personalized molecular/genetic diagnosis for common diseases such as osteoporosis and customized treatments that will be maximally effective in any given individual.
So currently how have rare bone diseases informed our understanding of complex traits? Rare bone disorders have revealed molecular pathways whose members are strong candidates for variation in normal bone traits. These monogenic trait gene discoveries have provided a focusing lens for prioritizing candidate genes for further biological validation in intervals of genome level significance that are identified in GWAS studies. Conversely, GWAS studies have also provided clues as to genes underlying rare bone disorders. Currently there are several genes that represent a convergence between having a mutation that causes a rare bone disease and variants that contribute to variation in bone density in the population (summarized in Figure 1). The rapid evolution of technologies that has occurred in the genome/post-genome era including DNA sequencing, bioinformatics and model systems to study gene/protein function has provided us with the tools needed to genetically define causal mutations for any monogenic disease. Partnered with the growing wealth of knowledge gained from GWAS studies, it seems highly likely that genetic risk assessment for complex diseases such as osteoporosis will be possible in the very near future for every individual in the population. This will be a paradigm shift for both prevention and management of complex diseases.
Acknowledgments
MLJ is supported by a grant from the NIH NIA P01 AG039355.
Footnotes
The author declares no conflict of interest.
References
- Boudin E, Fijalkowski I, Hendrickx G, Van Hul W. Genetic control of bone mass. Mol Cell Endocrinol 2015; 432: 3–13. [DOI] [PubMed] [Google Scholar]
- Karasik D, Rivadeneira F, Johnson ML. The genetics of bone mass and susceptibility to bone diseases. Nat Rev Rheumatol 2016; 12: 496. [DOI] [PubMed] [Google Scholar]
- Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM et al. LDL Receptor-Related Protein 5 (LRP5) Affects Bone Accrual and Eye Development. Cell 2001; 107: 513–523. [DOI] [PubMed] [Google Scholar]
- Little RD, Folz C, Manning SP, Swain PM, Zhao S-C, Eustace B et al. A Mutation in the LDL Receptor–Related Protein 5 Gene Results in the Autosomal Dominant High–Bone-Mass Trait. Am J Hum Genet 2002; 70: 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA et al. High Bone Density Due to a Mutation in LDL-Receptor–Related Protein 5. N Engl J Med 2002; 346: 1513–1521. [DOI] [PubMed] [Google Scholar]
- Rivadeneira F, Styrkarsdottir U, Estrada K, Halldorsson BV, Hsu YH, Richards JB et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat Genet 2009; 41: 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Choi HJ, Estrada K, Leo PJ, Li J, Pei YF et al. Multistage genome-wide association meta-analyses identified two new loci for bone mineral density. Hum Mol Genet 2014; 23: 1923–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet 2012; 44: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JB, Kavvoura FK, Rivadeneira F, Styrkársdóttir U, Estrada K, Halldórsson BV et al. Collaborative Meta-analysis: Associations of 150 Candidate Genes With Osteoporosis and Osteoporotic Fracture. Ann Intern Med 2009; 151: 528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp JP, Medina-Gomez C, Estrada K, Pourcain B St, Heppe DH, Warrington NM et al. Phenotypic dissection of bone mineral density reveals skeletal site specificity and facilitates the identification of novel loci in the genetic regulation of bone mass attainment. PLoS Genet 2014; 10: e1004423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem 2011; 286: 19489–19500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijalkowski I, Geets E, Steenackers E, Van Hoof V, Ramos FJ, Mortier G et al. A Novel Domain-Specific Mutation in a Sclerosteosis Patient Suggests a Role of LRP4 as an Anchor for Sclerostin in Human Bone. J Bone Miner Res 2016; 31: 874–881. [DOI] [PubMed] [Google Scholar]
- Boudin E, Steenackers E, de Freitas F, Nielsen TL, Andersen M, Brixen K et al. A common LRP4 haplotype is associated with bone mineral density and hip geometry in men-data from the Odense Androgen Study (OAS). Bone 2013; 53: 414–420. [DOI] [PubMed] [Google Scholar]
- Wang C, Zhang Z, Zhang H, He JW, Gu JM, Hu WW et al. Susceptibility genes for osteoporotic fracture in postmenopausal Chinese women. J Bone Miner Res 2012; 27: 2582–2591. [DOI] [PubMed] [Google Scholar]
- Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S et al. Bone Dysplasia Sclerosteosis Results from Loss of the SOST Gene Product, a Novel Cystine Knot–Containing Protein. Am J Hum Genet 2001; 68: 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet 2001; 10: 537–543. [DOI] [PubMed] [Google Scholar]
- Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 2002; 39: 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A et al. A 52- kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet 2002; 110: 144–152. [DOI] [PubMed] [Google Scholar]
- Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T et al. New sequence variants associated with bone mineral density. Nat Genet 2009; 41: 15–17. [DOI] [PubMed] [Google Scholar]
- Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet 2007; 39: 960–962. [DOI] [PubMed] [Google Scholar]
- Whyte MP, Totty WG, Novack DV, Zhang X, Wenkert D, Mumm S. Camurati-Engelmann disease: unique variant featuring a novel mutation in TGFbeta1 encoding transforming growth factor beta 1 and a missense change in TNFSF11 encoding RANK ligand. J Bone Miner Res 2011; 26: 920–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A et al. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet 2008; 83: 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralston SH, Albagha OM. Genetics of Paget's disease of bone. Curr Osteoporos Rep 2014; 12: 263–271. [DOI] [PubMed] [Google Scholar]
- Chong B, Hegde M, Fawkner M, Simonet S, Cassinelli H, Coker M et al. Idiopathic hyperphosphatasia and TNFRSF11B mutations: relationships between phenotype and genotype. J Bone Miner Res 2003; 18: 2095–2104. [DOI] [PubMed] [Google Scholar]
- Janssens K, de Vernejoul MC, de Freitas F, Vanhoenacker F, Van Hul W. An intermediate form of juvenile Paget's disease caused by a truncating TNFRSF11B mutation. Bone 2005; 36: 542–548. [DOI] [PubMed] [Google Scholar]
- Hsu YH, Zillikens MC, Wilson SG, Farber CR, Demissie S, Soranzo N et al. An integration of genome-wide association study and gene expression profiling to prioritize the discovery of novel susceptibility Loci for osteoporosis-related traits. PLoS Genet 2010; 6: e1000977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paternoster L, Lorentzon M, Lehtimäki T, Eriksson J, Kähönen M, Raitakari O et al. Genetic Determinants of Trabecular and Cortical Volumetric Bone Mineral Densities and Bone Microstructure. PLoS Genet 2013; 9: e1003247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 2001; 104: 205–215. [DOI] [PubMed] [Google Scholar]
- Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol 2013; 9: 522–536. [DOI] [PubMed] [Google Scholar]
- Duncan EL, Danoy P, Kemp JP, Leo PJ, McCloskey E, Nicholson GC et al. Genome-Wide Association Study Using Extreme Truncate Selection Identifies Novel Genes Affecting Bone Mineral Density and Fracture Risk. PLoS Genet 2011; 7: e1001372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura D, Yamaguchi A, Sasaki K, Deguchi K et al. Targeted disruption of Cbfa1 results in a complete lack of formation owing to maturbone ational arrest of osteoblasts. Cell 1997; 89: 755–764. [DOI] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton R, Denzel A, Gilmour KC, Rosewell IR et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997; 89: 765–771. [DOI] [PubMed] [Google Scholar]
- Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 1997; 89: 773–779. [DOI] [PubMed] [Google Scholar]
- Hsu YH, Kiel DP. Clinical review: Genome-wide association studies of skeletal phenotypes: what we have learned and where we are headed. J Clin Endocrinol Metab 2012; 97: E1958–E1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YJ, Zhang L, Papasian CJ, Deng HW. Genome-wide association studies for osteoporosis: a 2013 update. J Bone Metab 2014; 21: 99–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapunzina P, Aglan M, Temtamy S, Caparros-Martin JA, Valencia M, Leton R et al. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet 2010; 87: 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YF, Matsuo N, Sumiyoshi H, Yoshioka H. Sp7/Osterix is involved in the up-regulation of the mouse pro-alpha1(V) collagen gene (Col5a1) in osteoblastic cells. Matrix Biol 2010; 29: 701–706. [DOI] [PubMed] [Google Scholar]
- Masi L, Agnusdei D, Bilezikian J, Chappard D, Chapurlat R, Cianferotti L et al. Taxonomy of rare genetic metabolic bone disorders. Osteoporos Int 2015; 26: 2529–2558. [DOI] [PubMed] [Google Scholar]
- Kiel DP, Demissie S, Dupuis J, Lunetta KL, Murabito JM, Karasik D. Genome-wide association with bone mass and geometry in the Framingham Heart Study. BMC Med Genet 2007; 8(Suppl 1): S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Dong J, Wang X, Guo H, Wang H, Zhao J et al. JAG1 Mutation Spectrum and Origin in Chinese Children with Clinical Features of Alagille Syndrome. PLoS ONE 2015; 10: e0130355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti S, Canalis E. Notch regulation of bone development and remodeling and related skeletal disorders. Calcif Tissue Int 2012; 90: 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung AWC, Xiao S-M, Cherny S, Li GHY, Gao Y, Tso G et al. Association of JAG1 with Bone Mineral Density and Osteoporotic Fractures: A Genome-wide Association Study and Follow-up Replication Studies. Am J Hum Genet 2010; 86: 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med 1994; 331: 1056–1061. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang Z, An Y, Wu C, Xu Y, Fu Q et al. Exome sequencing reveals a novel PTHLH mutation in a Chinese pedigree with brachydactyly type E and short stature. Clin Chim Acta 2015; 446: 9–14. [DOI] [PubMed] [Google Scholar]
- Albers-Schonberg HE. Roentgenbilder einer seltenen Kochennerkrankung. Munch Med Wochenschr 1904; 51: 365–368. [Google Scholar]
- Cleiren E, Benichou O, Van Hul E, Gram J, Bollerslev J, Singer FR et al. Albers-Schonberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum Mol Genet 2001; 10: 2861–2867. [DOI] [PubMed] [Google Scholar]
- de Vernejoul MC. Sclerosing bone disorders. Best Pract Res Clin Rheumatol 2008; 22: 71–83. [DOI] [PubMed] [Google Scholar]
- Sly WS, Hewett-Emmett D, Whyte MP, Yu YS, Tashian RE. Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci USA 1983; 80: 2752–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venta PJ, Welty RJ, Johnson TM, Sly WS, Tashian RE. Carbonic anhydrase II deficiency syndrome in a Belgian family is caused by a point mutation at an invariant histidine residue (107 His----Tyr): complete structure of the normal human CA II gene. Am J Hum Genet 1991; 49: 1082–1090. [PMC free article] [PubMed] [Google Scholar]
- Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet 2000; 25: 343–346. [DOI] [PubMed] [Google Scholar]
- Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T et al. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet 2000; 9: 2059–2063. [DOI] [PubMed] [Google Scholar]
- Sobacchi C, Frattini A, Orchard P, Porras O, Tezcan I, Andolina M et al. The mutational spectrum of human malignant autosomal recessive osteopetrosis. Hum Mol Genet 2001; 10: 1767–1773. [DOI] [PubMed] [Google Scholar]
- Gelb BD, Shi GP, Chapman HA, Desnick RJ. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 1996; 273: 1236–1238. [DOI] [PubMed] [Google Scholar]
- Johnson MR, Polymeropoulos MH, Vos HL, Ortiz de Luna RI, Francomano CA. A nonsense mutation in the cathepsin K gene observed in a family with pycnodysostosis. Genome Res 1996; 6: 1050–1055. [DOI] [PubMed] [Google Scholar]
- Gong Y, Vikkula M, Boon L, Liu J, Beighton P, Ramesar R et al. Osteoporosis-pseudoglioma syndrome, a disorder affecting skeletal strength and vision, is assigned to chromosome region 11q12-13. Am J Hum Genet 1996; 59: 146–151. [PMC free article] [PubMed] [Google Scholar]
- Johnson ML, Gong G, Kimberling W, Recker SM, Kimmel DB, Recker RB. Linkage of a gene causing high bone mass to human chromosome 11 (11q12-13). Am J Hum Genet 1997; 60: 1326–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001; 107: 513–523. [DOI] [PubMed] [Google Scholar]
- Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet 2002; 70: 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA et al. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med 2002; 346: 1513–1521. [DOI] [PubMed] [Google Scholar]
- Wehrli M, Dougan ST, Caldwell K, O'Keefe L, Schwartz S, Vaizel-Ohayon D et al. Arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature 2000; 407: 527–530. [DOI] [PubMed] [Google Scholar]
- Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y et al. LDL-receptor-related proteins in Wnt signal transduction. Nature 2000; 407: 530–535. [DOI] [PubMed] [Google Scholar]
- Mao J, Wang J, Liu B, Pan W, Farr GH 3rd, Flynn C et al. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell 2001; 7: 801–809. [DOI] [PubMed] [Google Scholar]
- Semenov MV, Tamai K, Brott BK, Kuhl M, Sokol S, He X. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr Biol 2001; 11: 951–961. [DOI] [PubMed] [Google Scholar]
- Rijsewijk F, Schuermann M, Wagenaar E, Parren P, Weigel D, Nusse R. The Drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell 1987; 50: 649–657. [DOI] [PubMed] [Google Scholar]
- Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet 2001; 10: 537–543. [DOI] [PubMed] [Google Scholar]
- Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 2002; 39: 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A et al. A 52- kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet 2002; 110: 144–152. [DOI] [PubMed] [Google Scholar]
- Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet 2001; 68: 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem 2005; 280: 19883–19887. [DOI] [PubMed] [Google Scholar]
- Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem 2005; 280: 26770–26775. [DOI] [PubMed] [Google Scholar]
- Winkler DG, Sutherland MS, Ojala E, Turcott E, Geoghegan JC, Shpektor D et al. Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by bone morphogenetic proteins. J Biol Chem 2005; 280: 2498–2502. [DOI] [PubMed] [Google Scholar]
- Van Wesenbeeck L, Cleiren E, Gram J, Beals RK, Benichou O, Scopelliti D et al. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet 2003; 72: 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomes C, Bottomley HM, Jackson RM, Towns KV, Scott S, Mackey DA et al. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am J Hum Genet 2004; 74: 721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikopoulos K, Venselaar H, Collin RW, Riveiro-Alvarez R, Boonstra FN, Hooymans JM et al. Overview of the mutation spectrum in familial exudative vitreoretinopathy and Norrie disease with identification of 21 novel variants in FZD4, LRP5, and NDP. Hum Mutat 2010; 31: 656–666. [DOI] [PubMed] [Google Scholar]
- Choi HY, Dieckmann M, Herz J, Niemeier A. Lrp4, a novel receptor for Dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo. PLoS ONE 2009; 4: e7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MK, Kramer I, Huber T, Kinzel B, Guth-Gundel S, Leupin O et al. Disruption of Lrp4 function by genetic deletion or pharmacological blockade increases bone mass and serum sclerostin levels. Proc Natl Acad Sci USA 2014; 111: E5187–E5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styrkarsdottir U, Thorleifsson G, Sulem P, Gudbjartsson DF, Sigurdsson A, Jonasdottir A et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature 2013; 497: 517–520. [DOI] [PubMed] [Google Scholar]
- Jin YR, Yoon JK. The R-spondin family of proteins: emerging regulators of WNT signaling. Int J Biochem Cell Biol 2012; 44: 2278–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Zheng XF, Yang YH, Li B, Wang YR, Jiang SD et al. LGR4 acts as a key receptor for R-spondin 2 to promote osteogenesis through Wnt signaling pathway. Cell Signal 2016; 28: 989–1000. [DOI] [PubMed] [Google Scholar]
- Person AD, Beiraghi S, Sieben CM, Hermanson S, Neumann AN, Robu ME et al. WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev Dyn 2010; 239: 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods CG, Stricker S, Seemann P, Stern R, Cox J, Sherridan E et al. Mutations in WNT7A cause a range of limb malformations, including Fuhrmann syndrome and Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome. Am J Hum Genet 2006; 79: 402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Qattan MM. Molecular basis of the clinical features of Al-Awadi-Raas-Rothschild (limb/pelvis/uterus-hypoplasia/aplasia) syndrome (AARRS) and Fuhrmann syndrome. Am J Med Genet A 2013; 161a: 2274–2280. [DOI] [PubMed] [Google Scholar]
- Zheng HF, Tobias JH, Duncan E, Evans DM, Eriksson J, Paternoster L et al. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet 2012; 8: e1002745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ibarbia C, Perez-Nunez MI, Olmos JM, Valero C, Perez-Aguilar MD, Hernandez JL et al. Missense polymorphisms of the WNT16 gene are associated with bone mass, hip geometry and fractures. Osteoporos Int 2013; 24: 2449–2454. [DOI] [PubMed] [Google Scholar]
- Bollerslev J, Henriksen K, Nielsen MF, Brixen K, Van Hul W. Autosomal dominant osteopetrosis revisited: lessons from recent studies. Eur J Endocrinol 2013; 169: R39–R57. [DOI] [PubMed] [Google Scholar]
- Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML et al. Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem 2005; 280: 21162–21168. [DOI] [PubMed] [Google Scholar]
- Glass DA 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 2005; 8: 751–764. [DOI] [PubMed] [Google Scholar]
- Al-Dujaili SA, Lau E, Al-Dujaili H, Tsang K, Guenther A, You L. Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J Cell Biochem 2011; 112: 2412–2423. [DOI] [PubMed] [Google Scholar]
- Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med 2011; 17: 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 2011; 17: 1231–1234. [DOI] [PubMed] [Google Scholar]
- Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS ONE 2011; 6: e25900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-awadh AN, Delgado-Calle J, Tu X, Kuhlenschmidt K, Allen MR, Plotkin LI et al. Parathyroid hormone receptor signaling induces bone resorption in the adult skeleton by directly regulating the RANKL gene in osteocytes. Endocrinology 2014; 155: 2797–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, Piemontese M, Thostenson JD, Weinstein RS, Manolagas SC, O'Brien CA. Osteocyte-derived RANKL is a critical mediator of the increased bone resorption caused by dietary calcium deficiency. Bone 2014; 66: 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens K, Gershoni-Baruch R, Guanabens N, Migone N, Ralston S, Bonduelle M et al. Mutations in the gene encoding the latency-associated peptide of TGF-beta 1 cause Camurati-Engelmann disease. Nat Genet 2000; 26: 273–275. [DOI] [PubMed] [Google Scholar]
- Simpson MA, Hsu R, Keir LS, Hao J, Sivapalan G, Ernst LM et al. Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am J Hum Genet 2007; 81: 906–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurnberg P, Thiele H, Chandler D, Hohne W, Cunningham ML, Ritter H et al. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet 2001; 28: 37–41. [DOI] [PubMed] [Google Scholar]
- Reichenberger E, Tiziani V, Watanabe S, Park L, Ueki Y, Santanna C et al. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am J Hum Genet 2001; 68: 1321–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Chen IP, de Almeida S, Tiziani V, Do Amaral CM, Gowrishankar K et al. A novel autosomal recessive GJA1 missense mutation linked to Craniometaphyseal dysplasia. PLoS ONE 2013; 8: e73576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JJ, Esseltine J, Shao Q, Jabs EW, Sampson J, Auranen M et al. Specific functional pathologies of Cx43 mutations associated with oculodentodigital dysplasia. Mol Biol Cell 2016; 27: 2172–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vernejoul MC, Kornak U. Heritable sclerosing bone disorders: presentation and new molecular mechanisms. Ann NY Acad Sci 2010; 1192: 269–277. [DOI] [PubMed] [Google Scholar]
- Waterval JJ, Borra VM, Van Hul W, Stokroos RJ, Manni JJ. Sclerosing bone dysplasias with involvement of the craniofacial skeleton. Bone 2014; 60: 48–67. [DOI] [PubMed] [Google Scholar]
- Forlino A, Marini JC. Osteogenesis imperfecta. Lancet 2016; 387: 1657–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine CM, Joeng KS, Campeau PM, Kiviranta R, Tarkkonen K, Grover M et al. WNT1 Mutations in Early-onset Osteoporosis and Osteogenesis Imperfecta. N Engl J Med 2013; 368: 1809–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keupp K, Beleggia F, Kayserili H, Barnes AM, Steiner M, Semler O et al. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet 2013; 92: 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini JC, Reich A, Smith SM. Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation. Curr Opin Pediatr 2014; 26: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symoens S, Malfait F, D'Hondt S, Callewaert B, Dheedene A, Steyaert W et al. Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J Rare Dis 2013; 8: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]