

Abstract
Hereditary spastic paraplegias (HSPs) are a clinically and genetically heterogeneous group of disorders characterized by lower extremity spasticity and weakness. Recently, the first de novo mutations in KIF1A were identified in patients with an early onset severe form of complicated HSP. We report two additional patients with novel de novo mutations in KIF1A hereby expanding the genetic spectrum of KIF1A-related HSP. Both children presented with spastic paraplegia and additional findings of optic nerve atrophy, structural brain abnormalities, peripheral neuropathy, cognitive/language impairment, and never achieved ambulation. In particular, we highlight the progressive nature of cerebellar involvement as captured on sequential MRIs, therefore linking the neurodegenerative and spastic paraplegia phenotypes. Exome sequencing in Patient 1 and Patient 2 identified novel heterozygous missense mutations in KIF1A at c.902G>A (p.R307Q) and c.595G>A (p.G199R), respectively. Therefore our report contributes to the expanding the genotypic and phenotypic spectrum of HSP caused by mutations in KIF1A.
Keywords: Hereditary spastic paraplegia, Neuromuscular disorders, KIF1A, Genetics
INTRODUCTION
Hereditary spastic paraplegias (HSPs) are a clinically and genetically heterogeneous group of disorders characterized by progressive lower extremity spasticity and weakness, with onset ranging from early childhood through late adulthood. Complicated HSPs exhibit additional clinical features, including cognitive impairment, ataxia, optic nerve atrophy, retinopathy, dementia, peripheral neuropathy, or epilepsy.1 HSPs can be inherited in an autosomal dominant (AD), autosomal recessive (AR) or X-linked recessive manner. The pathophysiology of HSP involves a length-dependent, progressive, distal axonopathy and retrograde degeneration of the corticospinal tracts.1 This degeneration results from alterations in various neuronal cellular mechanisms including neuronal development, protein folding, endoplasmic reticulum membrane formation and shaping, mitochondrial function and maintenance, lysosomal and endosomal function, myelination, DNA repair, lipid metabolism, and axonal transport.1 To date, 55 HSP causing genes have been identified in these functional areas and new gene discoveries are rapidly emerging.1
One recently discovered HSP associated gene, KIF1A (2q37.3), encodes for Kinesin-like protein KIF1A (KIF1A), which is involved in neuron-specific axonal transport. While recessive KIF1A mutations are known to cause a rare form of childhood onset pure HSP, heterozygous de novo mutations in KIF1A were recently identified as the cause of a more severe phenotype of dominant, early-onset, complicated HSP characterized by cognitive impairment, spastic paraplegia, optic nerve atrophy, peripheral neuropathy, cerebellar atrophy and seizures.2,3 Here we report two additional patients with novel, de novo dominant, missense mutations in KIF1A, hereby providing further confirmation of de novo dominant mutations causing this new phenotype of complicated HSP.
MATERIALS AND METHODS
Patients were evaluated under protocol 12-N-0095 approved by the National Institute of Neurological Disease and Stroke Internal Review Board at the National Institutes of Health (NIH) Neurogenetics clinic. Informed consent was obtained from the family by a qualified investigator. Whole exome sequencing was performed at NIH Intramural Sequencing Center (NISC) using Illumina’s TruSeq Exome Enrichment Kit and Illumina HiSeq 2000 sequencing instruments. Results were confirmed with Sanger sequencing on an ABI 3130×1 capillary sequencer, in forward and reverse direction. Segregation was performed on the parents of both patients. Mutations were analyzed using GEM.app, x-browse, and PolyPhen-2 and searched for in dbSNP, NHLBI EVS, and Exac Browser. Exome sequencing data was processed through a pipeline based on Picard, using base quality score recalibration and local realignment at known indels. We used the BWA aligner for mapping reads to the human genome build 37 (hg19). Single Nucleotide Polymorphism (SNPs) and insertions/deletions (indels) were jointly called across all samples using Genome Analysis Toolkit (GATK) HaplotypeCaller package version 3.1. Default filters were applied to SNP and indel calls using the GATK Variant Quality Score Recalibration (VQSR) approach. Lastly, the variants were annotated using Variant Effect Predictor (VEP). Clinical MRI images and reports were also obtained with informed consent.
CLINICAL PRESENTATION
Patient 1 (P1), who was initially evaluated at 14-years-old, is a young man from Saudi Arabia with cognitive impairment, progressive cerebellar atrophy, optic nerve atrophy, peripheral neuropathy, seizures, and a combination of lower extremity spasticity and upper extremity hypotonia. First concerns arose at age 6 months when he was unable to roll over and had poor head control. He was then diagnosed with optic nerve hypoplasia at 10 months and visual evoked potentials (VEPs) at 1 year of age were consistent with optic nerve atrophy. He was able to stand with support by 2.5 years of age, but lost this ability by age 11. His language development was delayed so that he only spoke a few words. By follow-up report, he developed generalized tonic-clonic seizures at 15 years of age. On examination, he had distal wasting with small hands and feet. He had increased tone and spasticity in the lower extremities with extensor plantar responses bilaterally, but was hypotonic and areflexic in the upper extremities. His strength appeared grossly normal. Additionally, he had scoliosis, distal joint hyperlaxity, and multiple joint contractures.
Sequential MRI images show an initially normal cerebellum, with development of atrophy with time and a persistent thin corpus callosum (Figure 1). Family history is significant for consanguinity in the parents and a sister with congenital muscular dystrophy due to a homozygous pathogenic mutation in exon 4 of FKRP. P1 is heterozygous for the FKRP mutation and muscle biopsy at 5 years of age was normal.
Figure 1.
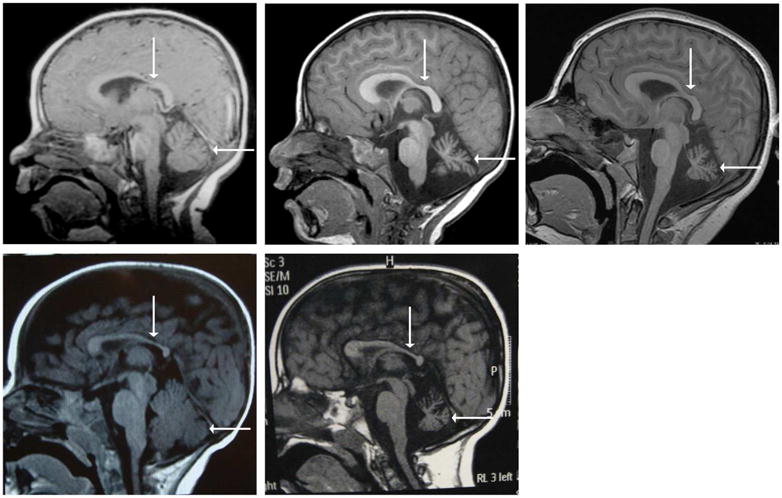
All images are T1-weighted sagittal MRIs. Images (a–c) are of Patient 1 at 11 months old (a), 3.6 years old (b), and 12.5 years old (c). Image (a) shows an age-appropriate size of the cerebellum. Images (b) and (d) illustrate cerebellar atrophy that progresses with age. All three images show thinning of the body of corpus callosum, particularly in the posterior aspect. Images (d–e) are of Patient 2 at 6 months old (a) and 6 years old (e). Again, image (d) shows an age-appropriate cerebellar size, whereas image (e) illustrates cerebellar atrophy with time. Thinning of the corpus callosum is also apparent in (d) and (e). The corpus callosum is designated by a vertical arrow and the cerebellum by a horizontal arrow.
Patient 2
Patient 2 (P2) is a 6-year-old boy from Brazil with cognitive impairment, cerebellar atrophy, optic nerve atrophy, distal neuropathy, spastic paraplegia, and axial hypotonia. He was born with bilateral clubfoot deformities. At 6 months of age, he was unable to roll over or sit and had poor head control. At 1 year of age, he was diagnosed with optic nerve atrophy. His language development was delayed and he had acquired about 20 words by 6 years of age. He made slow gains in cognitive function and had shown no regression or loss of functions previously obtained. On exam, he was unable to follow commands and had minimal speech. He had truncal hypotonia and a spastic increase in tone, more prominent in the lower extremities (Figure 2). He rolled from supine to prone to push himself into a sitting position, but could not stand or walk. Reflexes were hyperactive. His strength was within normal limits for his age, except for some apparent weakness in his hands. He had mild kyphosis and contractures in the ankles with persistent clubfoot deformities.
Figure 2.
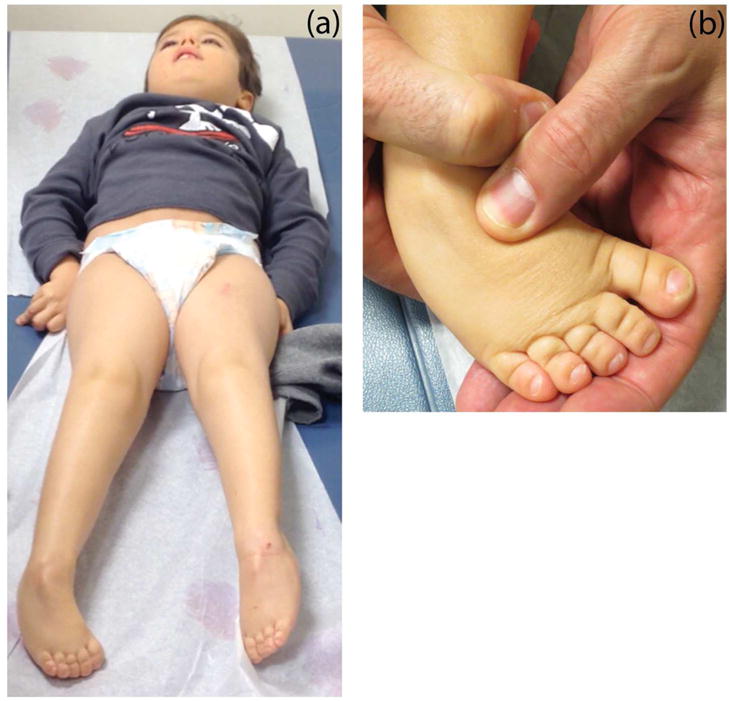
Clinical photos of P2 illustrating his extremity predominant spasticity (a) and club foot (b).
Muscle ultrasound showed a mixed pattern of increased echogenicity in a streak like pattern and fasciculations were seen in various muscles, further confirming a neurogenic etiology of the changes. The hands and distal lower extremity muscles were most involved. Brain MRI showed a thinning of the corpus callosum and cerebellar atrophy that developed over time (Figure 1).
GENETIC RESULTS
Exome sequencing in P1 and P2 identified novel heterozygous missense mutations in KIF1A at c.902G>A (p.R307Q) and c.595G>A (p.G199R), respectively. Parental segregation was negative, indicating that the mutations occurred de novo. These mutations were not reported in dbSNP, NHLBI EVS, and Exac Browser databases. Both mutations were designated as disease-causing in x-browse and were predicted to be probably damaging with a Polyphen2 score of 1.000. No other pathogenic mutations in known HSP genes were identified.
DISCUSSION
Here we report two novel de novo KIF1A mutations in patients presenting with early onset complicated HSP characterized by spastic paraplegia and additional findings: (1) cognitive impairment, (2) non-ambulation, (3) language impairment, (4) optic nerve atrophy, (5) peripheral neuropathy, and (6) progressive cerebellar atrophy. Additionally, patient 1 developed seizures as a teenager. This phenotype is consistent with the previously reported de novo KIF1A mutations with our patients representing an earlier onset and more severe end of the clinical spectrum compared to the pure HSP caused by recessive mutations in KIF1A.
In our patients, serial MRIs showed progressive cerebellar atrophy over a few years without overt clinical manifestations. Importantly, MRIs in the first year of life revealed appropriately sized cerebellums. Although the clinical course so far has appeared relatively static from a motor perspective, patient P1 developed seizures at 15 years of age, similarly to previous de novo patients. A recent report of de novo KIF1A mutations causing progressive encephalopathy and brain atrophy in 6 patients illustrates the central nervous system involvement associated with this new phenotype. Our patients firmly link the progressive and degenerative nature of brain involvement with the spastic paraplegia that is characteristic of HSP.
KIF1A is a kinesin (KIFs) that participates in axonal anterograde transport of synaptic vesicles. Though initially thought to act as a monomer, recent studies show it primarily acts as a highly processive dimer.4,5 KIF1A uses ATP hydrolysis to power its movement along microtubules (MTs). ATP hydrolysis produces conformational changes in three regions of the motor domain (amino acids (aa) 1–361): switch I region (aa 202–218), switch II cluster (aa 248–324), and the neck-linker region (aa 353–361).6 The four mutations in the initial report of recessive KIF1A and eleven recently reported de novo missense mutations all fall in the motor domain and many are in the regions that undergo conformational change (Figure 3).2,3,7 Similarly, both patients presented here were found to have missense mutations in the motor domain of KIF1A (Figure 3).2
Figure 3.

(a) Schematic representation of KIF1A. The motor domain (aa 1–361) is detailed on the left with pathogenic mutations identified. Mutations highlighted in green are the early reported homozygous mutations. Those in blue are the recently reported de novo mutations. The mutations in purple are those of P1 and P2 reported here. Additionally, the microtubule binding domains (MT1, MT2, MT3) are labeled. The schematic also includes distal mutations, not in the motor domain, that have been reported to cause Hereditary Sensory and Autonomic Neuropathy IIC (HSAN2C). Other important functional domains are identified: forkhead-associated domain (FHA) and pleckstrin homology domain (PH). (b) Illustration of the amino acid conservation amongst KIF1A orthologs at the residues altered in the two cases reported (G199 and R307, highlighted in purple). The amino acid alignment was generated using HomoloGene (NCBI) and includes human (H. sapiens; NP_001230937), mouse (M. musculus; NP_032466.2), chicken (G. gallus; XP_003641781.1), Zebrafish (D. rerio; XP_005166002.1), Drosophila (D. melanogaster; NP_001246373.1), roundworm (C. elegans; NP_001022041.1), and frog (X. tropicalis; XP_002933380.2).
Patient 1’s p.R307Q substitution falls within one of the three MT binding domains (Figure 3). This domain’s primary actor is the K-loop (aa 286–300), a unique feature of the KIF1 family. The high affinity of the K-loop for the MT is thought to contribute to KIF1A’s high processivity.8 This interaction is likely mediated by the strong positive charge of the K-loop (lysine rich) and the negative charge of tubulin (glutamate rich).8 Okada et al (2000) showed that a reduction in the K-loop’s positive charge decreased its affinity for MTs and subsequently decreased KIF1A’s processivity.8 P1’s mutation substitutes a positively charged arginine for an uncharged glutamine. This reduction in positive charge near the K-loop may therefore weaken KIF1A’s binding to MTs.
Patient 2’s G199R substitution involves the first amino acid in the loop L9, a secondary structure in the switch I region. L9 performs a β-to-α conformational change that allows for γ-phosphate to be released after ATP hydrolysis.8 This substitution in L9 may impact the successful turnover of ATP hydrolysis, which is crucial for KIF1A function. Additionally, Lee et al (2015) describes a patient with a mutation at A202 (also in the L9 loop) who presents with a similar phenotype.2 Furthermore, anterograde movement of KIF1A is impaired in A202P mutant mice as demonstrated by only a 9.9% accumulation of KIF1A in the distal neurite compared with 93.2% in the wild type.2 Since P2’s mutation affects a residue that is only three amino acids away, is part of the same secondary structure, and results in a similar phenotype, it is likely that p.G199R has a similar disease causing effect.
Given KIF1A’s functional requirements for efficient ATP hydrolysis, mutations in the regions involved in conformational change and MT binding are most likely to be disease causing. However, functional studies in cellular and animal models would be the next step to further support this assumption. Recent studies suggest that KIF1A is important in the transport of dense core vesicles carrying precursor proteins bound for the synapse.9 The transport of these vesicles, often over long distances, is crucial for neuronal development, survival, learning, and memory.9 The impairment of these functions likely contributes to the characteristic selective susceptibility and degeneration of long axons in HSP.
Our patients and other reports of dominantly acting KIF1A mutations suggest a more severe phenotype than recessively inherited KIF1A mutations. A recent study of MT gliding assays showed a decreased velocity of KIF1A with de novo mutations compared with recessive, consistent with the observed phenotypic variability.10 One possible explanation for this is a dominant negative effect, especially given KIF1A’s potential homodimerization in vivo.5 Additionally, the previously reported recessive mutations were all missense mutations, leaving the possibility that null mutations may be embryonically lethal. However, a recent report of a de novo KIF1A mutation resulting in pure HSP in a father and then his son also illustrates the still evolving phenotypic variability of KIF1A mutations.11 Along the same lines, more severe recessive KIF1A mutations causing complicated HSP or mild dominantly acting mutations may be discovered in the future. Therefore, these two case studies contribute to our expanding knowledge of genotype/phenotype correlations in HSP.
Table 1.
Clinical findings of P1 and P2, as well as a summary of clinical findings in the two previous reports of patients with de novo dominant KIF1A mutations.
| Patient 1 | Patient 2 | Lee et al. (2015) | Nieh et al. (2015) | |
|---|---|---|---|---|
| KIF1A mutation (de novo) | c.902G>A p.R307Q (p.Arg307Gln) |
c.595G>A p.G199R (p.Gly199Arg) |
Various de novo missense mutations | Various de novo missense mutations |
| Gender | Male | Male | 9 Female, 5 Male | 4 Female, 2 Male |
| Age (years) | 14 | 6 | 2–24 | 1.5–16 |
| Age of onset and initial findings | 6 mo – delayed milestones (head control, rolling over) | Congenital – b/l clubfoot 6 mo – delayed milestones (head control, rolling over, sitting unsupported) |
3/14 Congenital 8/14 Infancy 3/14 Young childhood |
NR |
| Cognition | Severe cognitive impairment with language delay | Severe cognitive impairment with language delay | 4/14 Mild ID 3/14 Moderate ID 3/14 Severe ID 4/14 ID NOS |
6/6 Severe global developmental delay |
| Language Development | Words and simple sentences | Approximately 20 words | 3/14 Normal 5/14 Delay-Sentences 2/14 Words 4/14 Non-verbal |
4/6 Severe language delay 1/5 Moderate language delay 1/6 Mild language delay |
| Max motor function | Get to seated (at 5 y/o) | Sit when placed (at 2.5 y/o) | 6/14 Independent ambulation 3/14 Ambulatory with assistance 5/14 Non-ambulatory |
2/6 Ambulatory with assistance 4/6 Non-ambulatory |
| Ophthalmologic involvement | Sluggish pupillary responses, does not track, roving eye movements, conjugate gaze, pale optic disks | Sluggish pupillary responses, fixates on light, does not track, minimal nystagmus, pale optic disks | 9/14 Optic Nerve Atrophy | 3/6 Optic nerve atrophy 4/6 Cortical visual impairment, 1/6 Abnormal eye movements 1/6 Cataracts |
| Microcephaly | Yes, 50.5cm (<3rd %ile) | No, 50cm (25th %ile) | 4/14 Yes | 4/6 Yes |
| Epilepsy | Yes, onset at 15 years old, generalized tonic-clonic | No | 3/14 Epilepsy 2/14 Abnormal EEG 9/14 No epilepsy |
2/6 Seizures 4/6 No seizures |
| EMG/NCS | Distal motor neuropathy with absent sensory responses in upper and lower extremities | Axonal sensory-motor polyneuropathy | 4/14 Neuropathy | NR |
| Spine | Scoliosis: 23° T11-L4 &16° T5–T11 | Minor kyphosis on exam | NR | NR |
| Contractures | Elbows (end-grade, wrists, ankles, knees (almost 90°) | Ankles (Achilles tendon release at 6 months of age) | NR | NR |
NR = not reported, ID = intellectual disability, EEG = electroencephalogram, EMG = electromyogram, NCS = nerve conduction studies
Acknowledgments
The study of these patients, under protocol 12-N-0095, was approved by the National Institute of Neurological Disease and Stroke Internal Review Board at the National Institutes of Health (NIH). All funding was provided by NIH.
Footnotes
DISCLOSURES
The authors declare no conflict of interest.
AUTHORSHIP CONTRIUBTIONS
Hotchkiss L was the primary creator and author of this manuscript. Donkervoort S interpreted the exome results, identified the mutations as causative, provided guidance throughout the writing and editing process. Leach ME and Bharucha-Goebel DX were involved in the clinical evaluation of patients and the editing of the manuscript. Mohassel P assisted with the brain MRI interpretation and editing. Bradley N, Nguyen B, and Hu, Ying were responsible for confirmation of the exome results by Sanger sequencing and assisted with the validation of the mutations. Gurgel-Giannetti J referred Patient 2 to NIH for evaluation, provided clinical assessment, and provided brain MRI images. Bönnemann CG is the principal investigator for the protocol, evaluated the patients clinically, provide guidance and editorial support throughout the drafting of this manuscript.
Contributor Information
Leslie Hotchkiss, National Institutes of Health and Weill Cornell Medical College.
Sandra Donkervoort, National Institutes of Health.
Meganne Leach, National Institutes of Health and Children’s National Medical Center.
Payam Mohassel, National Institutes of Health.
Diana X. Bharucha-Goebel, National Institutes of Health and Children’s National Medical Center.
Nathaniel Bradley, National Institutes of Health.
David Nguyen, National Institutes of Health.
Ying Hu, National Institutes of Health.
Juliana Gurgel-Giannetti, Universidade Federal de Minas Gerais.
Carsten G. Bönnemann, National Institutes of Health.
References
- 1.Blackstone C. Cellular Pathways of Hereditary Spastic Paraplegia. Annu Rev Neurosci. 2012;35:25–47. doi: 10.1146/annurev-neuro-062111-150400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JR, Srour M, Kim D, Hamdan FF, et al. De novo Mutations in the Motor Domain of KIF1A Cause Cognitive Impairment, Spastic Paraparesis, Axonal Neuropathy and Cerebellar Atrophy. Hum Mutat. 2014;36(1):69–78. doi: 10.1002/humu.22709. [DOI] [PubMed] [Google Scholar]
- 3.Erlich Y, Edvardson S, Hodges E, Zenvirt S, et al. Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res. 2011;21(5):658–664. doi: 10.1101/gr.117143.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okada Y, Hirokawa N. A processive single-headed motor: kinesin superfamily protein KIF1A. Science. 1999;283(5405):1152–7. doi: 10.1126/science.283.5405.1152. [DOI] [PubMed] [Google Scholar]
- 5.Hammond JW, Dawen C, Blasius TL, Li Z, et al. Mammalian Kinesin-3 motors are dimeric in vivo and move by processive motility upon release of autoinhibition. PLoS Biology. 2009;7(3):e1000072. doi: 10.1371/journal.pbio.1000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nitta R, Kikkawa M, Okada Y, Hirokawa N. KIF1A alternately uses two loops to bind microtubules. Science. 2004;305(5684):678–83. doi: 10.1126/science.1096621. [DOI] [PubMed] [Google Scholar]
- 7.Klebe S, Lossos A, Azzedine H, Mundwiller E, et al. KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Europ J Hum Genet. 2012;20(6):645–649. doi: 10.1038/ejhg.2011.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okada Y, Hirokawa N. Mechanism of the single-headed processivity: diffusional anchoring between the K-loop of kinesin and the C terminus of tubulin. Proc Natl Acad Sci. 2000;97(2):640–5. doi: 10.1073/pnas.97.2.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lo KY, Kuzmin A, Unger SM, Petersen JD, et al. KIF1A is the primary anterograde motor protein required for the axonal transport of dense-core vesicles in cultured hippocampal neurons. Neurosci Lett. 2011;491(3):168–173. doi: 10.1016/j.neulet.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 10.Nieh SE, Madou MR, Sirajuddin M, Fregeau B, et al. De novo mutations in KIF1A cause progressive encephalopathy and brain atrophy. Ann Clin Trans Neuro. 2015;2(6):623–35. doi: 10.1002/acn3.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ylikallio E, Kim D, Isohanni P, Auranen M, et al. Dominant transmission of de novo KIF1A motor domain variant underlying pure spastic paraplegia. Eur J Hum Genet. 2015 doi: 10.1038/ejhg.2014.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riviere JB, Ramalingam S, Lavastre V, et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet. 2011;89(2):219–230. doi: 10.1016/j.ajhg.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]