

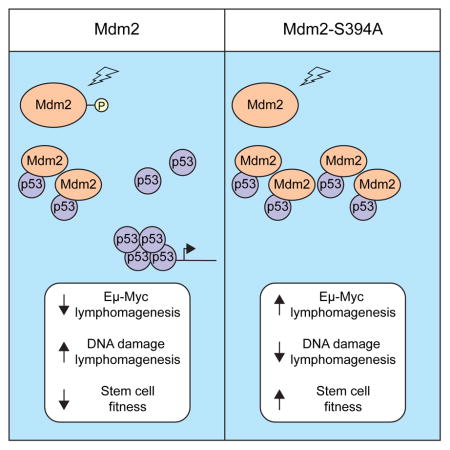
SUMMARY
ATM phosphorylation of Mdm2-S394 is required for robust p53 stabilization and activation in DNA damaged cells. We have now utilized Mdm2S394A knock-in mice to determine that phosphorylation of Mdm2-S394 regulates p53 activity and the DNA damage response in lymphatic tissues in vivo by modulating Mdm2 stability. Mdm2-S394 phosphorylation delays lymphomagenesis in Eμ-myc transgenic mice, and preventing Mdm2-S394 phosphorylation obviates the need for p53 mutation in Myc-driven tumorigenesis. However, irradiated Mdm2S394A mice also have increased hematopoietic stem and progenitor cell functions, and decreased lymphomagenesis in sub-lethally irradiated Mdm2S394A mice. These findings document contrasting effects of ATM-Mdm2 signaling on p53 tumor suppression, and reveal that destabilizing Mdm2 by promoting its phosphorylation by ATM would be effective in treating oncogene-induced malignancies, while inhibiting Mdm2-S394 phosphorylation during radiation exposure or chemotherapy would ameliorate bone marrow failure and prevent the development of secondary hematological malignancies.
Graphical abstract
INTRODUCTION
It is widely accepted that the p53 tumor suppressor protein functions primarily as a transcription factor, capable of promoting or repressing the transcription of a multitude of genes (Beckerman and Prives, 2010). Proper coordination of p53-responsive gene expression plays an important role in tumor suppression, as evidenced by the rapid development of tumors in mice lacking p53 (Donehower et al. 1992) and by the fact that most human cancers harbor mutations in p53 or in key regulators of p53 signaling (Soussi and Beroud, 2001). The tumor suppressive capacity of p53 has been traditionally attributed to its ability to inhibit cell proliferation or promote apoptosis, as limiting or eliminating cells bearing genetic lesions would obviously prevent the propagation and accumulation of genetic errors and the formation or progression of tumorigenesis. However, p53 tumor suppressive mechanisms distinct from p53-mediated growth arrest and apoptosis have been recently proposed, suggesting an even broader contribution of p53 activities to tumor suppression (Brady et al., 2011; Li et al., 2012).
Because deregulated growth arrest and apoptosis is detrimental to embryogenesis and normal cell growth, the activities of p53 are strictly regulated in non-damaged cells and tissues. Basal levels of p53 are low and this transcription factor is largely inactive under homeostatic conditions. In contrast, p53 is rapidly stabilized and activated in response to a multitude of stresses, including activated oncogenes, hypoxia, ribosomal stress and DNA damage. The chief negative regulator of p53 stabilization and activity is the Mdm2 oncoprotein, which can bind and mask the transactivation domain of p53 and function as an E3 ubiquitin ligase capable of directing p53 nuclear export and proteosomal degradation (Momand et al., 1992; Oliner et al., 1993; Haupt et al., 1997; Honda et al., 1997; Kubbutat et al., 1997). The central role of Mdm2 in regulating p53 activity is best illustrated by studies using Mdm2-conditional mouse models that identified roles for Mdm2 in regulating p53-dependent cell growth arrest or apoptosis in various tissues (Gannon and Jones, 2012), and p53-dependent lethality of Mdm2-null mice during early embryogenesis (Jones et al., 1995; Montes de Oca Luna et al., 1995).
Similar to Mdm2, the homologous protein MdmX (Mdm4) is also capable of binding p53 and inhibiting p53 transactivation of target genes (Shvarts et al., 1996), and mice null for MdmX display a similar p53-dependent embryonic lethality, albeit at a slightly later time during development (Parant et al., 2001; Migliorini et al., 2002). Unlike Mdm2, MdmX does not possess the ability to directly ubiquitinate p53 (Jackson and Berberich, 2000). However, Mdm2 and MdmX have been shown to interact via their C-terminal RING domains, and this heterodimerization promotes maximal Mdm2 E3 ligase activity towards p53 (Tanimura et al., 1999; Sharp et al., 1999, Kawai et al., 2007). Recently, a series of Mdm2 and MdmX knock-in mouse models have been generated that display altered Mdm2-MdmX interactions or Mdm2 E3 ligase activity (Itahana et al., 2007; Pant et al., 2011; Huang et al., 2011; Tollini et al., 2014). Analyses of these various models have revealed that Mdm2-MdmX interactions are critical in inhibiting p53 activity during development and tissue homeostasis, whereas the E3 ligase function of Mdm2 is critical in regulating p53 protein and activity levels in cellular and organismal response to DNA damage (Tollini et al., 2014).
Inhibition of p53 levels and activities by MDM proteins must be interrupted in order for p53 to become elevated and activated in response to DNA damage or other forms of stress (Meek, 2015). During the DNA damage response (DDR), Mdm2-p53 signaling is mediated by DNA damage activated kinases such as ATM (ataxia telangiectasia mutated). Upon sensing double stranded DNA breaks, the PI3K-related ATM becomes activated and directly or indirectly induces phosphorylation of a multitude of proteins involved in the damage response, including p53, Mdm2 and MdmX (Shieh et al. 1997; Shieh et al., 2000; Maya et al., 2001; Pereg et al., 2005; Chen et al., 2005). Initial biochemical and cell-based studies suggested that ATM-mediated phosphorylation of two key residues on p53, Ser18 and Ser23 (human Ser15 and Ser20), could account for p53 stabilization and activation following DNA damage. However, analysis of genetically engineered mouse models revealed that these phosphorylation events were insufficient to account for the full effects of DNA damage on Mdm2-p53 signaling or p53 tumor suppression (Wu et al., 2002; Chao et al., 2003; Sluss et al., 2004; Chao et al., 2006). We have previously reported the generation and initial characterization of a mouse model (Mdm2S394A mice) wherein ATM phosphorylation of Mdm2 at serine residue 394 was abolished (Gannon et al., 2012). Cells and tissues in Mdm2S394A mice display profound defects in DNA damage-induced p53 protein stabilization and p53 target gene activation. This failure to induce a robust p53 response translates to less p53-dependent apoptosis in hematopoietic tissues, radio-resistance, and increased spontaneous lymphomagenesis. Regulation of the amplitude and duration of the p53-induced DNA damage response in Mdm2S394A cells and mice underscores the importance of ATM phosphorylation of Mdm2 in the DNA damage response.
Several in vitro studies have suggested that ATM phosphorylation of Mdm2 governs p53 stability and/or activity following DNA damage by regulating p53 degradation and nuclear export, Mdm2 autodegradation, and Mdm2 oligomerization and E3 processivity (Maya et al., 2001; Stommel and Wahl, 2004; Cheng et al., 2009). In our present study, we explore in vivo the mechanism by which Mdm2 phosphorylation alters p53 functions in a radiosensitive tissue. Furthermore, we have sought to address whether ATM-Mdm2-p53 signaling in mice impacts tumorigenesis induced by activated oncogenes or ionizing radiation (IR), and explore the role of ATM-Mdm2 signaling in promoting IR-mediated bone marrow failure. Our results indicate that Mdm2 phosphorylation has dramatically different and stress-dependent effects in tumorigenesis.
RESULTS
ATM-Mdm2 signaling regulates the p53-response in lymphatic tissues
To further explore the effects of Mdm2 Ser394 phosphorylation on p53 protein levels and activity in vivo, we analyzed thymi of mice treated with a low dose (1.75 Gy) of IR. Although p53 protein levels were elevated in the thymus of Mdm2S394A mice at 3 hours and 6 hours following IR, there was considerably less total p53 protein and phosphorylated p53 phosphorylation (S18) in treated Mdm2S394A mice than in irradiated wild-type (WT) mice (Figure 1A). This is similar to what we observed previously in these mice using higher dosages of IR (Gannon et al., 2012). However, we also noted that basal levels of p53 appeared slightly lower in Mdm2S394A thymi than in WT thymi, and more Mdm2 protein appeared to be present in Mdm2S394A thymi in absence of acute DNA-damage and following IR treatment (Figure 1A). There was a significant decrease in WT Mdm2 protein levels in whole thymus following DNA damage, an observation that has been made in cell culture settings by several groups (Stommel and Wahl, 2004; Itahana et al., 2007; Inuzuka et al., 2010; Malonia et al., 2015). However, Mdm2 protein levels appeared to diminish after IR at a lesser rate in Mdm2S394A thymus than in WT thymus. To more definitively quantify the observed differences in thymic Mdm2 and p53 levels, we analyzed biological triplicates of untreated and irradiated WT and Mdm2S394A thymi. This confirmed that Mdm2 protein levels were higher in Mdm2S394A thymi both before and after treatment, and that the IR-induced relative decrease in Mdm2 protein levels was far less in Mdm2S394A thymi (Figure 1B). In contrast, the reduction of MdmX levels induced by DNA damage (Wang et al., 2009) appears to be similar in WT and Mdm2S394A thymi. Though not statistically significant, p53 levels are slightly lower in untreated Mdm2S394A thymi (Figure 1B, lower panel), and are significantly lower in irradiated Mdm2S394A thymi. Although p53 target gene expression was similar in non-damaged WT and Mdm2S394A thymi (Figure 1C), a reduction in IR-activation of p53 target genes was seen in Mdm2S394A thymi. Reduced expression levels of Mdm2, the cell cycle regulator Cdkn1a (p21), and the pro-apoptotic genes Puma, Noxa, and Bax are in agreement with reduced levels of p21, Puma, and cleaved Caspase-3 protein (Figure 1A), and with a clear reduction in DNA damage induced apoptosis in Mdm2S394A thymi following exposure of mice to low-level IR (Figure 1D).
Figure 1. ATM phosphorylation of Mdm2-S394 regulates Mdm2 levels and p53 activity.

(A) WT and Mdm2S394A mice were left untreated or exposed to 1.75 Gy ionizing radiation (IR) and thymi were harvested at 3 and 6 hours. Protein levels were analyzed by western blotting. TKO indicates Mdm2−/−, MdmX−/−, p53−/− control; Tg indicates Mdm2Tg/+ Mdm2 overexpressing control.
(B) Thymus protein levels of biological triplicates of WT and Mdm2S394A mice left untreated or treated as in (A) for 3 hours, were analyzed by western blotting. Band intensities were determined by densitometry. Mdm2 and p53 levels were normalized for Vinculin levels and average values plotted (±SEM). *P < 0.05, **P < 0.01 (Student’s t-tests).
(C) Mice were treated as in (A) and fold expression of p53-target genes was determined by real-time quantitative PCR, relative to untreated WT samples using Rplp0 as internal reference (n = 3, ±SEM). *P < 0.05, **P < 0.01 (Student’s t-tests).
(D) TUNEL staining of thymi from mice treated as in (A). Scale bars represent 100 μm.
To explore whether these effects were unique to the thymus, we also examined protein levels in spleens of WT and Mdm2S394A mice. As observed in the thymus, there appeared to be less total p53 in Mdm2S394A spleens before and after irradiation (Figure S1), and Mdm2 levels were higher in both untreated and irradiated Mdm2S394A spleens. Intriguingly, while no induction of Mdm2 protein was observed in thymi of WT and Mdm2S394A mice in response to IR, spleens of both genotype displayed a more ‘classic’ induction of Mdm2 often seen in cultured cells after genotoxic stress. Similar to what was observed in the thymus, decreased levels of p53 and increased levels of Mdm2 in irradiated Mdm2S394A spleens correlate with reduced p53 activation (phospho-S18 p53), reduced levels of Puma and p21, and reduced levels of cleaved Caspase-3 (Figure S1).
ATM phosphorylation of Mdm2-S394 governs Mdm2 levels and stability
In order to directly analyze the effects of Mdm2-S394 phosphorylation on the stability of Mdm2 in the presence and absence of exogenous DNA damage, we measured the half-life of Mdm2 proteins using the protein synthesis inhibitor cycloheximide. Although basal levels of Mdm2 transcription are similar in WT and Mdm2S394A thymi, Mdm2 protein levels are elevated in non-damaged Mdm2S394A mice (Figures 1A, 1B), suggesting that the mutant Mdm2 protein is more stable in the absence of exogenous DNA damage. However, we observed no significant difference in the half-lives of WT Mdm2 and Mdm2S394A in untreated thymocytes (80 and 65 minutes respectively, overlapping 95% confidence intervals) (Figure 2A). But following treatment of the thymocytes with 2.5 Gy IR, the half-life of WT Mdm2 decreased by more than 50 percent (29 minutes) whereas the half-life of Mdm2S394A remained unchanged (69 minutes) (Figure 2B). These data show that phosphorylation of Mdm2-S394 by ATM is a crucial event in DNA damage-induced destabilization of Mdm2 under physiological conditions. As previous studies have shown ATM phosphorylation of Mdm2 to impact the ability of Mdm2 to promote p53 degradation (Maya et al., 2001; Cheng et al., 2009; Cheng et al., 2011), we also examined whether p53 stability was affected in the presence and absence of DNA damage. We observed no difference in the half-life of p53 in non-treated WT versus Mdm2S394A thymocytes (Figure S2A). As expected, DNA damage stabilized p53 levels in WT cells (Figure S2B). Likewise, DNA damage stabilized p53 levels in Mdm2S394A thymocytes, albeit to a lower level than seen in WT thymocytes.
Figure 2. ATM phosphorylation of Mdm2-S394 regulates Mdm2 stability and levels of Mdm2-bound p53.

(A) Thymocytes harvested from WT and Mdm2S394A mice (n = 6) were treated with 100 μg/ml cycloheximide and harvested at the indicated time points. The levels of Mdm2 and a-tubulin were analyzed by western blotting. Band intensities were determined by densitometry and Mdm2 levels normalized to a-tubulin were plotted. One-phase decay curves were fitted using GraphPad Prism software.
(B) Thymocytes harvested from WT and Mdm2S394A mice (n = 6-8) were exposed to 2.5 Gy IR and treated as in (A).
(C) Thymus protein extracts from mice untreated or exposed to 5 Gy IR were immunoprecipitated with antibodies for Mdm2 (NBP1-02158 (Ab-I) and Ab-5 (Ab-II)) and p53. Immunoprecipitates were analyzed by western blotting for Mdm2 and p53. Total lysate (10% input) was analyzed by western blotting.
Since we observed no difference in the rate of p53 decay in Mdm2S394A and WT thymocytes, it is possible that phosphorylation of Mdm2-S394 upregulates p53 activity not only by altering p53 protein stability but by also inhibiting Mdm2-p53 complex formation and Mdm2-mediated steric inhibition of p53 transcriptional activation. Therefore, we examined the effects of Mdm2-S394 phosphorylation on Mdm2-p53 binding in the presence and absence of DNA damage in whole tissue extracts. In an effort to control for potential differences in antibody affinity following IR-induced modification of Mdm2, Mdm2 was immunoprecipitated from untreated and irradiated thymus lysates in separate experiments with two distinct antibodies whose epitopes reside in opposing termini of Mdm2 (Figure 2C left panel). In both cases, more p53 co-immunoprecipitated with Mdm2S394A than WT Mdm2 in untreated thymi, whereas similar amounts of p53 co-immunoprecipitated with Mdm2 in irradiated Mdm2S394A and WT thymi. As there was less p53 observed in the total lysates of untreated and irradiated Mdm2S394A thymi relative to WT thymi (Figure 2C right panel), this result reveals increased levels of Mdm2-bound p53 (relative to total p53) in undamaged and IR-treated Mdm2S394A thymi. We confirmed this finding by performing the reciprocal experiment using p53 immunoprecipitation, and observed that equivalent amounts of Mdm2 co-immunoprecipitated with lesser amounts of p53 in untreated and irradiated Mdm2S394A thymi. Additional immunoprecipitation experiments against MdmX detected no effect on the relative amounts of Mdm2-bound MdmX or p53-bound MdmX before or after DNA damage (Figure S3).
Collectively, these data reveal that phosphorylation of Mdm2-S394 under basal conditions and following acute IR exposure reduces Mdm2 stability, thereby reducing the relative amount of Mdm2-bound p53. This reduction in Mdm2-p53 complex negatively impacts Mdm2 inhibition of p53 target gene transactivation as well as Mdm2 destabilization of p53.
Accelerated Eμ-myc driven lymphomagenesis in Mdm2S394A mice
We have previously described an increased susceptibility to spontaneous tumorigenesis in Mdm2S394A mice (Gannon et al., 2012). We next sought to examine the effects of Mdm2-S394 phosphorylation on oncogene-induced tumorigenesis. Mdm2S394A mice were bred to Eμ-myc transgenic mice to generate Eμ-myc and Eμ-myc;Mdm2S394A mice. Eμ-myc mice succumb to pre-B/B-cell lymphomas within 3-6 months of age. We observed a median time to tumor presentation of 126 days in Eμ-myc mice, consistent with previous studies (Adams et al., 1985; Eischen et al., 1999; Sluss et al., 2010) (Figure 3A). In contrast, the median time to tumor presentation in Eμ-myc;Mdm2S394A mice was only 71 days. This represents a nearly 50% reduction in the time to Myc-induced tumorigenesis when ATM phosphorylation of Mdm2-S394 is inhibited. All tumor-bearing Eμ-myc mice and Eμ-myc;Mdm2S394A mice presented with enlarged lymph nodes and spleens, and representative tumors were examined histologically by hematoxylin and eosin (H&E) staining. Both genotypes developed similar, high-grade lymphomas, composed of monotonous populations of pre-B/B cells. Tumors displayed high levels of mitosis and apoptosis, and the characteristic “starry-sky” pattern resultant from abundant tingible body-laden macrophages. The cell type was further confirmed by immunohistochemistry (IHC) through positive staining for B220/CD45R (Figure 3B).
Figure 3. Accelerated Eμ-myc driven lymphomagenesis in Mdm2S394A mice.

(A) Kaplan-Meier survival curves of Eμ-Myc (n = 16) and Eμ-Myc;Mdm2S394A (n = 11) mice. Median survival times were as follows: Eμ-Myc (125.5 days) and Eμ-Myc;Mdm2S394A (71 days). Curves were compared by Log-rank test: P < 0.0001.
(B) Representative B cell lymphomas in lymph nodes of Eμ-Myc (98 days) and Eμ-Myc;Mdm2S394A (80 days) mice were stained by haematoxylin and eosin (top) and with an antibody specific for B220 (bottom). Scale bars represent 100 μm.
(C) The levels of Mdm2, p53 and Arf were analyzed by western blotting in a panel of Eμ-Myc;Mdm2S394A tumors. RT-PCR confirmed that full-length Arf message was absent in 3 of 10 tumors (2nd from bottom). p53 cDNA was sequenced for each tumor, revealing no mutations (bottom). LN, lymph node; Spl, spleen.
(D) Schematic outlining the proposed methods of p53 activation by Myc. Tumors in mice in which Mdm2 is WT select for loss of Arf or p53, or Mdm2 overexpression (Eischen et al., 1999). We propose that Mdm2S394A mice obviate the need for p53 loss or Mdm2 overexpression by mitigating the effects of the DDR arm of Myc signaling to p53.
Myc-driven B cell tumors face selective pressure to inactivate the p53 pathway through p53 mutation, Mdm2 overexpression, or by loss of Arf (Eischen et al., 1999). We examined the status of p53, Mdm2 and Arf in a panel of ten tumors that developed in Eμ-myc;Mdm2S394A mice (Figure 3C). No marked differences in p53 protein levels were observed in any of the ten tumors examined. However, Arf levels appeared more variable, with loss of detectable Arf protein seen in four of the ten tumors. RT-PCR confirmed that three of those four tumors did not express full length Arf mRNA. Arf levels have been shown previously to be elevated in cases where p53 is mutant and the negative feedback loop between p53 and Arf is disrupted, and we identified several tumors wherein Arf appeared to be increased. However, sequencing of the entire p53 coding sequence of all ten tumors revealed no mutations in p53 gene transcripts. Thus, the observed variability in Arf levels within the Myc tumors was not a result of p53 status. Furthermore, Mdm2 protein levels did not vary significantly between tumors, and qPCR analysis also failed to detect alterations in Mdm2 transcript levels in any tumor (data not shown). Thus, ATM phosphorylation of Mdm2-S394 strongly suppresses Myc oncogene-induced tumorigenesis in mice, and inhibition of this signaling event obviates the need for mutation of the Mdm2-p53 tumor suppressor axis in Myc-driven B cell lymphomagenesis (Figure 3D).
Mdm2S394A mice are resistant to IR-induced lymphomagenesis
We next sought to examine the effects of ATM phosphorylation of Mdm2-S394 on IR-induced tumorigenesis. Exposure of mice to repeated low-dose IR promotes the development of thymic lymphomas (Kaplan and Brown, 1952). This lymphomagenesis is significantly enhanced in the absence of p53 (Kemp et al., 1994; Labi et al., 2010; Michalak et al., 2010). As we have shown that IR-induced p53 activity is diminished in both the thymus and spleen of Mdm2S394A mice, we anticipated a heightened sensitivity to IR-induced lymphomagenesis in this model. Cohorts of WT, Mdm2S394A, p53+/− and p53−/− mice were subjected to four weekly doses of 1.75 Gy IR and monitored over time for tumor presentation. 89% of WT mice developed lymphomas by 400 days, with a median survival of 197 days (Figure 4A), consistent with a previous study employing this dosing strategy (Labi et al., 2010). Also consistent with previous studies was the significant acceleration of lymphomagenesis observed in the absence of p53 (Kemp et al., 1994; Labi et al., 2010; Michalak et al., 2010). All p53−/− mice developed lymphomas within 151 days, with a median survival of 131 days, whereas all p53+/− mice developed lymphomas within 167 days, with a median survival of 154 days. Surprisingly, Mdm2S394A mice proved to be highly resistant to IR-induced thymic lymphomagenesis. Although tumor presentation in Mdm2S394A mice followed similar initial kinetics as observed in WT mice, only 42% of Mdm2S394A mice developed lymphoma, with a median survival among tumor-bearing mice of 184 days. Tumor-bearing animals presented with profoundly enlarged thymi, as well as frequent splenomegaly and hepatomegaly. Histological analyses of H&E stained tissues showed disorganized, hyperplastic lymphatic tissues as well as significant lymphocyte infiltration in portal regions of the liver (Figure 4B). Immunohistochemistry confirmed that the lymphomas arising in IR-treated mice were T cell-derived, with positive staining for CD3 and negative staining for B220 (Figures 4C, S4). Thus, in contrast to their increased rate of spontaneous and oncogene-induced tumorigenesis, Mdm2S394A mice are actually more resistant to radiation-induced T cell lymphomagenesis than WT mice, despite having clear defects in p53-mediated thymic apoptosis.
Figure 4. Mdm2S394A mice are resistant to IR-induced lymphomagenesis.

(A) Kaplan-Meier survival curves of WT (n = 20), Mdm2S394A (n = 19), p53+/− (n = 9) and p53−/− (n = 7) mice exposed to 7 Gy cumulative IR. Median survival times were as follows: WT (185 days), Mdm2S394A (n.d.), p53+/− (154 days) and p53−/− (131 days). Curves were compared by log-rank test: WT to p53−/− (P < 0.0001), WT to p53+/− (P < 0.0001), WT to Mdm2S394A (P = 0.0007), Mdm2S394A to p53+/− (P < 0.0001), Mdm2S394A to p53−/− (P < 0.0001).
(B) Representative tissue sections of lymphomas that developed in the thymus, spleen and liver of WT and Mdm2S394A mice stained with haematoxylin and eosin at 10X (left) and 40X (right) magnification. Scale bars represent 100 μm.
(C) Representative tissue sections of lymphomas that developed in the thymus, spleen and liver of WT and Mdm2S394A mice stained with antibodies specific for CD3 (left) and B220 (right). Scale bars represent 100 μm. See also Supplemental Figure S4.
Radiation resistance in Mdm2S394A mice is dictated by improved bone marrow recovery following IR
We have previously reported that Mdm2S394A mice are resistant to threshold-lethal doses (8 Gy) of radiation. To better understand why ATM phosphorylation of Mdm2-S394 would promote radiation-induced lymphomagenesis yet provide resistance to acute radiation, we decided to examine further the response of these mice to whole-body IR. The primary cause of lethality in mice subjected to IR doses as high as 10 Gy is a p53-dependent ablation of the bone marrow compartment, known as “hematopoietic syndrome” (Komarova et al., 2004). Previous reports have revealed that even small changes in p53 activity can profoundly impact the hematopoietic system (Mendrysa et al., 2003; Terzian et al., 2007; Wang et al., 2011; Pant et al., 2013), and hematopoietic failure phenotypes have previously been described in mice bearing hypomorphic or reduced copies of functional Mdm2 alleles (Mendrysa et al., 2003; Terzian et al., 2007). Furthermore, several p53 target genes, including those encoding p21 or Puma have been implicated in governing the radiosensitivity of murine bone marrow (Cheng et al., 2000; van Os et al., 2007; Shao et al., 2010; Yu et al., 2010; Wang et al., 2011; Pant et al., 2013).
To elucidate the basis for the acute radioresistance of Mdm2S394A mice, we examined the expression of the p53-target genes Mdm2, p21, Puma and Noxa by qPCR in the bone marrow of WT, Mdm2S394A and p53−/− mice before and after irradiation (Figure 5A). As we observed in the thymus, no differences were present in the expression levels of any of the examined p53 target genes in untreated Mdm2S394A bone marrow. Similar expression levels of the target genes were observed in untreated p53−/− mice. Following whole body treatment of mice with 5 Gy IR, the expression levels of all four genes increased dramatically in WT bone marrow, indicative of a strong p53 response. However, lower levels of p21, Puma and Noxa transcripts were detected in Mdm2S394A bone marrow, indicating reduced p53 activity in this tissue. Although Mdm2 transcript levels did not appear to be overtly reduced in irradiated Mdm2S394A bone marrow, no induction of Mdm2, p21 or Puma transcription was observed in the bone marrow of p53-deficient mice, pointing to the p53-dependence of their induction following irradiation.
Figure 5. Threshold-lethal radiation resistance in Mdm2S394A mice is dictated by improved bone marrow recovery following IR.

(A) Fold expression of p53-target genes in bone marrow of WT, Mdm2S394A and p53−/− mice, untreated and 6 hours after 5 Gy IR, was determined by real-time quantitative PCR. Fold expression was calculated relative to untreated WT samples using Rplp0 as internal reference (n = 3-4 mice, ±SEM). *P < 0.05, **P < 0.01 (Student’s t-tests of Ct values).
(B) Haematoxylin and eosin stained bone marrow from WT, Mdm2S394A and p53−/− mice exposed to 8 Gy IR. Blue arrows indicate nascent hematopoietic cell colonies. Scale bars represent 100 μm.
(C) Bone marrow sections from untreated and 9 days post-IR mice described in (B) were stained with an antibody specific for TER-119. Scale bars represent 100 μm.
We next examined the bone marrow of WT, Mdm2S394A and p53−/− mice by H&E staining at intervals up to 9 days following exposure to 8 Gy IR (Figure 5B). To our surprise, all mice examined showed a dramatic decrease in cellularity at 1 and 3 days following irradiation, to the point where the three genotypes were phenotypically indistinguishable. Although WT bone marrow continued to display a progressive loss of cellularity at day 6 after IR, large colonies of cells had appeared in the bone marrow of Mdm2S394A and p53−/− mice at this time (Figure 5B-see arrows). By 9 days post-IR, the colonies present in Mdm2S394A and p53−/− mice had expanded significantly and often bridged the medullary cavity, whereas only a few smaller colonies were observed in WT bone marrow. Immunohistochemical staining for CD45 confirmed that the colonies were of hematopoietic origin (data not shown). The timing of the increased bone marrow cellularity in Mdm2S394A and p53−/− mice (but not in WT mice) is likely significant, as it precedes by one day the onset of mortality observed in WT mice treated with 8 Gy IR (Gannon et al., 2012).
It has been shown that mice undergoing hematopoietic recovery produce primarily myeloerythroid cells, and myeloerythroid-restricted progenitors are sufficient to confer radioprotection (Uchida et al., 1994; Na Nakorn et al., 2002). We further characterized the bone marrow colonies by IHC staining for the erythroid marker TER-119 (Figure 5C). Consistent with previous findings, we found that approximately 25% of nucleated bone marrow cells and all mature erythrocytes stained positive for TER-119 in the untreated bone marrow of mice irrespective of their genotype (Kina et al., 2000). However, at 9 days post-8 Gy IR treatment, only the few remaining mature erythrocytes in the WT bone marrow stained positive for TER-119, whereas all of the large colonies present in Mdm2S394A and p53−/− bone marrow were predominantly TER-119 positive. Thus, the p53-dependant resistance to hematopoietic syndrome observed in Mdm2S394A and p53−/− mice following whole body IR is linked to the increased capability of these models to repopulate their erythroid cell compartment.
Radiation resistance in Mdm2S394A mice is governed by hematopoietic stem and progenitor cells
In order to characterize the cell type responsible for the increase in bone marrow repopulation and subsequent radioresistance observed in Mdm2S394A mice, we utilized flow cytometry to examine total bone marrow harvested 24 hours after treatment with IR. No differences were observed in the numbers of lineage-defined, mature hematopoietic cells in untreated WT or Mdm2S394A mice (Figure 6A). Furthermore, no differences were observed in the numbers of mature hematopoietic cells in irradiated WT or Mdm2S394A bone marrow, save for slightly higher numbers of surviving B cells (p=0.046) in Mdm2 mutant mice. These results are in keeping with the lack of a histopathological difference in the initial loss of cellularity in irradiated WT and Mdm2S394A bone marrow. In addition, no difference was observed in the numbers of Lin-Sca1-cKit+ (L−S−K) common myeloid progenitors (CMPs) in untreated WT and Mdm2S394A bone marrow, or in WT and Mdm2S394A bone marrow following irradiation (Figure 6B). This was confirmed by in vitro colony forming assays performed using the bone marrow of untreated and irradiated WT and Mdm2S394A mice (Figure S5). While there was no difference in the numbers of Lin-Sca1+cKit+ (L−SK) hematopoietic stem and progenitor cells (HSPCs) in untreated WT and Mdm2S394A bone marrow, there were significantly more HSPCs present in Mdm2S394A bone marrow than in WT bone marrow following irradiation (Figure 6B). This finding indicates that the radioresistance observed in Mdm2S394A mice is due to reduced loss and/or increased function of HSPCs in this model following IR exposure.
Figure 6. Radiation resistance in Mdm2S394A mice is governed by hematopoietic stem and progenitor cells (HSPCs).

(A) Quantification of lineage-defined hematopoietic cells in bone marrow of WT and Mdm2S394A mice in the absence of treatment (top) and 24 hours after exposure to 5 Gy IR (bottom) (n = 3-4, ±SEM).
(B) Quantification of L−S−K (CMP) and L−SK (HSPC) hematopoietic progenitor cells in bone marrow of WT and Mdm2S394A mice described in (A).
(C) Schematic showing the experimental design of bone marrow repopulation assays. PB, peripheral blood; BM, bone marrow.
(D) Leukocyte marker analysis of peripheral blood of recipient mice described in (C) at 4 (n = 15), 8 (n = 15) and 10 (n = 5) weeks after bone marrow transplantation (±SEM).
(E) Leukocyte marker analysis of total bone marrow of recipient mice described in (C), untreated, 1 and 12 days after exposure to 5 Gy IR (n = 5, ±SEM). *P < 0.05, **P < 0.01 (Student’s t-tests).
We further examined the HSPCs of Mdm2S394A mice by performing in vivo competitive repopulation assays. Mdm2S394A bone marrow expressing the CD45.2 leukocyte marker and WT bone marrow expressing the CD45.1 leukocyte marker were transplanted in a 1:1 ratio into lethally irradiated WT (CD45.1) recipient mice (Figure 6C). Peripheral blood analysis at 4, 8 and 10 weeks following transplantation showed gradually increasing relative contributions to the hematopoietic lineage of Mdm2S394A bone marrow (62%, 69% and 72%, respectively) (Figure 6D). This indicated that Mdm2S394A HSPCs have an inherent advantage relative to WT HSPCs in their ability to repopulate the hematopoietic compartment of lethally irradiated mice. In addition, we irradiated a cohort of the same bone marrow-chimeric mice at 8 weeks post-transplantation and assayed the relative contributions of WT and Mdm2S394A bone marrow, 1 and 12 days later (Figure 6E). Relative contributions of 21% WT and 78% Mdm2S394A were observed in the untreated bone marrow of reconstituted mice. No significant changes were observed in the percentages of WT and Mdm2S394A bone marrow at 1 day after IR (18% and 81%, respectively). However, a significant shift towards a greater proportion of Mdm2S394A bone marrow was observed (8% WT, 91% Mdm2S394A) in the reconstituted mice 12 days after IR. These results confirm that Mdm2S394A HSPCs have a greater capacity to repopulate irradiated bone marrow and determine that Mdm2S394A HSPCs display greater stem cell function after genotoxic insult. Furthermore, increased Mdm2S394A HSPC cell function is cell-autonomous, as Mdm2S394A HSPCs outperform WT HSPCs in a WT microenvironment.
DISCUSSION
Upon exposure to ionizing radiation, the activated ATM effector kinase induces the phosphorylation of multiple signaling molecules involved in the DNA damage response (DDR), including the p53 tumor suppressor protein and its chief negative regulator, Mdm2. This signaling cascade has been proposed by many groups to activate p53 by interrupting Mdm2-inhibition of p53, thereby facilitating p53 transactivation of downstream target genes whose products manifest the cellular response to DNA damage. Previously, we have shown that phosphorylation of p53 serine 18 in mice following DNA damage has only a modest role in regulating p53-mediated apoptosis and in governing p53 tumor suppression, whereas ATM phosphorylation of Mdm2 serine 394 following DNA damage regulates the amplitude and the duration of the DDR in mice, and alters p53 suppression of spontaneous tumorigenesis (Sluss et al., 2004; Gannon et al., 2012). These studies demonstrate in vivo the importance of Mdm2 phosphorylation in ATM regulation of the p53 DNA damage response.
In our present study, we explore in vivo the mechanism by which Mdm2 phosphorylation alters p53 functions, and examine whether ATM-Mdm2-p53 signaling regulates tumorigenesis in mice induced by activated oncogenes or ionizing radiation. Mdm2 is present at higher levels in the thymus and spleen of Mdm2S394A mice following DNA damage (Figures 1A, 2A, S1). These elevated Mdm2 levels are the result of Mdm2S394A being more stable following DNA damage (Figure 2C), as Mdm2 transcript levels are equivalent or even slightly reduced in IR-treated Mdm2S394A tissues during this timeframe (Figure 1C). Although we previously found no difference in the stability of phosphorylated Mdm2 after DNA damage (Gannon 2012), using newer and well-validated Mdm2 antibodies and reduced radiation dosages (to promote recovery of the damaged cells), we now find that Mdm2-S394 phosphorylation indeed destabilizes Mdm2 in vivo. Our result is in keeping with previous in vitro studies that indicate a decrease in the half-life of Mdm2 after genotoxic stress (Stommel and Wahl, 2004; Itahana et al., 2007; Inuzuka et al. 2010). Our data provides direct, in vivo, evidence that DNA damage-induced phosphorylation of Mdm2 at this single residue by ATM induces Mdm2 destabilization. Furthermore, our data indicates that Mdm2 levels are elevated in thymi and spleens of Mdm2S394A mice even in the absence of acute, exogenous DNA damage (Figures 1A, S1). We interpret this finding to be indicative of some basal level of ATM activity in unstressed tissues that exerts influence on native Mdm2 protein stability.
It is presently unclear how Mdm2 phosphorylation facilitates Mdm2 destabilization. DNA damage-induced degradation of Mdm2 has been shown to occur even in the absence of Mdm2 E3 ligase activity (Itahana et al., 2007), and studies have identified the F-box proteins β-TRCP and FBXO31 as mediators of Mdm2 degradation by the SCF complex (Inuzuka et al. 2010; Malonia et al., 2015). Since FBXO31 is phosphorylated by ATM and was recently found to interact with Mdm2 in a manner dependent on ATM phosphorylation of Mdm2 (Santra et al., 2009; Malonia et al., 2015), it is plausible that ATM phosphorylation of Mdm2-S394 in IR-treated mice results in FBXO31-induced destabilization of Mdm2. Unfortunately, our attempts to assay Mdm2-FBXO31 interactions in whole tissues before and after DNA damage have proven unsuccessful. Thus, the precise mechanism of how ATM phosphorylation of Mdm2-S394 promotes Mdm2 degradation under physiologic conditions remains to be determined.
The increased levels of Mdm2 observed in the spleen and thymus of Mdm2S394A mice after DNA damage correlates with a decrease in p53 levels and p53 activity in these Mdm2S394A tissues. Previous studies have suggested that ATM phosphorylation of the analogous residue on human MDM2 (S395), either alone or in combination with several other ATM-target serine residues in the same region, impacts Mdm2’s ability to promote p53 degradation and nuclear export, and governs RING-domain oligomerization and polyubiquitination of p53 (Maya et al., 2001; Cheng et al., 2009). DNA damage resulted in p53 stabilization in WT thymocytes, albeit after a brief period of p53 degradation (Figure S2B). While the initial rate of p53 degradation does not appear affected in irradiated Mdm2S394A thymocytes, we observed a prolonged period of p53 destabilization, ultimately resulting in lower relative levels of p53. This prolonged p53 destabilization may reflect the increased stability of Mdm2S394A after DNA damage. This is supported by the increased relative amounts of Mdm2-bound p53 after IR observed in Mdm2S394A thymi (Figure 2C). However, it remains possible that increased DNA damage-induced p53 activity in WT thymi is caused not only by reduced Mdm2-mediated p53 degradation (due to destabilization of Mdm2) but also by reduced Mdm2 steric inhibition of p53. As Mdm2 binds to the amino-terminal, transcriptional activation domain of p53 and inhibits p53 target gene expression, reduced Mdm2-p53 complex formation after Mdm2 phosphorylation by ATM may account for the increase in p53 activity even when p53 protein stability is only modestly altered (Momand et al., 1992; Oliner et al., 1993).
The reduced level of p53 activity in Mdm2S394A mice facilitates more rapid B cell lymphomagenesis in Eμ-myc mice (Figure 3). It has been previously demonstrated that activated oncogenes such as Myc result in elevated Arf expression, a result of hyperproliferative signaling (Zindy et al., 1998; Sherr et al., 2005). The importance of disrupting the Arf-Mdm2-p53 pathway in Myc-driven lymphomagenesis is evidenced by the fact that Eμ-myc driven tumors in mice face a selective pressure to inactivate the p53 pathway, by either p53 mutation, Mdm2 overexpression, or loss of Arf (Eischen et al., 1999). However, we observed a significant acceleration in the median time of tumor presentation in Eμ-myc;Mdm2S394A mice in the absence of Mdm2 overexpression or p53 mutation in tumors (Figures 3A, 3C). This suggests that an absence of Mdm2-S394 phosphorylation is sufficient to diminish p53 activity in response to oncogene activation and reduces the selective pressure to genetically disrupt the Mdm2-p53 signaling axis (Figure 3D). Although loss of Arf was observed in a subset of Eμ-myc;Mdm2S394A tumors, this finding likely reflects the ability of Arf to promote tumorigenesis in Eμ-myc mice through an Mdm2/p53 independent mechanism.
Although elevated Myc levels have been shown to result in increased levels of cellular DNA damage, the relative contributions of the pro-proliferative effects and DNA damage-induced effects of Myc on tumor formation remain uncertain. (Halazonetis et al., 2008; Meek, 2015). To further explore a role for Mdm2 phosphorylation in the DDR and in regulation of p53 tumor suppression, we examined lymphomagenesis induced by low level exposure of mice to ionizing radiation (Figure 4). In contrast to our results in Myc-driven lymphomagenesis, Mdm2S394A mice proved to be more resistant to DNA damage-induced T cell lymphomagenesis, highlighting a stark difference in the effects of ATM-Mdm2-p53 signaling in lymphomas induced by different types of DNA damage-related stress.
The cell of origin in IR-induced lymphomas has historically been viewed as a stem/progenitor cell residing within the bone marrow (Kaplan, 1964), and IR-induced lymphomagenesis is significantly enhanced in the absence of functional p53 (Kemp et al., 1994). Subsequent studies examining the contribution of the p53-dependent pro-apoptotic genes Puma and Noxa made the paradoxical observation that Puma−/− mice develop fewer IR-induced lymphomas (Labi et al., 2010; Michalak et al., 2010). This was attributed to increased survival of leukocytes in the bone marrow, which reduced the proliferative stress and/or propagation of lesions within progenitor cells tasked with repopulating the bone marrow. That Noxa−/− mice displayed only modest radioprotection of LSK cells and developed more lymphomas was interpreted as a failure to clear damaged progenitors, thereby promoting the survival of damaged stem/progenitor cells (Michalak et al., 2010). Despite a reduction of p53-dependent gene expression of Puma and Noxa in total bone marrow of IR-damaged Mdm2S394 mice (Figure 5A), we observed no defects in the attrition of mature hematopoietic cells or lineage-defined progenitor cells (CMPs) (Figure 6A, 6B). Only the most primitive HSPCs display resistance to IR (Figure 6B). Our finding that Mdm2S394A mice are resistant to IR-induced lymphomas reveals that the effects of a reduced p53-dependent damage response in Mdm2S394A mice does not mirror the ablation of either Puma or Noxa alone. Interestingly, a recent study by Kirsch and coworkers suggests that the tumor-initiating cell in IR-induced lymphomas is thymic in origin (Lee et al. 2015). Using mice in which p53 activity was temporally blocked during total-body irradiation, the authors propose that the IR-induced p53 response in bone marrow promotes lymphomagenesis by reducing HSPC fitness, thereby reducing the competition of cells originating from the bone marrow with thymocytes containing oncogenic lesions. The results we have observed with Mdm2S394A mice align favorably with this model, as we have observed less p53 activity and increased HSPC fitness in Mdm2S394A mice following threshold-lethal doses of radiation, and reduced incidence of T cell lymphomagenesis in Mdm2S394A mice after IR exposure (Figures 5, 6). The p53 dependence of the increased HSPC fitness in Mdm2S394A mice is further intimated by studies which have observed increased bone marrow repopulation potential in bone marrow deficient for p53, both in the presence and absence of IR (reviewed in Pant et al., 2012). Interestingly, a recent study has linked Mdm2 to enhanced stem-ness via association with the Polycomb Repressor Complex 2 (PRC2) (Wienken et al., 2015). Further studies into the relative p53-dependent and p53-independent contributions of Mdm2 to HSPC fitness are clearly warranted.
We conclude that ATM phosphorylation of Mdm2-S394 is critical for Mdm2 degradation and robust p53 activation after DNA damage. Our results show that while ATM’s regulation of p53 activity through Mdm2 is critical in preventing oncogene-induced tumorigenesis, disrupting this regulation imparts protection from bone marrow ablation and lymphomagenesis resulting after DNA damage. These observations are of clinical significance, as they suggest that the temporal inhibition of Mdm2 phosphorylation by short-acting kinase inhibitors or by use of excess decoy ATM target-sites (a molecular sponge of sorts) would reduce p53 activation and alleviate the toxicity associated with radiotherapies. Our results further indicate that such treatment would likely protect against the subsequent development of secondary hematological malignancies induced by exposure to ionizing radiation.
EXPERIMENTAL PROCEDURES
Mice and Animal Studies
All animals described in this study were on a C57Bl/6 background. Mice and cells were irradiated with a cesium-137 source (Gammacell 40). The generation of Mdm2S394A mice has been previously described (Gannon et al, 2012). Eμ-Myc mice were a gift from Christine Eischen (Vanderbilt University). Mice in Eμ-Myc tumor assays were virgin and inherited the Eμ-Myc allele paternally. For IR-induced tumor assays, mice 31±3 days of age were irradiated weekly with 1.75 Gy for 4 weeks (7 Gy cumulative dose). For bone marrow transplantation experiments, recipient CD45.1 mice received 11 Gy of whole body irradiation in a split dose (2 × 5.5 Gy, 4 hours apart). Irradiated recipients were reconstituted by i.v. injection of 2 × 106 bone marrow cells (1:1 mixture of WT and Mdm2S394A). All animals used in this study were maintained and assayed in accordance with federal guidelines and those established by the Institutional Animal Care and Use Committee at the University of Massachusetts Medical School.
Protein Analysis
Tissues and cells were lysed in NP-40 lysis buffer or in CelLytic MT Cell Lysis Reagent (Sigma), supplemented with protease and phosphatase inhibitors. Protein extracts were analyzed by western blotting or immunoprecipitation/western blotting. A detailed description of the methods employed, including antibodies and clones, is provided in the Supplemental Experimental Procedures.
Gene Expression Analysis and Sequencing
Total RNA was isolated from tissues by RNeasy mini kit (QIAGEN) and cDNA synthesized by Superscript III First Strand Synthesis System (Invitrogen). Quantitative PCR was performed using SYBR Select Master Mix (Applied Biosystems) in conjunction with a 7300 Real-Time PCR System (Applied Biosystems). A detailed description of the methods employed, including primer sequences, for qPCR, RT-PCR and sequencing is provided in the Supplemental Experimental Procedures.
Histopathology
Tissues samples were fixed in 10% formalin for 24 hr. The UMMS Diabetes and Endocrinology Research Center Morphology Core performed embedding, sectioning, and staining. TUNEL staining was performed using the In Situ Cell Death Detection Kit, POD (Roche) according to manufacturer’s instructions. Immunohistochemistry was performed with antibodies specific for B220 (550286; BD Pharmingen), CD3 (A0452; Dako) and TER-119 (553671; BD Pharmingen). Stained tissue was analyzed using an Olympus CX41 microscope fitted with a PixeLINK camera and software.
Flow Cytometry
Total bone marrow from both hind limbs was harvested, RBCs were lysed, and single-cell suspensions were stained with cell-surface antibodies for Gr-1, CD11B, CD3, and B220. For LSK analysis, bone marrow cells were stained with a biotin lineage mixture, and antibodies for Sca-1, c-Kit, CD34, and Flk2. To distinguish between WT and Mdm2S394A hematopoietic cells in the reconstitution studies, peripheral blood and bone marrow was stained with antibodies specific for CD45.1 and CD45.2. All samples were run on a BD LSRII flow cytometer (BD Bioscience) and analyzed using FlowJo software (Tree Star). A complete list of antibodies including clone numbers is given in Table S1.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism software, version 6.0d. Kaplan–Meier survival curves were analyzed by log-rank test. A P-value < 0.05 was considered statistically significant for Student’s t-tests.
Supplementary Material
Acknowledgments
This research was supported by grants from the NIH: R01-CA077735 (S.N.J.) and R01-CA096899 (M.A.K.). J.E.R. was supported by Post-doctoral fellowship 125087-PF-13-247-01-LIB from the American Cancer Society.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, M.I.C, and S.N.J.; Methodology M.I.C., H.S.G. and S.N.J.; Investigation, M.I.C. and J.E.R.; Resources, M.A.K. and S.N.J.; Writing – Original Draft, M.I.C. and S.N.J.; Writing – Review & Editing, M.I.C., J.E.R., H.S.G., M.A.K. and S.N.J.; Funding Acquisition, S.N.J.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Beckerman R, Prives C. Transcriptional Regulation by P53. Cold Spring Harb Perspect Biol. 2010;2 doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Broz DK, Basak S, Park EJ, McLaughlin ME, et al. Distinct p53 Transcriptional Programs Dictate Acute DNA-Damage Responses and Tumor Suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C, Hergenhahn M, Kaeser MD, Wu Z, Saito S, Iggo R, Hollstein M, Appella E, Xu Y. Cell Type- and Promoter-specific Roles of Ser18 Phosphorylation in Regulating p53 Responses. J Biol Chem. 2003;278:41028–41033. doi: 10.1074/jbc.M306938200. [DOI] [PubMed] [Google Scholar]
- Chao C, Herr D, Chun J, Xu Y. Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J. 2006;25:2615–2622. doi: 10.1038/sj.emboj.7601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005;24:3411–3422. doi: 10.1038/sj.emboj.7600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic Stem Cell Quiescence Maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Chen L, Li Z, Lane WS, Chen J. ATM activates p53 by regulating MDM2 oligomerization and E3 processivity. EMBO J. 2009;28:3857–3867. doi: 10.1038/emboj.2009.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Cross B, Li B, Chen L, Li Z, Chen J. Regulation of MDM2 E3 Ligase Activity by Phosphorylation after DNA Damage. Mol Cell Biol. 2011;31:4951–4963. doi: 10.1128/MCB.05553-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CAJ, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon HS, Jones SN. Using Mouse Models to Explore MDM-p53 Signaling in Development, Cell Growth, and Tumorigenesis. Genes Cancer. 2012;3:209–218. doi: 10.1177/1947601912455324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon HS, Woda BA, Jones SN. ATM phosphorylation of Mdm2 Ser394 regulates the amplitude and duration of the DNA damage response in mice. Cancer Cell. 2012;21:668–679. doi: 10.1016/j.ccr.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Huang L, Yan Z, Liao X, Li Y, Yang J, Wang ZG, Zuo Y, Kawai H, Shadfan M, Ganapathy S, et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc Natl Acad Sci. 2011;108:12001–12006. doi: 10.1073/pnas.1102309108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka H, Tseng A, Gao D, Zhai B, Zhang Q, Shaik S, Wan L, Ang XL, Mock C, Yin H, et al. Phosphorylation by Casein Kinase I Promotes the Turnover of the Mdm2 Oncoprotein via the SCF(β-TRCP) Ubiquitin Ligase. Cancer Cell. 2010;18:147–159. doi: 10.1016/j.ccr.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindström MS, Bhat KP, Godfrey VL, Evan GI, Zhang Y. Targeted Inactivation of Mdm2 RING Finger E3 Ubiquitin Ligase Activity in the Mouse Reveals Mechanistic Insights into p53 Regulation. Cancer Cell. 2007;12:355–366. doi: 10.1016/j.ccr.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Jackson MW, Berberich SJ. MdmX Protects p53 from Mdm2-Mediated Degradation. Mol Cell Biol. 2000;20:1001–1007. doi: 10.1128/mcb.20.3.1001-1007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- Kaplan HS, Brown MB. A Quantitative Dose-Response Study of Lymphoid-Tumor Development in Irradiated C57 Black Mice. J Natl Cancer Inst. 1952;13:185–208. [PubMed] [Google Scholar]
- Kaplan HS. The Role of Radiation on Experimental Leukemogenesis. Natl Cancer Inst Monogr. 1964;14:207–220. [PubMed] [Google Scholar]
- Kawai H, Lopez-Pajares V, Kim MM, Wiederschain D, Yuan ZM. RING Domain–Mediated Interaction Is a Requirement for MDM2’s E3 Ligase Activity. Cancer Res. 2007;67:6026–6030. doi: 10.1158/0008-5472.CAN-07-1313. [DOI] [PubMed] [Google Scholar]
- Kemp CJ, Wheldon T, Balmain A. p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat Genet. 1994;8:66–69. doi: 10.1038/ng0994-66. [DOI] [PubMed] [Google Scholar]
- Kina T, Ikuta K, Takayama E, Wada K, Majumdar AS, Weissman IL, Katsura Y. The monoclonal antibody TER-119 recognizes a molecule associated with glycophorin A and specifically marks the late stages of murine erythroid lineage. Brit J Haematol. 2000;109:280–287. doi: 10.1046/j.1365-2141.2000.02037.x. [DOI] [PubMed] [Google Scholar]
- Komarova EA, Kondratov RV, Wang K, Christov K, Golovkina TV, Goldblum JR, Gudkov AV. Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene. 2004;23:3265–3271. doi: 10.1038/sj.onc.1207494. [DOI] [PubMed] [Google Scholar]
- Kubbutat MHG, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Labi V, Erlacher M, Krumschnabel G, Manzl C, Tzankov A, Pinon J, Egle A, Villunger A. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev. 2010;24:1602–1607. doi: 10.1101/gad.1940210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-L, Castle KD, Moding EJ, Blum JM, Williams N, Luo L, Ma Y, Borst LB, Kim Y, Kirsch DG. Acute DNA damage activates the tumour suppressor p53 to promote radiation-induced lymphoma. Nat Commun. 2015;6 doi: 10.1038/ncomms9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malonia SK, Dutta P, Santra MK, Green MR. F-box protein FBXO31 directs degradation of MDM2 to facilitate p53-mediated growth arrest following genotoxic stress. Proc Natl Acad Sci. 2015;112:8632–8637. doi: 10.1073/pnas.1510929112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya R, Balass M, Kim ST, Shkedy D, Leal JFM, Shifman O, Moas M, Buschmann T, Ronai Z’ev, Shiloh Y, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–1077. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek DW. Regulation of the p53 response and its relationship to cancer. Biochem J. 2015;469:325–346. doi: 10.1042/BJ20150517. [DOI] [PubMed] [Google Scholar]
- Mendrysa SM, McElwee MK, Michalowski J, O’Leary KA, Young KM, Perry ME. mdm2 Is Critical for Inhibition of p53 during Lymphopoiesis and the Response to Ionizing Irradiation. Mol Cell Biol. 2003;23:462–472. doi: 10.1128/MCB.23.2.462-473.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak EM, Vandenberg CJ, Delbridge ARD, Wu L, Scott CL, Adams JM, Strasser A. Apoptosis-promoted tumorigenesis: -irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev. 2010;24:1608–1613. doi: 10.1101/gad.1940110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliorini D, Denchi EL, Danovi D, Jochemsen A, Capillo M, Gobbi A, Helin K, Pelicci PG, Marine JC. Mdm4 (Mdmx) Regulates p53-Induced Growth Arrest and Neuronal Cell Death during Early Embryonic Mouse Development. Mol Cell Biol. 2002;22:5527–5538. doi: 10.1128/MCB.22.15.5527-5538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- Na Nakorn T, Traver D, Weissman IL, Akashi K. Myeloerythroid-restricted progenitors are sufficient to confer radioprotection and provide the majority of day 8 CFU-S. J Clin Invest. 2002;109:1579–1585. doi: 10.1172/JCI15272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–860. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- Pant V, Xiong S, Iwakuma T, Quintás-Cardama A, Lozano G. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc Natl Acad Sci. 2011;108:11995–12000. doi: 10.1073/pnas.1102241108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant V, Quintás-Cardama A, Lozano G. The p53 pathway in hematopoiesis: lessons from mouse models, implications for humans. Blood. 2012;120:5118–5127. doi: 10.1182/blood-2012-05-356014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant V, Xiong S, Jackson JG, Post SM, Abbas HA, Quintás-Cardama A, Hamir AN, Lozano G. The p53–Mdm2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes Dev. 2013;27:1857–1867. doi: 10.1101/gad.227249.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–95. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- Pereg Y, Shkedy D, de Graaf P, Meulmeester E, Edelson-Averbukh M, Salek M, Biton S, Teunisse AFAS, Lehmann WD, Jochemsen AG, et al. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc Natl Acad Sci. 2005;102:5056–5061. doi: 10.1073/pnas.0408595102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra MK, Wajapeyee N, Green MR. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature. 2009;459:722–725. doi: 10.1038/nature08011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L, Sun Y, Zhang Z, Feng W, Gao Y, Cai Z, Wang ZZ, Look AT, Wu WS. Deletion of proapoptotic Puma selectively protects hematopoietic stem and progenitor cells against high-dose radiation. Blood. 2010;115:4707–4714. doi: 10.1182/blood-2009-10-248872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp DA, Kratowicz SA, Sank MJ, George DL. Stabilization of the MDM2 Oncoprotein by Interaction with the Structurally Related MDMX Protein. J Biol Chem. 1999;274:38189–38196. doi: 10.1074/jbc.274.53.38189. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Bertwistle D, Den Besten W, Kuo ML, Sugimoto M, Tago K, Williams RT, Zindy F, Roussel MF. p53-Dependent and -independent functions of the Arf tumor suppressor. Cold Spring Harb Symp Quant Biol. 2005;70:129–137. doi: 10.1101/sqb.2005.70.004. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RC, van der Houven van Oordt W, Hateboer G, van der Eb AJ, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996;15:5349–5357. [PMC free article] [PubMed] [Google Scholar]
- Sluss HK, Armata H, Gallant J, Jones SN. Phosphorylation of Serine 18 Regulates Distinct p53 Functions in Mice. Mol Cell Biol. 2004;24:976–984. doi: 10.1128/MCB.24.3.976-984.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluss HK, Gannon H, Coles AH, Shen Q, Eischen CM, Jones SN. Phosphorylation of p53 Serine 18 Upregulates Apoptosis to Suppress Myc-Induced Tumorigenesis. Mol Cancer Res. 2010;8:216–222. doi: 10.1158/1541-7786.MCR-09-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soussi T, Beroud C. Assessing TP53 status in human tumours to evaluate clinical outcome. Nat Rev Cancer. 2001;1:233–239. doi: 10.1038/35106009. [DOI] [PubMed] [Google Scholar]
- Stommel JM, Wahl GM. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J. 2004;23:1547–1556. doi: 10.1038/sj.emboj.7600145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 1999;447:5–9. doi: 10.1016/s0014-5793(99)00254-9. [DOI] [PubMed] [Google Scholar]
- Terzian T, Wang Y, Van Pelt CS, Box NF, Travis EL, Lozano G. Haploinsufficiency of Mdm2 and Mdm4 in Tumorigenesis and Development. Mol Cell Biol. 2007;27:5479–5485. doi: 10.1128/MCB.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollini LA, Jin A, Park J, Zhang Y. Regulation of p53 by Mdm2 E3 Ligase Function Is Dispensable in Embryogenesis and Development, but Essential in Response to DNA Damage. Cancer Cell. 2014;26:235–247. doi: 10.1016/j.ccr.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Aguila HL, Fleming WH, Jerabek L, Weissman IL. Rapid and sustained hematopoietic recovery in lethally irradiated mice transplanted with purified Thy-1.1lo Lin-Sca-1+ hematopoietic stem cells. Blood. 1994;83:3758–3779. [PubMed] [Google Scholar]
- van Os R, Kamminga LM, Ausema A, Bystrykh LV, Draijer DP, van Pelt K, Dontje B, de Haan G. A Limited Role for p21Cip1/Waf1 in Maintaining Normal Hematopoietic Stem Cell Functioning. Stem Cells. 2007;25:836–843. doi: 10.1634/stemcells.2006-0631. [DOI] [PubMed] [Google Scholar]
- Wang YV, Leblanc M, Wade M, Jochemsen AG, Wahl GM. Increased radio-resistance and accelerated B-cell lymphomas in mice with Mdmx mutations that prevent modifications by DNA damage-activated kinases. Cancer Cell. 2009;16:33–43. doi: 10.1016/j.ccr.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YV, Leblanc M, Fox N, Mao JH, Tinkum KL, Krummel K, Engle D, Piwnica-Worms D, Piwnica-Worms H, Balmain A, et al. Fine-tuning p53 activity through C-terminal modification significantly contributes to HSC homeostasis and mouse radiosensitivity. Genes Dev. 2011;25:1426–1438. doi: 10.1101/gad.2024411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienken M, Dickmanns A, Nemajerova A, Kramer D, Najafova Z, Weiss M, Karpiuk O, Kassem M, Zhang Y, Lozano G, et al. MDM2 Associates with Polycomb Repressor Complex 2 and Enhances Stemness-Promoting Chromatin Modifications Independent of p53. Mol Cell. 2016;61:68–83. doi: 10.1016/j.molcel.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Earle J, Saito S, Anderson CW, Appella E, Xu Y. Mutation of Mouse p53 Ser23 and the Response to DNA Damage. Mol Cell Biol. 2002;22:2441–2449. doi: 10.1128/MCB.22.8.2441-2449.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Shen H, Yuan Y, XuFeng R, Hu X, Garrison SP, Zhang L, Yu J, Zambetti GP, Cheng T. Deletion of Puma protects hematopoietic stem cells and confers long-term survival in response to high-dose -irradiation. Blood. 2010;115:3472–3480. doi: 10.1182/blood-2009-10-248278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.