

Abstract
Protein flexibility and conformational diversity is well known to be a key characteristic of the function of many proteins. Human blood coagulation proteins have multiple substrates, and various protein-protein interactions are required for the smooth functioning of the coagulation cascade to maintain blood hemostasis. To address how a protein may cope with multiple interactions with its structurally diverse substrates and the accompanied structural changes that may drive these changes, we studied human Factor X. We employed 20 ns of molecular dynamics (MD) and steered molecular dynamics (SMD) simulations on two different conformational forms of Factor X, open and closed, and observed an interchangeable conformational transition from one to another. This work also demonstrates the roles of various aromatic residues involved in aromatic-aromatic interactions which make this dynamic transition possible.
Keywords: protein flexibility, molecular dynamics, steered molecular dynamics, simulations, coagulation, Factor Xa
INTRODUCTION
Evolution has induced a large diversity within the serine proteases of the blood coagulation cascade. This evolution has been driven primarily by the efficiency of catalysis, presence of zymogenic forms, multiple substrates, and regulation by a variety of positive and negative activators, inhibitors, and co-factors. Since most of these factors are directly or indirectly related to the binding sites of protein-protein interaction, it would be logical to infer that the spatial organization and flexibility of the coagulation serine protease binding sites has been a major contributor in this evolutionary process.1–3
The blood coagulation proteases are central to physiological processes and are involved in maintaining organism hemostasis. These coagulation proteins are strongly regulated and synchronized in this process, and failure at any step in this process leads to various diseases and dysfunctions such as hemophilia and von Willebrand disease.4,5 Factor X, a vitamin K dependent serine protease, is part of this human coagulation cascade. Structurally, it is a two chain and four domain protein. The first three domains consist of an N-terminal γ-carboxyglutamic acid rich Gla domain followed by two epidermal growth factor (EGF) like domains EGF1 and EGF2 constituting the light chain which is joined by a disulfide linkage to the heavy chain, the serine protease domain. Factor X is synthesized in liver or human hepatoma cells in zymogen form (FX) and is converted into its active form (FXa) by a proteolytic cleavage between amino acids Arg15-Ile16 (chymotrypsinogen nomenclature is used throughout the study), resulting in the release of an activation peptide segment of 52 amino acids from the N-terminus of the heavy chain.6–8 After activation, FXa combined with FVa (as a cofactor) on activated platelet membranes in a Ca2+-dependent association and makes the prothrombinase complex which is responsible for activating prothrombin (zymogen) to thrombin (active).9–11
The substrate recognition and cleavage sites in FXa are mainly characterized by: an active site containing the catalytic triad residues His-57, Asp-102, and Ser-195; and two binding subsites, S1 and S4, along with several other small subsites, all located on the serine protease domain of FXa. The S1 subsite is a narrow 8.0 Å deep recess near the active site. It has hydrophobic walls and contains a negatively charged aspartate (Asp-189) at the bottom of the cavity which makes a salt bridge with the positively charged moiety of lysine or arginine side chains from the substrates. In contrast, the S4 subsite is a large and well defined hydrophobic surface cleft. It is comprised of aromatic residues Tyr-99, Phe-174 and Trp-215 on three sides of its surface and can bind isoleucine, proline, and glutamic acid residues of the substrates.12,13
There has been a tremendous growth in the number of published structures of FXa since the time when Padmanabhan et al (1993) reported the first crystal structure. Currently there are 35 crystal structures available in the PDB databank, out of which two are unliganded FXa structures and 33 are FXa bound with small chemical-compounds/ligands.14–24 However, no reported structure of FXa in complex with any of its natural substrates is currently known.
A comparison of all of these known structures reveals that there is very little difference in the general topology and overall shape of FXa upon the binding of small compounds. This is especially true for the size and shape of the binding sites. The calculated backbone RMSD of all of the reported structures (focusing only on the serine protease domain) is in the range of 1.3 to 2.0 Å, whereas, the all-atom RMSD of the binding site residues is only 0.2 to 0.6 Å. This shows that there is little, if any, induced fit in response to the small molecule binding, suggesting rigidity in FXa. Since there are various substrates (Table I) that act on FXa in the human blood coagulation pathway, like a). prothrombin - conversion to meizothrombin, b). meizothrombin - conversion to thrombin,25,26 c). FVII - conversion to FVIIa as a form of positive feedback in the extrinsic pathway,27 d). protease-activated receptor 2 (PAR2)28, and e). autocatalysis - autocleavage of its own 148 loop,29,30 it raises the question of how the multiple protein-protein interactions and their efficient catalytic reactions occur and what structural roles various binding site and surrounding residues play to aid in this protein-protein interaction, given that the protein remains unchanged with small molecule binding.
Table I.
Sequence of Various Substrate Showing the Cleavage Site Between P1 and P1′ Residues (In Bold)
| Substrate | P6 | P5 | P4 | P3 | P2 | P1 | P1′ | P2′ | P3′ | P4′ | P5′ | P6′ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pt1a | S | Y | I | D | G | R | I | V | E | G | S | D |
| Pt2b | S | K | P | Q | G | R | I | V | G | G | K | V |
| FVIIc | R | A | I | E | G | R | T | A | T | S | E | Y |
| FXd | T | H | E | K | G | R | Q | S | T | R | L | K |
Prothrombin.
Meizothrombin.
Factor VII.
Factor Xa showing autocatalytic loop residues.
We propose a hypothesis of conformational diversity as a possible explanation for this phenomenon. According to this hypothesis, the substrates capture an active conformation from among the ensemble of available conformations of FXa, something which is not triggered by small molecule binding – this general idea is gaining traction in the literature and has been shown to exist in systems like the Arc repressor31, plasmepsin II32, glutathione transferase33 and germline 48G7 antibody34. Under this mechanism, FXa would exist in many different conformational states and each substrate could capture its most suitable conformation for binding.35
To test if our hypothesis is valid for FXa, we studied a unique crystal structure published by Wang et al in 2003.36 This FXa structure (pdb code 1P0S) is bound with ecotin - a serine protease inhibitor from Escherichia coli, and is unique in its arrangement of binding site residues. It has a backbone RMSD of 1.8 Å (of the serine protease domain) compared to the unliganded structure of FXa, which is in the range of backbone RMSD differences of other small molecule bound structures (1.3–2 Å). However, the peculiar thing to note here is that, the all-atom RMSD of binding site residues in this system is 1.42 Å which is very different from the binding site differences of all other available structures (where the range was between 0.2 – 0.6 Å). Renderings of the crystal structure of the three conformations (liganded, unliganded, and ecotin-bound FXa) and their structural differences are shown in Figure 1. Among the binding subsites, the main difference was observed in the S4 binding subsite. This subsite, which is open in all of the known structures (the minimum distance between the two gating aromatic residues Tyr-99 and Phe-174, not including hydrogens, is in the range of 7.0 to 9.5 Å in both the liganded and unliganded structures) is in a collapsed or closed state in the ecotin-bound crystal structure (where the minimum distance between the two gating aromatic residues Tyr-99 and Phe-174, not including hydrogens, is 3.7 Å) and is physically not accessible to any substrate residue that must bind in the S4 subsite.
Figure 1.
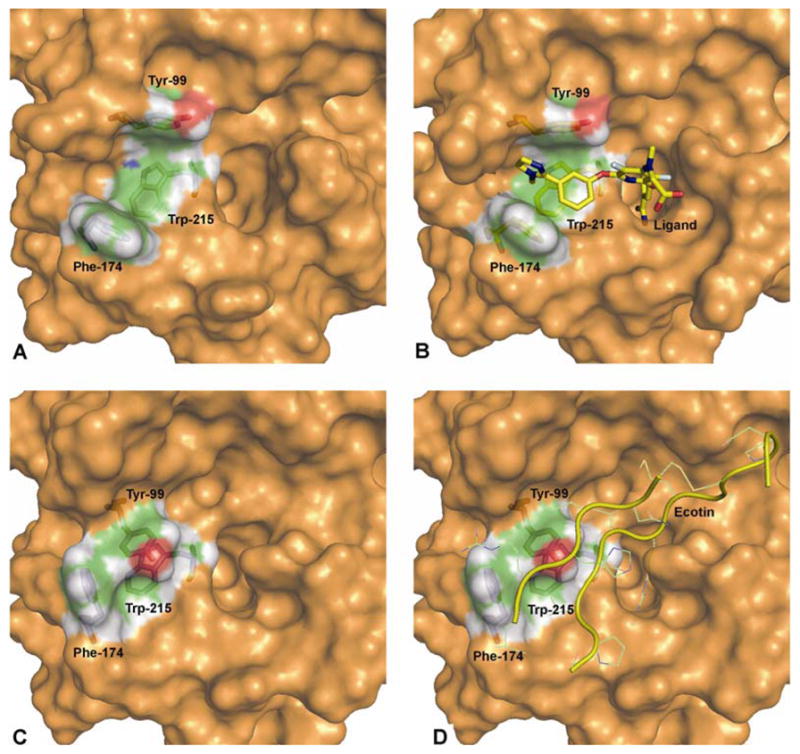
Surface models of the three different crystal structure conformations: A). unliganded, B). liganded, C). ecotin-bound (without ecotin) and D). ecotin-bound (with ecotin) FXa. The three S4 subsite residues Tyr-99, Phe-174 and Trp-215 are shown as stick models along with the ligand (in model B). The model D shows ecotin in yellow ribbon with ecotin residues interacting with FXa in line model. The pdb codes for crystal structures used are 1C5M, 1FJS and 1P0S for unliganded, liganded and ecotin-bound FXa, respectively.
Although a wealth of information can be obtained from the small molecule bound and unbound crystal structures, nevertheless it is important to consider that FXa can exhibit other significant dynamic motions under physiological conditions. In this study, we tested the hypothesis of conformational diversity by performing MD simulations on two different conformational states of FXa in an aqueous environment at physiological temperature. This study provided important insights into binding site dynamics of FXa. The mechanistic structural role of Glu-97 and Lys-96 and the importance of aromatic-aromatic interactions of various aromatic residues His-57, Tyr-60, Phe-94, Tyr-99, Phe-194 and Trp-215 which causes the opening and closing of the S4 binding subsite was shown (histidine was considered as an aromatic residue, as shown by Meurisse et al37). Finally, we will also discuss the ‘interchangeable capability’ of FXa from an open state to a closed state and closed to open state and the residues that aid in this process.
MATERIALS AND METHODS
Calculations were performed on a 152-node HP Itanium2 based Atlantis cluster in the Texas Learning and Computation Center (TLC2) at UH. All of the simulations were performed using the NAMD2 program with the CHARMM 29b2 force field.38,39 For structure visualization and simulation analyses, the visual molecular dynamics (VMD)40 and PyMOL41 programs were used.
System Setup
Two simulation systems were prepared, one for the unliganded FXa system (open system) and another for the ecotin-bound FXa system (closed system). A 20 ns of MD simulation was carried out for the open system while 15 ns of SMD was performed for the closed system.42
Open System
The chosen structure for this study (PDB entry 1C5M; unliganded FXa crystal structure) was solved to a resolution of 1.95 Å and contains 293 residues (241 heavy-chain residues and 52 light chain residues) along with 440 crystal waters. The system setup procedure was initiated by adding hydrogen atoms using the HBUILD procedure in CHARMM followed by a short 50 step conjugate gradient (CG) energy minimization. All hydrogen atoms were added according to the predicted protonation states of all the ionizable residues using a pKa method implemented in the University of Houston Brownian Dynamics (UHBD) program at pH 7.0, using a dielectric constant of 80.0 for water and 20.0 for the protein.43,44 The protein was solvated in a box of TIP3P water molecules, such that there was at minimum 13.0 Å of water between the surface of the protein and the edge of the simulation box, using the Solvate plugin in VMD. Crystal waters were retained and any added bulk water molecules within 2.5 Å of the protein were excluded. To maintain electrical neutrality of the system, a single chloride ion was added using the Autoionize plugin in VMD, which was initially at least 7.0 Å away from the surface of the protein. The final resulting periodic box was 104 Å × 104 Å × 84 Å in dimension and contained a total of 90,979 atoms from 293 protein residues, 440 crystal waters, 28,362 TIP3P bulk waters and one chloride ion.
Closed System
The chosen structure for this study (PDB entry 1P0S; Ecotin-bound FXa crystal structure) was solved to a resolution of 2.80 Å and contains 473 residues (235 heavy-chain residues, 99 light chain residues and 139 ecotin residues) along with 51 crystal waters. Since our aim was to compare the dynamics of each of the two systems, the system sizes were kept as similar as possible. Hence, only 52 light chain residues (the first 47 residues which were part of Gla domain and absent in open system were deleted) in the ecotin-bound structure (closed system) were kept. Only six residues of ecotin were retained in the system (Val-81, Ser-82, Thr-83, Arg-84, Met-85 and Ala-86). The rest of the system setup procedure was the same as for the unliganded system. The final resulting periodic box was 97 Å × 91 Å × 98 Å and contained a total of 86,173 atoms derived from 293 protein residues, 31 crystal waters and 27,519 TIP3P bulk water molecules.
Simulation Setup
A stepwise energy minimization of water molecules, side chains, and finally the entire system was performed to relieve bad contacts and to accommodate the protein to its newly solvated environment. First, only the hydrogen atoms were energy minimized for 300 steps of CG, followed by minimization of water and protein side chains while keeping the protein backbone fixed for 400 CG steps, and finally the entire system was energy minimized for 500 steps of CG. The NAMD program with the CHARMM 29b2 force field were used to slowly heat the system from 0 to 310 K in 10 K/ps increments for a total of 50 ps under constant volume conditions. During the simulation, periodic boundary conditions were used to avoid edge effects and a 12.0 Å cut-off was used to truncate non-bonded interactions. Long range electrostatic interactions were treated using the smooth particle mesh Ewald (PME) method.45 All bonds involving hydrogen atoms were constrained with the SHAKE algorithm46 in conjunction with a 2.0 fs time step.
A short 500 ps of equilibration simulation was carried out on both systems in the NPT (isothermal-isobaric) ensemble to allow the protein and water to adapt properly in their environment. Finally, a 20 ns of production run was performed on the open system and data were collected every 1.0 ps during the simulation.
Steered Molecular Dynamics
A constant-velocity SMD was used in the closed system to pull the ecotin out from its bound FXa conformation. The direction of ecotin departure was determined using the criterion that the ecotin could be pulled out of the binding site with the least collision with the neighboring protein residues. Force was applied to the atom closest to the center of mass of the ecotin, along a vector defined outward direction from the binding subsites of FXa, to avoid the possibility of its clash with the binding subsite wall of the protein. The pulling force was assigned to backbone nitrogen atom of Arg-84 that was closest to the center of mass of the ecotin molecule. The velocity was fixed at 10.0 Å/ns. In a few hundred picoseconds (~400–450 ps) after starting the simulation, ecotin moved beyond van der Waal’s contact distance (~4–4.2 Å) with Factor Xa. During the remainder of the simulation, once ecotin was 8 Å away from the surface of the protein, it was constrained in that position and a total of 15 ns of simulation was performed on the system. Conformations of the system were saved every 1.0 ps during the simulation.
RESULTS AND DISCUSSION
Molecular dynamics simulations were performed on the open and closed conformations of FXa. A 20 ns of MD simulation was performed on the open state system and 15 ns of SMD simulation was performed on the closed state system. SMD was employed in the ecotin-bound structure of FXa because we wanted to slowly accommodate the FXa alone to its solvent environment instead of simply removing the ecotin and starting the simulation of FXa.
Since the binding subsite of FXa is open in all of its unliganded and liganded forms, except in the ecotin-bound form, it is reasonable to assume that this is the preferred conformation of FXa. By this reasoning, ecotin binds to less populated conformation exhibiting a closed binding site. If ecotin is not bound, FXa should sample open and closed states, given sufficient conformational sampling.
Conformational Changes
To support our view, we started our analysis by monitoring the structural changes occurring throughout the simulation of FXa. The root mean square deviation (RMSD) plots in Figure 2 show the comparison of these two simulated systems (only the serine protease domain). In this figure, showing the backbone RMSD of both the open and closed systems with respect to their starting equilibrated structures, it is clear that both the systems are relatively stable with RMSDs in the range of 1.7 – 2.5 Å.
Figure 2.
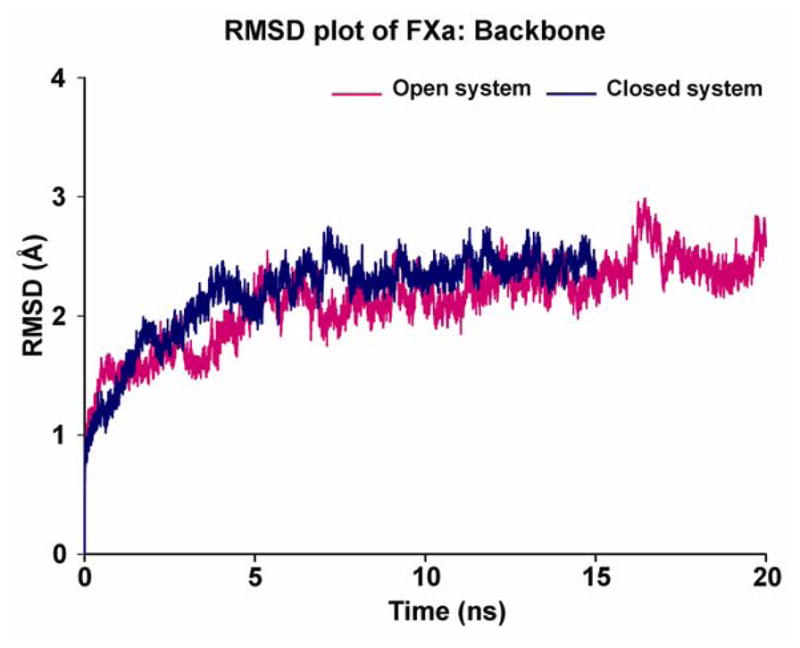
RMSD time course for backbone atoms of the serine protease domain of the two different conformations of FXa (magenta color - open system of unliganded protein with respect to its starting structure; blue color - closed system of ecotin-bound protein with respect to its starting structure). The time courses are shown for 20 ns for open system and 15 ns for closed system.
Since our primary objective was to monitor the dynamical behaviors of the binding site, an all-atom RMSD plot was constructed of just the aromatic residues Tyr-99, Phe-174 and Trp-215 that comprise the S4 binding site and are major contributors to open and closed conformations of this binding site (Figure 3). The first two traces of RMSD differences shown in this figure are 1). open system and 2). closed system, both with respect to their starting equilibrated structures. These traces clearly show major structural deviations in the 15–20 ns range in the open system and in the 13–15 ns range in the closed system. These dynamic changes in the S4 binding site are localized to sidechains and do not affect the overall deviations of the rest of the protein since the backbone deviation of these systems remain stable at the time points where the binding site RMSD exhibits major changes. To test whether the closed conformation tries to fall back into the open one, a third plot was constructed in Figure 3, where the RMSD of the closed system is shown with respect to the starting conformation of the open system. This graph reveals that the closed conformation explores the open conformational states (i.e. the closer the 3rd trace to the X-axis, the closer it gets to the open state conformation) for short periods of time up to 13 ns after which the S4 subsite moves toward the open state and remains there through the end of the simulation at 15 ns.
Figure 3.
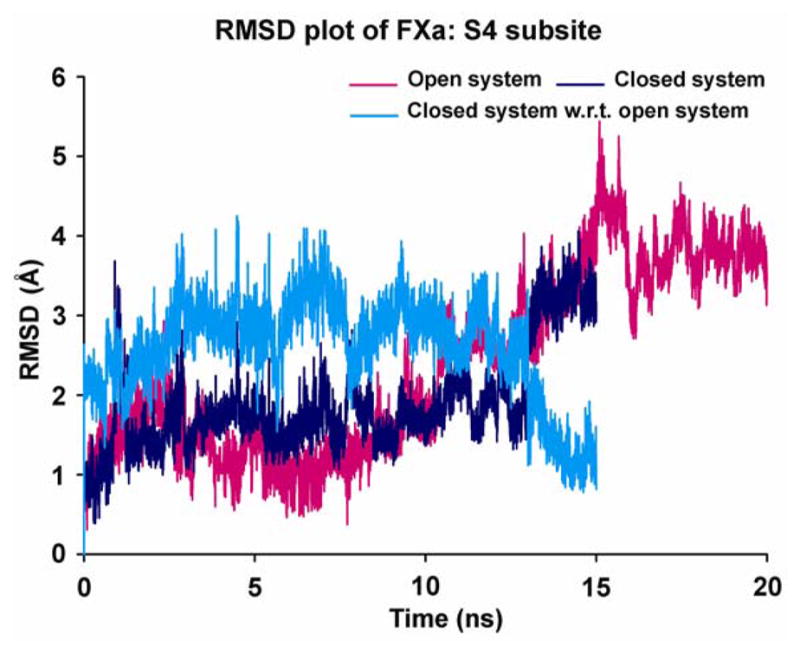
RMSD time course for all-atoms of S4 subsite (Tyr-99, Phe-174 and Trp-215) of the two different conformations of FXa (magenta color - open system of unliganded protein with respect to its starting structure; blue color - closed system of ecotin-bound protein with respect to its starting structure; cyan color - closed system of ecotin-bound protein with respect to the starting structure of open system conformation). The time courses are shown for 20 ns for open system and 15 ns for closed system.
Protein Flexibility
The B-factor values of all residues of the serine protease domain were calculated by using the Debye-Waller formula (B=(8/3)π2RMSF2) to further dissect the protein flexibility. B-factors, called Debye-Waller temperature factors, are a measure of thermal fluctuations of each atom per residue. The B-factors were averaged for each residue from each frame of the trajectory and plotted against residue number (Figure 4). The simulated B-factors are in general agreement with the experimentally determined B-factors from the crystal structures and suggest three major findings: 1). Large fluctuations are exhibited in the loops (with the exception of the loop from 100–120, which exhibited subdued fluctuations because of steric contacts with the light chain of FXa). 2). The most mobile part in this plot is the autocatalysis loop from residue number 140–156, showing fluctuations up to 1.7 Å and 3). Most of the thermal fluctuations of the two different conformations match well except for the loops that contain the two gating residues Tyr-99 and Phe-174 in the S4 binding site. The thermal fluctuations are 36% and 53% greater in the closed state as compared to the open state for Tyr-99 and Phe-174, respectively. This observation suggests that these residues explore more conformations in the closed state while adjusting itself back to its unliganded open state conformation.
Figure 4.
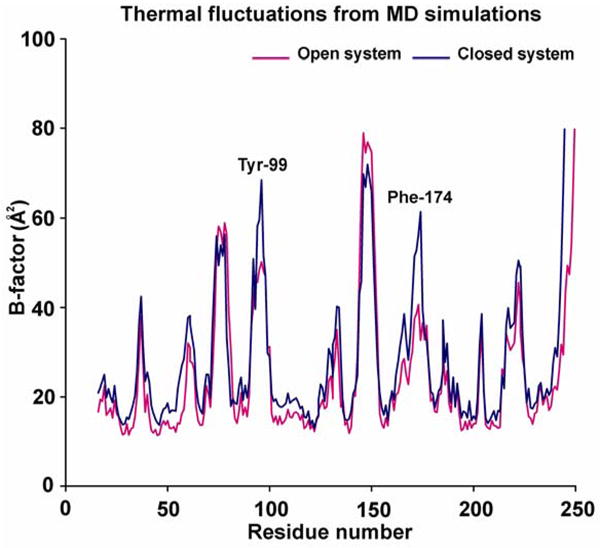
Thermal fluctuations (B-factor) plot of the two different conformations of FXa (magenta color - open system of unliganded protein; blue color - closed system of ecotin-bound protein). The B-factors shown in the plot were calculated by Debye-Waller formula (B=(8/3)π2RMSF2) and the values are for all-atoms averaged per residue. The two major areas of distinction in two systems are centered around two gating residues Tyr-99 and Phe-174.
Gating Residues
As shown in Figure 1, the S4 binding site is a U-shaped cavity which is surrounded by three aromatic residues Tyr-99, Phe-174 and Trp-215 on its three sides. In all of the available crystal structures for FXa, only the S1 and S4 subsites are distinct and visible. The S2 binding subsite is comparatively exposed only in the ecotin-bound crystal structure and is blocked by Tyr-99 in all other structures. This observation is supported by the fact that this site only binds glycine (P2) from its substrates. However, in 2002 this view was challenged by Le Bonniec et al. who experimentally showed in their fluorescence-quenched assays that cleavage is faster when a phenylalanine is substituted for the P2 residue glycine under similar conditions.47 Tryptophan also exhibits a strong preference for the P2 position. Since both of these bulky aromatic residues presumably bind in the S2 subsite for cleavage to take place, Tyr-99 must be able to move out of the way so that these bulky aromatic residues may bind in the newly exposed S2 subsite. Also a range of substrate P4 amino acids like isoleucine, proline and glutamic acid can bind at the S4 subsite. This indicates that the S4 subsite is not residue specific and is conformationally accessible to the physiological demands in the blood coagulation cascade which requires different substrates to be processed by FXa under different conditions.
In our analysis of the simulations, we have provided evidence for the roles that certain residues play in triggering the cascade of events that allows the S4 binding subsite to adopt a different conformational form, which changes its structure. Monitoring the distance between the two gating residues (Tyr-99 and Phe-174) during the simulation provided us with important evaluation of a conformational drift that occurs in the S4 subsite. We have also analyzed the structural hindering effect of Tyr-99 on blocking and unblocking the S2 subsite.
Figure 5 shows the distance between the pairs of atoms (excluding hydrogens) that are closest to each other between selected pairs of residues (Tyr-99 and Phe-174) throughout the simulation. In the open system, there is a clear indication of S4 binding site structural drift from the open state (0–14 ns) to the closed state (14–20 ns). In the beginning of the simulation, the distances between the gating residues which was ca. 10 Å, signifying an open state, which mostly explores and stays in the range of 4–10 Å with an average distance of 6.9 Å. In this system, after the first 14 ns of simulation S4 shrinks to just 2–5 Å, with an average of 2.7 Å, during the final phase of the simulation (14–20 ns) indicating a closed state. In contrast, in the closed state simulation where the minimum distance between the gating residues was 3.7 Å in the crystal structure, it fluctuated mostly between 2.3–4.0 Å with short bursts (at 1, 8 and 11 ns) to distances up to 8 Å in the first 13 ns of simulation time. This distance constantly increased from 4 Å to 8.5 Å, indicating an open state system in the last 2 ns of simulation time.
Figure 5.
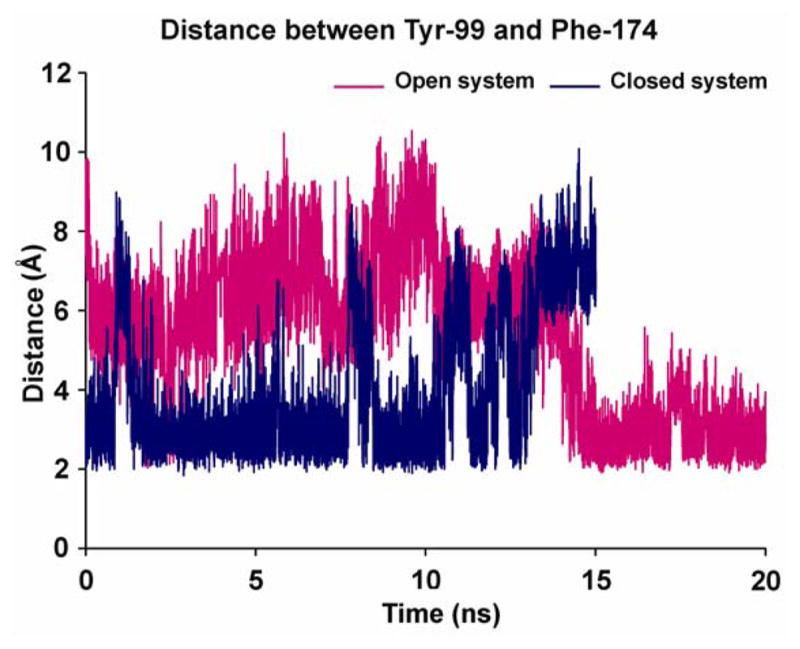
Conformational transitions in the S4 binding site shown by the distance spanned by the two gating residues (Tyr-99 and Phe-174) in the two different conformations of FXa (magenta color - open system of unliganded protein; blue color - closed system of ecotin-bound protein). A clear structural drift is from open to closed state is observable in open system after 14 ns, whereas the opposite is true for closed system which shows a drift from closed to open binding site after 13 ns.
The question arises again about what specific adjunct structural changes induce the protein to change its S4 binding site conformation. Also what impact might this change have on our view of the biological function of FXa. To answer these questions, we visually inspected the binding site and found that both the systems undergo a similar set of changes, involving extensive sets of aromatic-aromatic interactions between binding site and surrounding aromatic residues, which aids the protein in exploring both open and closed conformations.
Open System
In the open system, selected conformational states are shown in a series of snapshots in Figure 6a. The loop regions containing residues Tyr-99 (loop 91–103) and Phe-174 (loop 171 to 194) undergo significant changes in their conformation during the simulation, which leads to changes in the S4 binding site from the original open structure up to 14 ns. At this time a partially or completely closed cavity emerges during the last 6 ns of the simulation, resembling the crystal structure of the ecotin-bound FXa complex. This fist-like opening and closing of the binding site involves a series of aromatic-aromatic interactions from aromatic residues in and around the S4 binding site. As shown in the crystal structure (Figure 6a), the open state conformation is maintained by T-shaped (edge to face) stacking of Phe-94 and offset-stacking (face to face stacking in staggered manner) of His-57 on residue Tyr-99 which also keeps the S2 binding site blocked. In this situation, the T-shaped stacking is stabilized by both the electrostatic and dispersive Lennard-Jones interactions through interactions between partially positively charged hydrogens of Phe-94 which point toward the negatively charged π-electron cloud of the ring of Tyr-99. On the other hand, offset-stacked His-57 exhibits mainly Lennard-Jones interactions. A number of studies have investigated the suitability of molecular mechanics force fields for describing aromatic-aromatic and to some extent aromatic-cation interactions as a part of van der Waals and Coulomb interactions.48–55 The open state conformation in the crystal structure is also bolstered by the Lys-96, and Tyr-60 ring which in a cone-like shape, sterically blocks Phe-94 in its position, which, in turn, also stabilizes Tyr-99. The rest of the core binding site residues (Tyr-99, Phe-174 and Trp-215) do not exhibit any interactions.
Figure 6.

Figure 6A and 6B. The various snapshots are shown here, along the trajectory, depicting structural drift from A). open to closed state of S4 binding subsite from unliganded crystal structure simulation B). from closed state to open state of S4 binding subsite from ecotin-bound crystal structure simulation. The various residues participating in this process are shown in stick model (Glu-97, Lys-96), van der Waals (vdW) model in light magenta (Phe-94, His-57), vdW model with atom color (Tyr-99, Phe-174, Trp-215), ribbon model in yellow (protein) and part of ecotin molecule in stick (only in panel b).
During the simulation, the outward movement of loop 91–103 causes the Lys-96 and Tyr-60 residues to slowly drift apart, creating more space for Phe-94 to explore. This event provides Tyr-99 the chance to explore π-π interactions with other aromatic residues, namely, Phe-174 and Trp-215 as shown in a snapshot at 15 ns (Figure 6a). During this time, His-57 remains relatively stable and maintains its position because of its two hydrogen-bonds with Asp-102 on one side and Ser-195 on other side. In the 14–20 ns time range, when the binding site is closing, the two dominant types of aromatic-aromatic interactions that play significant roles are 1). T-shaped interaction of His-57 with Trp-215, and offset-stacked interactions of Tyr-99 and Phe-174 with Trp-215 (snapshot at 18 ns) and 2). offset-stacked interaction of Phe-174 with Tyr-99 and T-shaped interaction of Trp-215 with Tyr-99 (snapshot at 20 ns). Both of these conformations are also sterically stabilized in their closed form by Lys-96 and Glu-97, both on the outside.
Closed System
The cascade of events involved in the simulation of the closed system exhibits a drift from a closed to an open conformation of the binding site, as depicted in a series of snapshots (Figure 6b). In the crystal structure of this system, the closed conformation is maintained by the sterically suppressed Tyr-99 surrounded by ecotin residues Leu-52, Arg-54 and Val-81. Residues Leu-52 and Arg-54 also restrict the movement of loop 91–103. The two important aromatic-aromatic interactions seen in this system are Tyr-99 and Phe-174 (T-shaped) and Tyr-99 and Trp-215 (offset-stacked). In loop 91–103, Phe-94 exhibits aromatic-aromatic interactions with both His-57 and Tyr-60. His-57 is also maintained in its position by a hydrogen-bond with Thr-98 of FXa and Arg-54 of ecotin. During the SMD simulation in which ecotin is pulled out, three important events occur 1). steric hindrance of Glu-97 and Lys-96 is removed, 2). which causes loop 91–103 to move outside (snapshot at 6 ns in Figure 6b) and 3). the hydrogen-bond between His-57 and Thr-98 breaks, which causes His-57 to move toward Trp-215 to make a T-shaped aromatic-aromatic interaction. The outside movement of loop 91–103 causes Phe-94 to open up like a lid allowing for the rearrangement of Tyr-99 and Phe-174, causing the S4 cavity to partially open (snapshots at 13 and 13.5 ns). In the final conformation of the simulation (15 ns snapshot), the distance between the gating residues was 8 Å. This fully open conformation, which is similar to that of an open system crystal structure, is well maintained by three T-shaped aromatic-aromatic interactions between Tyr-99 and His-57, Tyr-99 and Trp-215, and Phe-174 and Trp-215.
Biologically, these interconversions between open and closed forms that were observed in the simulation studies may represent viable FXa conformations available for various structurally different substrate molecules essential for the maintenance of blood coagulation.
CONCLUSIONS
Protein flexibility, evident by presence of multiple conformational states, is an inherent and essential property of biomolecules.56 Along with playing important roles in crucial and basic functions like catalysis, motility and transport etc. it is also known to participate in diseases like AIDS, encephalopathies and many protein-misfold diseases. Thus, gaining knowledge about protein flexibility is important not only to understand protein function but also for the rational design of new drugs and novel therapeutics in structure-based drug design efforts.57–59
Our study of MD simulations allowed us to gain important insights into the flexible behavior of one such physiologically important blood coagulation protease, FXa. Though there is an abundance of literature available for this protein, this is the first time a detailed opening and closing behavior of FXa subsites has been reported. The two previously reported MD simulation studies of FXa, in spite of being very informative in their objectives, failed to observe the conformational drift behavior due to their short time scales of simulation, which we observed to occur only after 13 ns of simulation time. Among the first of these two articles, a total of 1.5 ns MD simulation was presented by Daura et al in 200060 while a 6.2 ns of MD simulation was presented by Venkateswarlu et al in 2002.61
In this study, we have theoretically demonstrated the dynamic interconversion of two separate and distinctly different binding site conformations of FXa by using computational techniques, which are otherwise difficult to distinguish from the available static X-ray structures alone. This study supports our hypothesis that under physiological conditions, FXa can exist in distinct conformational states that may facilitate different specific protein-protein interactions of FXa with its structurally diverse substrates like prothrombin, FVII, Protease-activated receptor 2 (PAR2) and itself (autocatalysis). With multiple conformations available, the substrates can then select the FXa one that most favorably complements their binding and eliminates the need to induce changes in FXa for binding. We have also described the potential mechanistic roles of Glu-97, Lys-96 and the importance of aromatic-aromatic interactions of various aromatic residues His-57, Tyr-60, Phe-94, Tyr-99, Phe-194 and Trp-215 in the opening and closing dynamics of the FXa subsites.
These studies may aid in a better understanding of various protein-protein interactions involved in the coagulation cascade with are otherwise difficult to elucidate experimentally. The strategically important location of FXa in the coagulation cascade makes it a therapeutically attractive target and we believe that our detailed analyses of the FXa binding subsite may have impact on the design and development of anticoagulant/antithrombotic drugs.
Gratitude is expressed to the National Institutes of Health for partial support of this project through grant AI055883. The NASA University Research Center at Texas Southern University of also acknowledged for partial support of this project. We gratefully acknowledge computer time kindly provided through the Texas Learning and Computation Center at the University of Houston.
References
- 1.Tang J, Wong RN. J Cell Biochem. 1987;33:53–63. doi: 10.1002/jcb.240330106. [DOI] [PubMed] [Google Scholar]
- 2.Irwin DM. Nature. 1988;336:429–430. doi: 10.1038/336429b0. [DOI] [PubMed] [Google Scholar]
- 3.Rose T, Di Cera E. J Biol Chem. 2002;277:19243–19246. doi: 10.1074/jbc.C200132200. [DOI] [PubMed] [Google Scholar]
- 4.Caldwell SH, Hoffman M, Lisman T, Macik BG, Northup PG, Reddy KR, Tripodi A, Sanyal AJ. Hepatology. 2006;44:1039–1046. doi: 10.1002/hep.21303. [DOI] [PubMed] [Google Scholar]
- 5.Soliman DE, Broadman LM. Anesthesiol Clin. 2006;24:549–578. vii. doi: 10.1016/j.atc.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 6.Di Scipio RG, Hermodson MA, Yates SG, Davie EW. Biochemistry. 1977;16:698–706. doi: 10.1021/bi00623a022. [DOI] [PubMed] [Google Scholar]
- 7.Leytus SP, Foster DC, Kurachi K, Davie EW. Biochemistry. 1986;25:5098–5102. doi: 10.1021/bi00366a018. [DOI] [PubMed] [Google Scholar]
- 8.Padmanabhan K, Padmanabhan KP, Tulinsky A, Park CH, Bode W, Huber R, Blankenship DT, Cardin AD, Kisiel W. J Mol Biol. 1993;232:947–966. doi: 10.1006/jmbi.1993.1441. [DOI] [PubMed] [Google Scholar]
- 9.Tracy PB, Nesheim ME, Mann KG. J Biol Chem. 1981;256:743–751. [PubMed] [Google Scholar]
- 10.Krishnaswamy S, Jones KC, Mann KG. J Biol Chem. 1988;263:3823–3834. [PubMed] [Google Scholar]
- 11.Ahmad SS, Rawala-Sheikh R, Walsh PN. Semin Thromb Hemost. 1992;18:311–323. doi: 10.1055/s-2007-1002570. [DOI] [PubMed] [Google Scholar]
- 12.Bode W, Turk D, Karshikov A. Protein Sci. 1992;1:426–471. doi: 10.1002/pro.5560010402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katz BA, Mackman R, Luong C, Radika K, Martelli A, Sprengeler PA, Wang J, Chan H, Wong L. Chem Biol. 2000;7:299–312. doi: 10.1016/s1074-5521(00)00104-6. [DOI] [PubMed] [Google Scholar]
- 14.Maignan S, Guilloteau JP, Pouzieux S, Choi-Sledeski YM, Becker MR, Klein SI, Ewing WR, Pauls HW, Spada AP, Mikol V. J Med Chem. 2000;43:3226–3232. doi: 10.1021/jm000940u. [DOI] [PubMed] [Google Scholar]
- 15.Brandstetter H, Kuhne A, Bode W, Huber R, von der Saal W, Wirthensohn K, Engh RA. J Biol Chem. 1996;271:29988–29992. doi: 10.1074/jbc.271.47.29988. [DOI] [PubMed] [Google Scholar]
- 16.Adler M, Davey DD, Phillips GB, Kim SH, Jancarik J, Rumennik G, Light DR, Whitlow M. Biochemistry. 2000;39:12534–12542. doi: 10.1021/bi001477q. [DOI] [PubMed] [Google Scholar]
- 17.Nar H, Bauer M, Schmid A, Stassen JM, Wienen W, Priepke HW, Kauffmann IK, Ries UJ, Hauel NH. Structure. 2001;9:29–37. doi: 10.1016/s0969-2126(00)00551-7. [DOI] [PubMed] [Google Scholar]
- 18.Guertin KR, Gardner CJ, Klein SI, Zulli AL, Czekaj M, Gong Y, Spada AP, Cheney DL, Maignan S, Guilloteau JP, Brown KD, Colussi DJ, Chu V, Heran CL, Morgan SR, Bentley RG, Dunwiddie CT, Leadley RJ, Pauls HW. Bioorg Med Chem Lett. 2002;12:1671–1674. doi: 10.1016/s0960-894x(02)00213-5. [DOI] [PubMed] [Google Scholar]
- 19.Mueller MM, Sperl S, Sturzebecher J, Bode W, Moroder L. Biol Chem. 2002;383:1185–1191. doi: 10.1515/BC.2002.130. [DOI] [PubMed] [Google Scholar]
- 20.Matter H, Defossa E, Heinelt U, Blohm PM, Schneider D, Muller A, Herok S, Schreuder H, Liesum A, Brachvogel V, Lonze P, Walser A, Al-Obeidi F, Wildgoose P. J Med Chem. 2002;45:2749–2769. doi: 10.1021/jm0111346. [DOI] [PubMed] [Google Scholar]
- 21.Adler M, Kochanny MJ, Ye B, Rumennik G, Light DR, Biancalana S, Whitlow M. Biochemistry. 2002;41:15514–15523. doi: 10.1021/bi0264061. [DOI] [PubMed] [Google Scholar]
- 22.Maignan S, Guilloteau JP, Choi-Sledeski YM, Becker MR, Ewing WR, Pauls HW, Spada AP, Mikol V. J Med Chem. 2003;46:685–690. doi: 10.1021/jm0203837. [DOI] [PubMed] [Google Scholar]
- 23.Haginoya N, Kobayashi S, Komoriya S, Yoshino T, Suzuki M, Shimada T, Watanabe K, Hirokawa Y, Furugori T, Nagahara T. J Med Chem. 2004;47:5167–5182. doi: 10.1021/jm049884d. [DOI] [PubMed] [Google Scholar]
- 24.Kamata K, Kawamoto H, Honma T, Iwama T, Kim SH. Proc Natl Acad Sci U S A. 1998;95:6630–6635. doi: 10.1073/pnas.95.12.6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker RK, Krishnaswamy S. J Biol Chem. 1994;269:27441–27450. [PubMed] [Google Scholar]
- 26.Petrovan RJ, Govers-Riemslag JW, Nowak G, Hemker HC, Tans G, Rosing J. Biochemistry. 1998;37:1185–1191. doi: 10.1021/bi971948h. [DOI] [PubMed] [Google Scholar]
- 27.Parker ET, Pohl J, Blackburn MN, Lollar P. Biochemistry. 1997;36:9365–9373. doi: 10.1021/bi970599o. [DOI] [PubMed] [Google Scholar]
- 28.Camerer E, Huang W, Coughlin SR. Proc Natl Acad Sci U S A. 2000;97:5255–5260. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jesty J, Spencer AK, Nemerson Y. J Biol Chem. 1974;249:5614–5622. [PubMed] [Google Scholar]
- 30.Jesty J, Spencer AK, Nakashima Y, Nemerson Y, Konigsberg W. J Biol Chem. 1975;250:4497–4504. [PubMed] [Google Scholar]
- 31.Chien P, Weissman JS. Nature. 2001;410:223–227. doi: 10.1038/35065632. [DOI] [PubMed] [Google Scholar]
- 32.Lee AY, Gulnik SV, Erickson JW. Nat Struct Biol. 1998;5:866–871. doi: 10.1038/2306. [DOI] [PubMed] [Google Scholar]
- 33.Stella L, Caccuri AM, Rosato N, Nicotra M, Lo Bello M, De Matteis F, Mazzetti AP, Federici G, Ricci G. J Biol Chem. 1998;273:23267–23273. doi: 10.1074/jbc.273.36.23267. [DOI] [PubMed] [Google Scholar]
- 34.Wedemayer GJ, Patten PA, Wang LH, Schultz PG, Stevens RC. Science. 1997;276:1665–1669. doi: 10.1126/science.276.5319.1665. [DOI] [PubMed] [Google Scholar]
- 35.Kumar S, Ma B, Tsai CJ, Sinha N, Nussinov R. Protein Sci. 2000;9:10–19. doi: 10.1110/ps.9.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang SX, Hur E, Sousa CA, Brinen L, Slivka EJ, Fletterick RJ. Biochemistry. 2003;42:7959–7966. doi: 10.1021/bi027320a. [DOI] [PubMed] [Google Scholar]
- 37.Meurisse R, Brasseur R, Thomas A. Biochim Biophys Acta. 2003;1649:85–96. doi: 10.1016/s1570-9639(03)00161-4. [DOI] [PubMed] [Google Scholar]
- 38.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comp Chem. 1982;4:187–217. [Google Scholar]
- 40.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 41.DeLano WL. 2002 http://wwwpymolorg.
- 42.Izrailev S, Stepaniants S, Isralewitz B, Kosztin D, Lu H, Molnar F, Wriggers W, Schulten K. Computational Molecular Dynamics: Challenges, Methods, Ideas. Lecture Notes in Computational Science and Engineering. 1998;4:39–65. [Google Scholar]
- 43.Antosiewicz J, Briggs JM, McCammon JA. Eur Biophys J. 1996;24:137–141. doi: 10.1007/BF00180270. [DOI] [PubMed] [Google Scholar]
- 44.Davis ME, Madura JD, Luty BA, McCammon JA. Comput Phys Commun. 1991;62:187–197. [Google Scholar]
- 45.Essmann U, Perera M, Berkowitz ML, Darden T, Lee H, Pedersen LG. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 46.Ryckaert JP, Ciccotti G, Berendsen HJC. J Compt Phys. 1977;23:327–341. [Google Scholar]
- 47.Bianchini EP, Louvain VB, Marque PE, Juliano MA, Juliano L, Le Bonniec BF. J Biol Chem. 2002;277:20527–20534. doi: 10.1074/jbc.M201139200. [DOI] [PubMed] [Google Scholar]
- 48.Norberg J, Nilsson L. Biophys J. 1995;69:2277–2285. doi: 10.1016/S0006-3495(95)80098-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Norberg J, Nilsson L. J Am Chem Soc. 1995;117:10832–10840. [Google Scholar]
- 50.Norberg J, Nilsson L. Biophys J. 1998;74:394–402. doi: 10.1016/S0006-3495(98)77796-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McGaughey GB, Gagne M, Rappe AK. J Biol Chem. 1998;273:15458–15463. doi: 10.1074/jbc.273.25.15458. [DOI] [PubMed] [Google Scholar]
- 52.Banavali NK, MacKerell AD., Jr J Mol Biol. 2002;319:141–160. doi: 10.1016/S0022-2836(02)00194-8. [DOI] [PubMed] [Google Scholar]
- 53.Hart K, Nystrom B, Ohman M, Nilsson L. RNA. 2005;11:609–618. doi: 10.1261/rna.7147805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Priyakumar UD, MacKerell AD. J Chem Theory Comput. 2006;2:187–200. doi: 10.1021/ct0501957. [DOI] [PubMed] [Google Scholar]
- 55.Beautrait A, Karaboga AS, Souchet M, Maigret B. Proteins. 2008 doi: 10.1002/prot.21977. [DOI] [PubMed] [Google Scholar]
- 56.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 57.Teague SJ. Nat Rev Drug Discov. 2003;2:527–541. doi: 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- 58.Wong CF, McCammon JA. Annu Rev Pharmacol Toxicol. 2003;43:31–45. doi: 10.1146/annurev.pharmtox.43.100901.140216. [DOI] [PubMed] [Google Scholar]
- 59.Rauh D, Klebe G, Stubbs MT. J Mol Biol. 2004;335:1325–1341. doi: 10.1016/j.jmb.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 60.Daura X, Haaksma E, van Gunsteren WF. J Comput Aided Mol Des. 2000;14:507–529. doi: 10.1023/a:1008120005475. [DOI] [PubMed] [Google Scholar]
- 61.Venkateswarlu D, Perera L, Darden T, Pedersen LG. Biophys J. 2002;82:1190–1206. doi: 10.1016/S0006-3495(02)75476-3. [DOI] [PMC free article] [PubMed] [Google Scholar]