

Abstract
Development of the preterm infant gut microbiota is emerging as a critical research priority1. Since preterm infants almost universally receive early and often extended antibiotic therapy2, it is important to understand how these interventions alter gut microbiota development3-6. Analysis of 401 stools from 84 longitudinally sampled preterm infants demonstrates that meropenem, cefotaxime and ticarcillin–clavulanate are associated with significantly reduced species richness. In contrast, vancomycin and gentamicin, the antibiotics most commonly administered to preterm infants, have non-uniform effects on species richness, but these can be predicted with 85% accuracy based on the relative abundance of only two bacterial species and two antibiotic resistance (AR) genes at treatment initiation. To investigate resistome development, we functionally selected resistance to 16 antibiotics from 21 faecal metagenomic expression libraries. Of the 794 AR genes identified, 79% had not previously been classified as AR genes. Combined with deep shotgun sequencing of all stools, we find that multidrug-resistant members of the genera Escherichia, Klebsiella and Enterobacter, genera commonly associated with nosocomial infections, dominate the preterm infant gut microbiota. AR genes that are enriched following specific antibiotic treatments are generally unique to the specific treatment and are highly correlated with the abundance of a single species. The most notable exceptions include ticarcillin–clavulanate and ampicillin, both of which enrich for a large number of overlapping AR genes, and are correlated with Klebsiella pneumoniae. We find that all antibiotic treatments are associated with widespread collateral microbiome impact by enrichment of AR genes that have no known activity against the specific antibiotic driver.
Using a combination of metagenomic shotgun sequencing and functional selections, we deeply interrogated the gut microbiota and resistome of 401 stools from 84 preterm infants sampled longitudinally and stratified by antibiotic use during their hospitalization (Table 1, Supplementary Figs 2-3 and Supplementary Table 1a–c). All infants included were born prematurely (<33 weeks gestational age; median (interquartile range (IQR)) 27 weeks (25–29 weeks)) and of low birthweights (median (IQR) of 865 g (718–1,141 g)). In contrast to term infants7-10, we found the preterm infant gut microbiota is colonized by approximately tenfold fewer bacterial species, compositionally distinct, and established independent of delivery mode (Supplementary Fig. 1). While all but two infants amongst the 84 in our cohort received antibiotic therapy within 24 h of birth, 39% received no additional antibiotics after their first week of life (Table 1) and were used as controls to study the effect of specific antibiotic treatments administered between 1 and 10 weeks of life. Infants who were treated with antibiotics after the first week of life received a median (IQR) of 21 (16–32) total days of additional therapy during their hospitalization. Samples were analysed over time, ranging from 6.1 to 157.7 days of life, with a median (IQR) of 3 (2–6) samples analysed per individual. An extensive set of clinical covariates were considered for their impact on the developing gut microbiota (Supplementary Table 1a–b).
Table 1.
Details of preterm infant cohort analysed in this study.
| Early antibiotic exposure only (n = 33) | Early and subsequent antibiotic exposure (n = 51) | |
|---|---|---|
| Birth weight†, g, median (IQR) | 1,147 (1,000–1,330) | 790 (675–855) |
| Gestational age at birth†, weeks, median (IQR) | 29 (27–30) | 25 (24–27) |
| Gender†, M/F | 10/23 | 25/26 |
| Route of delivery†, C-section/vaginal | 21/12 | 30/21 |
| CRIB score†, median (IQR) | 5 (3–8) | 11 (8–13) |
| Total antibiotic exposure*,‡, days, median (IQR), n | ||
| Gentamicin | 2 (1–2), 24 | 8 (4–13), 49 |
| Ampicillin | 2 (2–2), 30 | 2 (2–2), 49 |
| Vancomycin | 0 (0–0), 5 | 8 (5–13), 48 |
| Meropenem | None | 0 (0–6) 19 |
| Cefotaxime | None | 0 (0–1), 15 |
| Ticarcillin–clavulanate | None | 0 (0–2), 15 |
| Clindamycin | None | 0 (0–0), 12 |
| Cefazolin | None | 0 (0–0), 9 |
| Trimethoprim-sulfamethoxazole | None | 0 (0–0), 4 |
| Feedings‡, days, median (IQR), n | ||
| Human milk (All) | 37 (32–47), 33 | 31 (21–58), 51 |
| Human milk (Maternal) | 35 (12–47), 33 | 30 (20–53), 50 |
| Human milk (Donor) | 0 (0–0), 5 | 0 (0–0), 5 |
| Formula | 6 (0–26), 23 | 15 (1–35), 38 |
| Fortification | 44 (32–50), 33 | 44 (22–56), 48 |
| Bacterial culture-positive‡,#, n, Gram+/Gram− (individuals) | ||
| Blood | None | 7/5 (10) |
| Tracheal | None | 31/60 (23) |
| Urine | None | 2/2 (4) |
Supplementary Table 1d includes antibiotic exposure days for exposed individuals only;
Additional information summarized in Supplementary Table 1b;
Additional information summarized in Supplementary Table 1a;
Includes bacterial culture positive tests from multiple time points within an individual.
We profiled the bacterial composition of the preterm infant gut with metagenomic shotgun sequencing using unique clade-specific marker genes (see Methods). Only six bacterial species are both highly prevalent (at least one of these species is present in 99.8% of samples) and highly abundant with median (IQR) relative abundance of 51% (25–72%) across all preterm infant gut microbiota (Fig. 1a). The majority of these species contain multidrug-resistant (MDR) members capable of causing human infections11,12. Of gut microbial communities where >50% of the population consists of one of these species, we observed a significant developmental trend in control individuals, driven primarily by species of the genera Klebsiella, Escherichia, and Enterobacter replacing species of the genera Enteroccoccus and Staphylococcus (Fig. 1b). 16S rRNA gene profiling studies have identified class-level developmental progression of the preterm infant gut microbiota from Bacilli to Gammaproteobacteria3; our data extend these predictions to the species level.
Figure 1. Development of the preterm infant gut microbiota and antibiotic use.
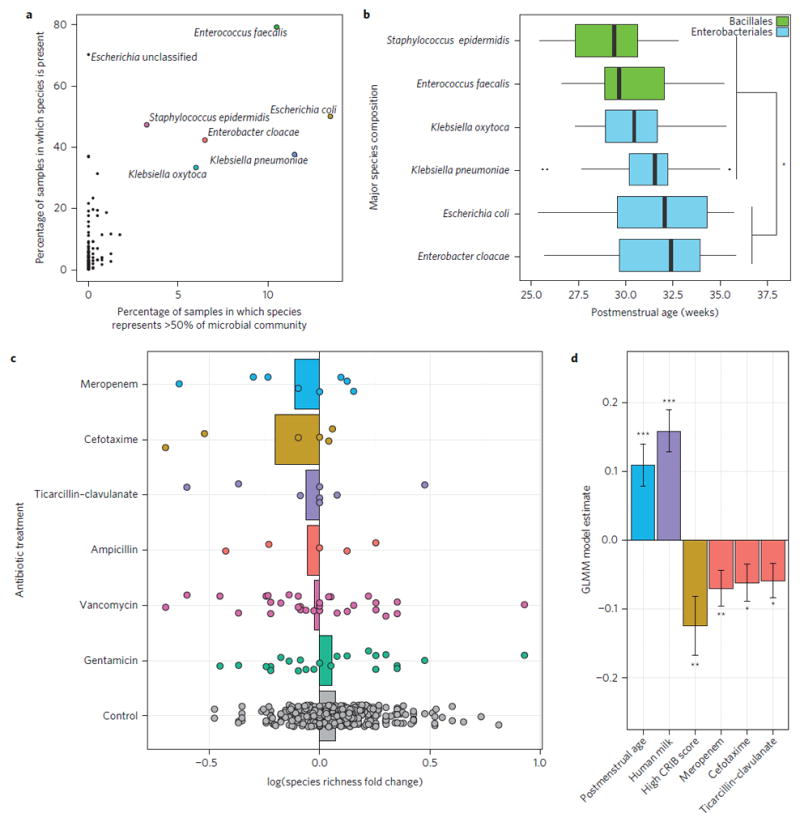
a, Six bacterial species are each prevalent (at least one of these species is present in 99.8% of samples) and highly abundant (more than one of these species comprise more than 50% of the community in more than 50% of samples) in the preterm infant gut microbiota. b, Significant developmental trend of abundant species composition over time in control individuals (P < 0.001, Kruskal–Wallis; *P < 0.05, Dunn’s test with correction for multiple comparisons). Significant for all pair-wise comparisons indicated. c, Species richness log fold-change of the preterm infant gut microbiota from directly before to directly after six different antibiotic treatments. Bars represent average species richness log fold-change. Control samples represent the species richness log fold-change over time in individuals with no antibiotic treatment outside the first week of life. Control samples represent similar mean and standard deviation of days between samples as in antibiotic treatment sets. d, Postmenstrual age and human milk significantly increase gut microbiota species richness, while high CRIB (Clinical Risk Index for Babies) score and three specific antibiotics, meropenem, cefotaxime, and ticarcillin–clavulanate, significantly decrease species richness (***P < 0.001, **P < 0.01; *P < 0.05; GLMM with individual as random effect using 401 preterm infant gut samples). Error bars indicate s.e.
Total antibiotic exposure in preterm infants was associated with significantly reduced species richness (P < 0.001; ANOVA). Decreased gut species richness and diversity in infancy and throughout life have been associated with a number of host pathologies13-16. To investigate the impact of specific antibiotic treatments on species richness, we calculated the log fold-change of species richness from immediately before to immediately after antibiotic treatment (within 48 h of initiating or terminating antibiotics) compared with the log fold-change of species richness in individuals who received no antibiotic treatment outside the first week of life, but over intervals representing typical treatment durations (Fig. 1c). On average, each antibiotic except gentamicin is associated with reduced species richness, while the gut microbiota of control individuals increases in richness over similar intervals. To identify significant effects of specific antibiotics while controlling for other factors that might contribute to preterm infant gut microbiota development, we used a generalized linear mixed model with individual as a random effect (Fig. 1d). All variables listed in Supplementary Table 1a and b were used in model selection. We identified postmenstrual age and human breast milk as significant contributors to increased species richness over time. In contrast, while controlling for these variables as well as for overall health of the infant as determined by CRIB II (Clinical Risk Index for Babies) score, our model identified three broad-spectrum antibiotic therapies that are significantly associated with decreased species richness of the preterm infant gut microbiota: meropenem, cefotaxime, and ticarcillin–clavulanate. Our model, with only six fixed-effects predictors, explains 33% of the variance in species richness and, when including individual as the random effect, explains 67% of the variance.
To further investigate preterm infant gut resistome development with antibiotic use, we used functional metagenomic selections17-22 to deeply characterize the reservoir of AR genes associated with preterm infant gut microbiota. This technique provides culture-unbiased interrogation of resistomes, identifying full-length, functionally verified AR genes without reliance on sequence similarity to previously established AR genes23,24. We constructed meta-genomic libraries from 21 representative preterm infant fecal samples, varying in host age and total antibiotic exposure, totaling approximately 107 Gb of bacterial DNA, and screened these libraries against 16 clinically relevant antibiotics (Supplementary Table 2), including those commonly used in neonatal intensive care units (NICUs)2 and later in childhood25. We assembled 2,004 contigs containing 794 AR genes as annotated using Resfams26, identifying extensive resistance to β-lactams, amphenicols, tetracyclines, and polymyxins (Supplementary Fig. 4a). Culture unbiased approaches have emphasized the immense sequence novelty of bacterial AR genes in every habitat investigated, including the adult gut and soil19,22, however, the preterm infant gut resistome defies this trend. While most AR genes we identified are similar (median 95.8% amino acid identity) to previously sequenced proteins (Fig. 2a), these functionally identified AR genes are surprisingly rare (median 25.4% amino acid identity) in current AR databases27. In other words, while the sequences of the functionally selected AR genes had been in databases, the proteins they encode had yet to be assigned AR functions.
Figure 2. Functional metagenomic selections identify novel antibiotic resistance gene clusters in Proteobacterial species in the preterm infant gut.
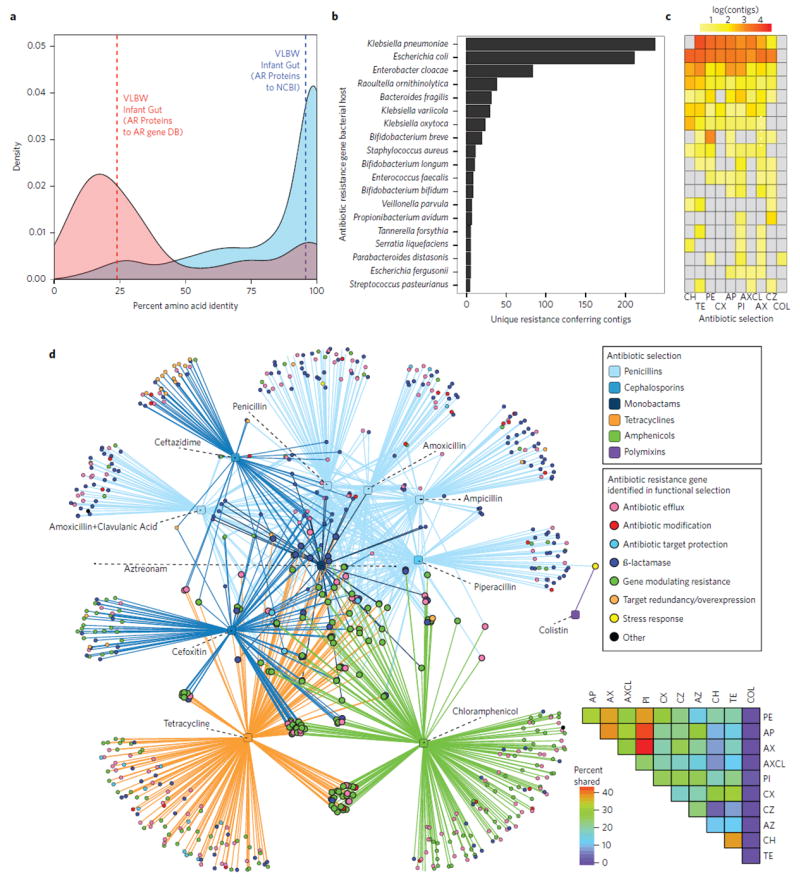
a, Amino acid identity between all functionally selected antibiotic resistance (AR) genes (identified using Resfams) from preterm infant gut resistomes and their top hit in the NCBI protein database (blue, n = 710). Functionally identified AR genes for preterm infant gut resistomes and top hit in AR databases (red, n = 707). Dashed lines represent mean amino acid identity for each group. While we identified known proteins, they have rarely been previously ascribed an AR function. b, The number of unique resistance-conferring fragments originating from MDR species identified from functional metagenomic selections. c, Heatmap representing the number of unique resistance-conferring fragments that originated from each bacterial species to each antibiotic (CH, chloramphenicol; TE, tetracycline; PE, penicillin; CX, cefoxitin; AP, ampicillin; PI, piperacillin; AXCL, amoxicillin-clavulanate; AX, amoxicillin; CZ, ceftazidime; COL, colistin). d, Co-selection of MDR proteins and protein clusters. Circles represent unique AR proteins coloured according to resistance mechanism, where circles representing AR proteins that result in resistance to more than one antibiotic class are twice as large as those representing all other AR proteins. Squares represent antibiotics that were included in functional metagenomic selections coloured on the basis of antibiotic class. Lines connect antibiotic selections to the resistance proteins that conferred resistance to that antibiotic. The heatmap in the lower right corner depicts the percentages of AR genes shared between antibiotic selections.
The three species (Escherichia coli, Enterobacter cloacae and Klebsiella pneumoniae) encoding the highest number of AR genes include organisms belonging to the ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella species, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) pathogen family28,29 and are widely distributed in NICU environments30 (Fig. 2b,c). Of the total unique AR-conferring fragments assembled from all functional selections, 42% are from one of these species, each of which is predicted to be resistant to over 50% of antibiotics tested (Fig. 2c). Of 794 functionally identified AR genes, 33% either conferred resistance to multiple antibiotics or were co-selected with another AR gene that encodes resistance to a different antibiotic (Fig. 2d). The most common co-selection was exhibited with piperacillin, which shared >50% of identified co-selected AR proteins or protein clusters with both ampicillin and amoxicillin (Fig. 2d), indicating broad-spectrum β-lactam resistance genotypes. 21% of functionally selected AR genes exhibit multidrug-resistance across multiple drug classes, including all β-lactams, amphenicols and tetracyclines (Fig. 2d).
To extend functional AR gene analysis to all shotgun-sequenced preterm infant gut microbiomes, we used ShortBRED to generate short unique markers for all AR gene families identified in functional selections and AR-specific gene databases31 (see Methods). Shotgun reads were mapped to the resulting AR-specific markers and normalized across samples to generate AR-gene profiles for all 401 infant gut metagenomes. We found a substantial number of genes encoding resistance to antibiotics that are not used in NICUs2, including chloramphenicol and tetracycline (Supplementary Fig. 4b). Hence, the preterm infant resistome established very early in life probably represents antibiotic selection of the colonizing bacteria by exposure in other habitats, rather than only by direct antibiotic selection in infants, just as we have proposed for the gut resistomes of healthy term infants20,21. The preterm infant gut resistome is also relatively stable: only 21% of AR gene classes changed significantly during the period of observation (P < 0.05, Supplementary Fig. 5).
We calculated the average change in relative abundance of each of the six high-prevalence/abundance species (Fig. 1a) from directly before to directly after administration of each treatment (Fig. 3a). For each such intervention in which we observed significantly reduced species richness (Fig. 1b), we find either significantly enriched (Staphylococcus epidermidis after meropenem-treated and K. pneumoniae after ticarcillin–clavulanate-treated individuals) or depleted (E. coli depleted in cefotaxime-treated individuals) species compared with these species in controls over similar intervals (Fig. 3a). In addition to species enrichment or depletion, we also interrogated the fraction of the resistome that is enriched following antibiotic treatment (Fig. 3b). We identified 50 unique AR genes that were enriched more than tenfold in the majority of treated individuals from directly before initiation to directly after termination of a specific agent. The fraction of the preterm gut-associated resistome that is highly enriched following specific antibiotic treatments is both antibiotic- and species-specific (Fig. 3b). The specific AR genes enriched following antibiotic treatment are largely limited to that specific agent, except for overlapping genes enriched during ticarcillin–clavulanate and ampicillin treatments. The extensive overlap in genes enriched following both gentamicin and vancomycin is expected as they are predominantly administered synchronously to preterm infants. Further, the collection of AR genes enriched following each specific antibiotic treatment is largely contributed by a single species. For example, in meropenem and ticarcillin–clavulanate treated individuals, most enriched AR genes are highly correlated (P < 0.001, Pearson Correlation) with either S. epidermidis or K. pneumoniae, which are also each significantly enriched following each respective treatment (Fig. 3, P < 0.05, Wilcoxon Rank Sum). For cefotaxime, we observe a significant depletion in the relative abundance of E. coli, resulting in enrichment of AR genes associated with E. cloacae (Fig. 3).
Figure 3. Species and antibiotic resistance gene enrichment following specific antibiotic treatments.
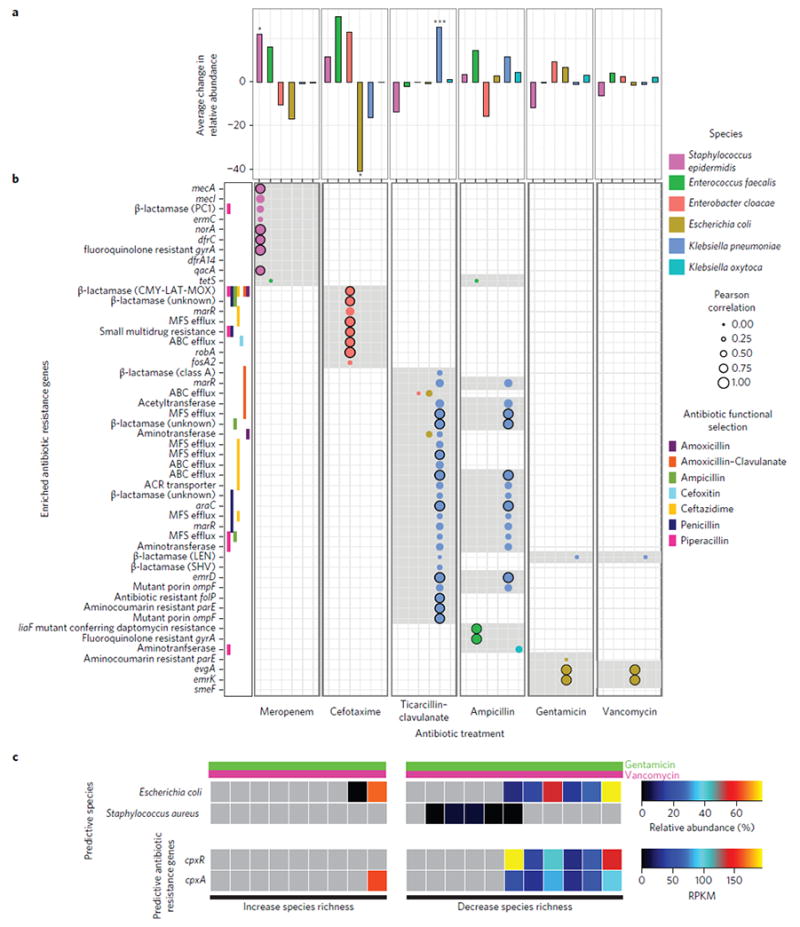
a, Average change in relative abundance of six high-abundance species from directly before to directly after treatment with six different antibiotics. (*P < 0.05, ***P < 0.001; Wilcoxon Rank Sum, Holm–Bonferronni correction for multiple hypotheses; significance tested for averages with absolute value greater than 20%). b, Grey shading represents antibiotic resistance (AR) genes enriched from directly before to directly after treatment at least tenfold in over half of the individuals treated with the specified antibiotic. Pearson correlation coefficient was calculated for all species by all AR genes. Circles represent Pearson correlation for the specified antibiotic by the six high-abundance species across all 401 preterm infant gut microbiota where correlation is greater than 0.2 (P < 0.001, Pearson Correlation, n = 401). Black-outlined circles denote instances where the Pearson correlation coefficient is greater than 0.8. AR genes considered include those identified in functional metagenomic selections. For AR genes identified by functional selection, each antibiotic selection for which the protein was identified is indicated directly to the left of the AR mechanism or class. c, Predictable response of preterm infant gut microbiota species richness to gentamicin and vancomycin combination treatment. Relative abundance of predictive species and AR genes directly before antibiotic treatment with a combination of gentamicin and vancomycin. RPKM, reads per kilobase per million reads.
xWhile many AR genes that are enriched after a specific antibiotic treatment correspond to the cognate function (e.g. β-lactamases enriched following β-lactam treatments), we also find collateral enrichment against a wide range of non-cognate antibiotics. For example, while we identify no AR genes that encode carbapenemases, treatment with meropenem is associated with increased relative abundance of AR genes providing resistance to fluoroquinolones, macrolides, tetracyclines, trimethoprim, and other β-lactams (Fig. 3b). We observed this type of collateral resistome change following every specific antibiotic treatment studied, probably resulting from a single organism encoding a diversity of AR genes. Supporting this finding, metagenome assembly revealed prevalent MDR clusters encoded by the preterm infant gut microbiota, predicted to be both plasmid-mediated and genomeencoded (Supplementary Fig. 6). Supplementary Fig. 6a represents a MDR cluster identified in 8% of samples and 11% of preterm infants, with high identity to Klebsiella oxytoca plasmid pKOX_R1, K. pneumoniae plasmid pKPN5, and Salmonella enterica plasmid pHCM1. Supplementary Figure 6b represents a MDR cluster genomically connected to a broad-spectrum β-lactam resistance gene identified in 8% of samples and 14% of preterm infants, with high identity to that of K. pneumoniae.
We demonstrated that a subset of antibiotics (meropenem, cefotaxime, and ticarcillin–clavulanate) each consistently are associated with reduced species richness (Fig. 1d). In contrast, co-therapy with vancomycin and gentamicin, one of the most common regimens in preterm infants, either substantially increases or decreases richness of different preterm infant gut species (Fig. 1c). We sought to determine if key features of the preterm infant gut microbiota and/or resistome before treatment might predictably mediate this large variance. Using random forests classification32, we predicted the species richness change following vancomycin and gentamicin treatment with only 15% out-of-bag error rate based on the relative abundance of only two species and two AR genes before treatment (Supplementary Fig. 7a,b). Based on this model, we hypothesize that this variance is mediated by two species: S. aureus, which is probably responding to vancomycin, and E. coli, which probably carries sequences encoding the two-component CpxR/CpxA protein misfolding stress response system, and is probably affected by gentamicin33. Across all preterm infant gut microbiomes, relative abundance of cpxR and cpxA is highly correlated with relative abundance of species of the genus Escherichia (P < 0.001; Pearson correlation coefficient: 0.93 and 0.74, respectively). Importantly, absence of cpxR in this two-component stress response system appears to abolish gentamicin resistance in E. coli33. Interestingly, in the single case in which our model incorrectly predicts species richness to decrease, we observe high abundance of E. coli and cpxA, but complete absence of the cpxR component (Fig. 3c).
In this study we extensively controlled for confounding factors by using individuals as their own controls in before- and after-treatment comparisons, and including many and diverse clinical covariates related to the health, diet, and treatment of infants and their mothers in our models (Supplementary Table 1a,b). However, even with these precautions, we cannot exclude the possibility that changes in the preterm infant gut microbiota are caused by factors that we can neither recognize nor quantify. For example, infants treated with ticarcillin–clavulanate and meropenem had been exposed to more discrete treatments with vancomycin and gentamicin than infants treated with other antibiotic therapies (Supplementary Fig. 8). However, cumulative previous antibiotic exposure of gentamicin and vancomycin did not significantly affect species richness of the preterm infant gut microbiota (P > 0.05). Nonetheless, this does not exclude the possibility that these antibiotics ‘prime’ the gut microbiota to respond differently to the next antibiotic treatment. A randomized control study of antibiotic treatment in preterm infant populations is not possible, but animal models to test hypotheses generated by this study might be informative. Our data represent an important first step in advancing and informing precision, evidence-based recommendations for use of antibiotics early in life in preterm infants so as to limit disruptions in gut microbiota development.
Methods
Sample collection
All samples were collected as part of preterm infant neonatal microbiome cohort project at Washington University School of Medicine and approved by the Human Research Protection Office (approval number 201205152). Samples were obtained from infants hospitalized in the Neonatal Intensive Care Unit (NICU) at Saint Louis Children’s Hospital. Infants were enrolled after parents provided informed consent. Details outlining the human subjects and clinical study protocols have been published previously3. Subjects and samples were selected to be included in our study to maximize ranges of both antibiotic exposure and of postmenstrual age. To achieve this, we stratified our selected cohort by antibiotic exposure, with a subset of individuals with early antibiotic exposure only (n = 33) with no antibiotic exposure outside the first week of life and a subset of individuals with early and subsequent antibiotic exposure (n = 51) with a range of antibiotic exposures reflective of total antibiotic exposure in the greater neonatal microbiome cohort (Table 1). All individuals included in our study were born premature (<33 weeks) and 82 individuals had very low birth weight (VLBW; <1,500 g). Two siblings of VLBW preterm infants were also included in our study with birth weights of 1,530 and 1,710 g. Samples were selected from subjects included in the early and subsequent antibiotic exposure subset with stool samples available within 48 h prior to antibiotic initiation as well as within 48 h after completion of antibiotic therapy. See Table 1 for more details on individual antibiotic exposure for the entire cohort analysed in this study, as well as the ‘Early Exposure Only’ and the ‘Early + Subsequent Exposure’ subsets. All stools produced were collected and stored as previously described3. In total, 401 samples collected longitudinally from 84 infants were shotgun sequenced and included in all metagenomic analysis.
Metagenomic DNA extraction
Total metagenomic DNA was extracted from approximately 0.1 g of preterm infant fecal samples using either the PowerMax Soil DNA Isolation Kit (MoBio Laboratories) following suggested protocols or using phenol/chlorophorm extraction methods following published protocols20 (extraction method for each sample is listed in Supplementary Table 1a). Extraction method did not significantly affect species composition or species richness and was explicitly controlled for in all data analysis (see Methods, Statistical modelling). DNA was quantified using a Qubit fluorometer BR assay kit (Life Technologies).
Comparison of term to preterm gut microbiota using 16S rRNA marker gene sequencing
We performed sequencing of the variable region (V4) of the 16S rRNA gene on a subset of 124 samples from 36 infants in preterm infant neonatal microbiome cohort (Supplementary Table 1e) in order to appropriately compare species richness and composition of the gut microbiota to age-matched term infants, which had previously been sequenced using similar methods (Supplementary Figure 1a,b). The 515F/806R PCR primers including Illumina flowcell adapter sequences were used to amplify the V4 region using the Earth Microbiome Protocols34 (described in more detail here: http://www.earthmicrobiome.org/emp-standard-protocols/16s/):
The following 25 μl reaction was prepared in a 96-well plate: 13 μl H20, 10 μl Five Primer Hot Master Mix (5Prime catalogue number 2200410), 0.5 μl forward primer (10 μM), 0.5 μl reverse primer mix (10 μM), 1.0 μl template DNA
PCR cycle temperatures were as follows: 94 °C for 3 min, then 35 cycles of [94 °C for 45 s, 50 °C for 60 s, 72 °C for 90 s], then 72 °C for 10 min.
PCR products were cleaned using the Agencourt AMPure XP PCR Purification kit (Beckman Coulter catalogue number A63880) and quantified using Picogreen (Invitrogen catalogue number #P11496), both following the manufacturers’ protocols. 16S rRNA gene amplicons were sequencing by 250 bp paired-end sequencing on the Illumina MiSeq platform using custom primers (read 1: 5′-TAT GGT AAT TGT GTG CCA GCM GCC GCG GTA A-3′; read 2: 5′-AGT CAG TCA GCC GGA CTA CHV GGG TWT CTA AT-3′; and index: 5′-ATT AGA WAC CCB DGT AGT CCG GCT GAC TGA CT-3′;) at a loading concentration of 8pM with 25% PhiX spike-in.
Sequencing data was de-multiplexed by sample and operational taxonomic units (OTUs) were generated following the UPARSE pipeline35 and USEARCH v7. Specifically, forward and reverse reads were merged (usearch –fastq_mergepairs – fastq_maxmergelen 258 –fastq_minmergelen 248 –fastq_truncqual 3 –fastq_maxdiffs 0), the merged reads were quality filtered (usearch –fastq_filter –fastq_maxee 0.5), de-replicated (usearch -derep_ fulllength –sizeout), sorted and singletons removed (usearch –sortbysize –minsize 2), clustered (usearch –cluster_otus), checked for chimeric sequences using the Gold database (usearch –uchime_ref –db gold.fa –strand plus –nonchimeras), OTUs were renamed (uparse/fasta_number.py), reads were mapped back to OTUs at 97% identity (usearch –usearch_global –strand both –id 0.97), and converted to the final OTU table (uparse/uc2otutab.py). Taxonomy was assigned using uclust against the Greengenes database (retrieved August 2013). All OTUs that were not identified by Greengenes were discarded from downstream analysis (in order to compare appropriately to closed reference OTU picking described below).
For comparison with age-matched term infants, previously generated 16S rRNA gene sequencing of human gut microbiota was downloaded from MG-RAST (Project 98)7. A total of 10 gut microbiota samples collected from term infants in the Saint Louis Neonatal Microbiome Initiative at 1 and 2 months of age were used in our comparison. OTUs were picked as using the QIIME pipeline36 and closed reference OTU picking (pick_closed_reference_otus.py).
Both OTU tables were individually subsampled to 2,750 reads per sample (single_rarefaction.py). After subsampling, each table was summarized at both the family and genus level (summarize_taxa.py). As OTUs were picked independently between groups, this summarization is required for comparison. Significance of bacterial family enrichment between term and preterm infant gut microbiota in the first two months of life was performed using non-parametric Student’s t-test with 9,999 permutations (group_significance.py).
Multiplexed shotgun metagenomic sequencing
Sequencing libraries were prepared from 500 ng total metagenomic DNA per sample. DNA was sheared to a target size range of approximately 500–600 bp using either the Covaris E210 sonicator with the following settings: intensity, 4; duty cycle, 10%; cycles per burst, 200; treatment time, 75 s; temperature, 4 °C; sample volume, 130 μl, or the Covaris E220 sonicator with the following settings: peak incident power, 140; duty cycle, 10%; cycles per burst, 200; treatment time, 75 s; temperature, 7 °C; sample volume, 130 μl. Sheared DNA was purified and concentrated using a MinElute PCR Purification Kit (Qiagen), eluting in 63 μl pre-warmed nuclease-free H2O. Purified sheared DNA was then end-repaired and Illumina adapters were ligated using the following protocol:
A 25 μl reaction volume was prepared (three reactions were performed reactions per sample in order to have a 3/1 ratio of barcode/sample) containing 20 μl of purified sheared DNA, 2.5 μl T4 DNA ligase buffer with 10 mM ATP (10×, New England BioLabs), 1 μl dNTPs (1 mM, New England BioLabs), 0.5 μl T4 polymerase (3 U μl−1, New England BioLabs), 0.5 μl T4 PNK (10 U μl−1, New England BioLabs), and 0.5 μl Taq Polymerase (5 U μl−1, New England BioLabs).
The reaction was incubated at 25 °C for 30 min followed by 20 min at 75 °C.
For the barcode mix forward and reverse sequencing adapters were stored in TES buffer (10 mM Tris, 1 mM EDTA, 50 mM NaCl, pH 8.0) and annealed by heating the 1 mM mixture to 95 °C followed by slow cooling (0.1 °C per second) to a final holding temperature of 4 °C.
A 2 μl volume of prepared barcode mix and 0.8 μl of T4 DNA ligase (New England BioLabs) were added to each end-repair reaction and the reaction was incubated at 16 °C for 40 min followed by 10 min at 65 °C.
Reactions were pooled (3–8 samples per pool) and purified using a MinEluted PCR Purification Kit (Qiagen), eluting in 16 μl pre-warmed elution buffer. The pooled, adaptor-ligated, sheared DNA was then size-selected to a target range of 400–900 bp on a 2% agarose gel in 0.5× Tris-Borate-EDTA (TBE), stained with GelGreen dye (Biotium) and extracted using a MinElute Gel Extraction Kit (Qiagen). The purified DNA was enriched using the following protocol:
A 25 μl reaction volume was prepared containing 2 μl of purified DNA, 12.5 μl 2× Phusion HF Master Mix (New England BioLabs), 1 μl of 10 mM Illumina PCR Primer Mix (5′-AAT GAT ACG GCG ACC ACC GAG ATC-3′ and 5′-CAAGCAGA A GAC GGC ATA CGA GAT-3′), and 9.5 μl of nuclease-free H2O.
The PCR cycle temperatures were as follows: 98 °C for 30 s, then 18 cycles of [98 °C for 10 s, 65 °C for 30 s, 72 °C for 30 s], then 72 °C for 5 min.
Amplified DNA was measured using the Qubit fuorometer HS assay kit (Life Technologies) and 10 nM of each sample were pooled for sequencing. Subsequently, samples were submitted for paired-end 150-bp sequencing using the Illumina HiSeq 2500 platform at GTAC (Genome Technology Access Center, Washington University in St Louis, USA). In total, 401 samples across 10 HiSeq lanes were sequenced to sufficient depth (1 million reads) for microbiome and resistome analyses. Technical replicates (two or three) of each fecal metagenomic DNA sample were individually barcoded and pooled for sequencing, and total reads per biological sample were used for analysis.
Prior to all downstream data analysis, Illumina paired-end shotgun metagenomic sequence reads were binned by barcode (exact match required), quality filtered using Trimmomatic v0.3037 (java -Xms1024m -Xmx1024m -jar trimmomatic-0.30.jar PE -phred33 ILLUMINACLIP: TruSeq3-PE.fa:2:30:10 LEADING:6 TRAILING:6 SLIDINGWINDOW:4:15 MINLEN:60), and human DNA was removed using DeconSeq38 using build 38 of the human genome using default parameters.
Calculating relative abundance of species from shotgun sequencing
Relative abundance of species was calculated from shotgun sequencing of samples using MetaPhlAn 2.039 downloaded from Bitbucket with repository tag 2.0.0. MetaPhlAn 2.0 was run with the following parameters: –bowtie2db metaphlan2/db_v20/mpa_v20_m200 –bt2_ps sensitive –input_type multifastq –mpa_pkl metaphlan2/db_v20/mpa_v20_m200.pkl g_viruses –ignore_eukaryotes –ignore_archea. Individual MetaPhlAn 2.0 relative abundance tables were merged using the metaphlan2/utils/merge_metaphlan_tables.py script. Species were classified as the major or dominant species in a sample if they displayed greater than 50% relative abundance (e.g. Figure 1a). The number of observed species in a sample was calculated from MetaPhlAn summary as the number of unique species-level bacterial organisms that were identified in a sample. For the subset of samples for which we also sequenced the V4 region of the 16S rRNA gene, we calculated the number of observed species and calculated a significant correlation (P < 0.0001, ANOVA) between these two methods.
Calculating relative abundance of antibiotic resistance genes from shotgun sequencing
Relative abundance of antibiotic resistance genes was calculated using ShortBRED31. A total of 6,731 proteins associated with antibiotic resistance were used as proteins of interest for identification of marker families using shortbred_identify.py with the following non-default parameters: –clustid 0.95. These proteins included all antibiotic resistance genes in the Comprehensive Antibiotic Resistance Database (CARD)27 (retrieved 2014 October 20), a curated list of β-lactamases from Lahey Clinic (http://www.lahey.org/Studies/), and antibiotic resistance proteins identified using functional metagenomic selections performed in the current study on preterm infant gut microbiota as well as from healthy infant microbiota21, adult gut microbiota22, and soil17,19. The reference protein database used for identification of antibiotic resistance specific markers included 24,874,711 proteins encoded in 6,629 microbial genomes downloaded from the Integrated Microbial Genomes (IMG) database40. A complete list of microbial proteomes included has been previously reported26. Of the 2,425 antibiotic resistance protein families created after clustering at 95% identity, 2,419 had at least one marker for quantification of antibiotic resistance from short reads. In order to calculate relative abundance of resistance genes using short markers, shortbred_quantify.py was used with the following non-default parameters: –avgreadBP 142.
Construction of metagenomic libraries from preterm infant gut samples for functional selection
Purified extracted total metagenomic DNA (11.7 ± 3.9 μg) was used as starting material for creation of metagenomic libraries. To create small-insert metagenomic libraries, DNA was sheared to a target size of 3,000 bp using the Covaris E210 sonicator following manufacturer’s recommended settings (http://covarisinc.com/wp-content/uploads/pn_400069.pdf). Sheared DNA was size-selected to a range of 1,000–5,000 bp by electrophoresis through a 1% low-melting-point agarose gel in 0.5× TBE buffer stained with GelGreen dye (Biotium). Size-selected fragments were gel extracted using a QIAquick Gel Extraction Kit (Qiagen), eluting in 30 μl of warm nuclease-free H20. Purified DNA was then end-repaired using the End-It DNA End Repair kit (Epicentre).
A 50 μl reaction volume was prepared containing 30 μl of purified DNA, 5 μl dNTP mix (2.5 mM), 5 μl 10× End-Repair buffer, 1 μl End-Repair Enzyme Mix and 4 μl nuclease-free H2O, mixed gently and incubated at room temperature for 45 min.
The reaction was heat-inactivated at 70 °C for 15 min.
End-repaired DNA was then purified using the QIAquick PCR purification kit (Qiagen) and quantified using the Qubit fluorometer BR assay kit (Life Technologies) and ligated into the pZE21-MCS-1 vector at the HincII site. The pZE21 vector was linearized at the HincII site using inverse PCR with PFX DNA polymerase (Life Technologies):
A 50 μl reaction volume was prepared containing 10 μl of 10× PFX reaction buffer, 1.5 μl of 10 mM dNTP mix (New England Biolabs), 1 μl of 50 mM MgSO4, 5 μl of PFX enhancer solution, 1 μl of 100 pg μl−1 21 circular pZE21, 0.4 μl PFX DNA polymerase, 0.75 μl forward primer (5′ GAC GGT ATC GAT AAG CTT GAT 3′), 0.75 μl reverse primer (5′ GAC CTC GAG GGG GGG 3′) and 29.6 μl of nuclease free H2O to a final volume of 50 μl. The PCR cycle temperatures were as follows: 95 °C for 5 min, then 35 cycles of [95 °C for 45 s, 55 °C for 45 s, 72 °C for 2.5 min], then 72 °C for 5 min.
Linearized pZE21 was size-selected (~2,200 bp) on a 1% low melting point agarose gel (0.5× TBE) stained with GelGreen dye (Biotium) and purified as described above. Pure vector was dephosphorylated using calf intestinal alkaline phosphatase (CIP, New England BioLabs) by adding one-tenth reaction volume of CIP, one-tenth reaction volume of New England BioLabs Buffer 3, and nuclease-free H2O to the vector eluate and incubating this at 37 °C overnight before heat inactivation from 15 min at 70 °C. End-repaired metagenomic DNA and linearized vector were ligated together using the Fast-Link Ligation Kit (Epicentre) at a 5/1 ratio of insert/vector using the following protocol:
A 15 μl reaction volume was prepared containing 1.5 μl 10× Fast-Link buffer, 0.75 μl ATP (10 mM), 1 μl FastLink DNA ligase (2 U μl−1), 5:1 ratio of metagenomic DNA to vector, and nuclease-free H2O to the final reaction volume, incubated at room temperature overnight and heat inactivated for 15 min at 70 °C.
After heat inactivation, ligation reactions were dialysed for 30 min using a 0.025 μm cellulose membrane (Millipore catalogue number VSWP09025) and the full reaction volume used for transformation by electroporation into 25 μl E. coli MegaX (Invitrogen) according to manufacturer’s recommended protocols (http://tools.invitrogen.com/content/sfs/manuals/megax_man.pdf). Cells were recovered in 1 ml Recovery Medium (Invitrogen) at 37 °C for one hour. Libraries were titered by plating out 0.1 and 0.01 μl of recovered cells onto Luria–Bertani (LB) agar (5 g yeast extract, 5 g NaCl, 10 g tryptone, 12 g agar in 1 liter of water) plates containing 50 μg ml−1 kanamycin. For each library, insert size distribution was estimated by gel electrophoresis of PCR products obtained by amplifying the insert from 12 randomly picked clones using primers flanking the HincII site of the multiple cloning site of the pZE21 MCS1 vector (which contains a selectable marker for kanamycin resistance). The average insert size across all libraries was determined to be 3,000 bp, and library size estimates were calculated by multiplying the average PCR-based insert size by the number of titered colony forming units (c.f.u.s) after transformation recovery. The rest of the recovered cells were inoculated into 50 ml of LB containing 50 μg kanamycin ml−1 and grown overnight. The overnight culture was frozen down with 15% glycerol and stored at −80 °C for subsequent screening.ml.
Functional selections for antibiotic resistance
For each preterm infant gut metagenomic library, selections for resistance to each of 16 antibiotics (at concentrations listed in Supplementary Table 2b plus 50 μg kanamycin ml−1 for plasmid library selection) was performed using Mueller-Hinton (MH) agar (2 g beef infusion solids, 1.5 g starch, 17 g agar, 17.5 g casein hydrolysate, pH 7.4, in a final volume of 1 l). It is of note that as our library host, E. coli, is intrinsically resistant to vancomycin, we are unable to functionally screen for this antibiotic. Furthermore, the use of kanamycin as the selective marker for the metagenomic plasmid library results in low-level cross-resistance with other aminoglycoside antibiotics, resulting in a higher required minimum inhibitory concentration for gentamicin. For each metagenomic library, the number of cells plated on each antibiotic selection represented ten times the number of unique c.f.u.s in the library, as determined by titers during library creation. Depending on the titer of live cells following library amplification and storage, the appropriate volume of freezer stocks were either diluted to 100 μl using MH broth + 50 μg kanamycin ml−1 or centrifuged and reconstituted in this volume for plating. After plating (using sterile glass beads), antibiotic selections were incubated at 37 °C for 18 h to allow the growth of clones containing an antibiotic resistant DNA insert. After overnight growth, all colonies from a single antibiotic plate (gut microbiota by antibiotic selection) were collected by adding 750 μl of 15% LB-glycerol to the plate and scraping with an L-shaped cell scraper to gently remove colonies from the agar. The liquid ‘plate scrape culture’ was then collected and this process was repeated a second time for a total volume of 1.5 ml to ensure that all colonies were removed from the plate. The bacterial cells were then stored at −80 °C before PCR amplification of antibiotic-resistant metagenomic fragments and Illumina library creation.
Amplification and sequencing of functionally selected fragments
Freezer stocks of antibiotic-resistant transformants were thawed and 300 μl of cells pelleted by centrifugation at 16,200g for 2 min and gently washed with 1 ml of nuclease-free H2O. Cells were subsequently pelleted a second time and re-suspended in 30 μl of nuclease-free H2O. Re-suspensions were then frozen at −20 °C for one hour and thawed to promote cell lysis. The thawed re-suspension was pelleted by centrifugation at 16,200g for 2 min and the resulting supernatant was used as template for amplification of resistance-conferring DNA fragments by PCR with Taq DNA polymerase (New England BioLabs):
A 25 μl reaction volume was prepared containing 2.5 μl of template, 2.5 μl of ThermoPol reaction buffer (New England BioLabs), 0.5 μl 10 mM deoxynucleotide triphosphates (dNTPs, New England Biolabs), 0.5 μl Taq polymerase (5 U μl−1), 3 μl of a custom primer mix, and 16 μl of nuclease-free H2O. The PCR cycle temperatures were as follows: 94 °C for 10 min, then 25 cycles of [94 °C for 45 s, 55 °C for 45 s, 72 °C for 5.5 min], then 72 °C for 10 min.
The custom primer mix consisted of three forward and three reverse primers, each targeting the sequence immediately flanking the HincII site in the pZE21 MCS1 vector, and staggered by one base pair. The staggered primer mix ensured diverse nucleotide composition during early Illumina sequencing cycles and contained the following primer volumes (from a 10 mM stock) in a single PCR reaction: (primer F1, CCGAATTCATTAAAGAGGAGAAAG, 0.5 μl); (primer F2, CGAATT CA TTAAAGAGGAGAAAGG, 0.5 μl); (primer F3, GAATTCATTAAAGAGGAGAAA GGTAC, 0.5 μl); (primer R1, GATATCAAGCTTATCGATACCGTC, 0.21 μl); (primer R2, CGATATCAAGCTTATCGATACCG, 0.43 μl); (primer R3, TCGATA TCAAGCTTATCGATACC, 0.86 μl). The amplified metagenomic inserts were then cleaned using the Qiagen QIAquick PCR purification kit and quantified using the Qubit fluorometer HS assay kit (Life Technologies).
For amplified metagenomic inserts from each antibiotic selection, elution buffer was added to PCR template for a final volume of 200 μl and sonicated in a half-skirted 96-well plate on a Covaris E210 sonicator with the following settings: duty cycle, 10%; intensity, 5; cycles per burst, 200; sonication time, 600 s. Following sonication, sheared DNA was purified and concentrated using the MinElute PCR Purification kit (Qiagen) and eluted in 20 μl of pre-warmed nuclease-free H2O.In the first step of library preparation, purified sheared DNA was end-repaired. A 25 μl reaction volume containing 20 μl of elute, 2.5 μl T4 DNA ligase buffer with 10 mM ATP (10×, New England BioLabs), 1 μl dNTPs (1 mM, New England BioLabs), 0.5 μl T4 polymerase (3 U μl−1, New England BioLabs), 0.5 μl T4 PNK (10 U μl−1, New England BioLabs), and 0.5 μl Taq Polymerase (5 U μl−1 l, New England BioLabs) was prepared and incubated at 25 °C for 30 min followed by 20 min at 75 °C.
Next, to each end-repaired sample, 5 μl of 1 μM pre-annealed, barcoded sequencing adapters were added (adapters were thawed on ice). Barcoded adapters consisted of a unique 7-bp oligonucleotide sequence specific to each antibiotic selection, facilitating the de-multiplexing of mixed-sample sequencing runs. Forward and reverse sequencing adapters were stored in TES buffer (10 mM Tris, 1 mM EDTA, 50 mM NaCl, pH 8.0) and annealed by heating the 1 μM mixture to 95 °C followed by slow cooling (0.1 °C per second) to a final holding temperature of 4 °C. After the addition of barcoded adapters, samples were incubated at 16 °C for 40 min and then for 10 min at 65 °C. Before size-selection, 10 μl volumes of each of the adapted-ligated samples were combined into pools of 12 and concentrated by elution through a MinElute PCR Purification Kit (Qiagen), eluting in 14 μl of elution buffer (10 mM Tris-Cl, pH 8.5). The pooled, adaptor-ligated, sheared DNA was then size-selected to a target range of 300–400 bp on a 2% agarose gel in 0.5× TBE, stained with GelGreen dye (Biotium) and extracted using a MinElute Gel Extraction Kit (Qiagen). The purified DNA was enriched using the following protocol. A 25 μl reaction volume was prepared containing 2 μl of purified DNA, 12.5 μl 2× Phusion HF Master Mix (New England BioLabs), 1 μl 10 mM Illumina PCR Primer Mix (5′-AAT GAT ACG GCG ACC ACC GAG ATC-3′ and 5′-CAAGCAGA A GAC GGC ATA CGA GAT-3′), and 9.5 μl of nuclease-free H2O. The PCR cycle temperatures were as follows: 98 °C for 30 s, then 18 cycles of [98 °C for 10 s, 65 °C for 30 s, 72 °C for 30 s], then 72 °C for 5 min.
Amplified DNA was measured using the Qubit fluorometer HS assay kit (Life Technologies) and 10 nM of each sample were pooled for sequencing. Subsequently, samples were submitted for paired-end 101-bp sequencing using the Illumina HiSeq 2,500 platform at GTAC. In total, three sequence runs were performed at 10 pM per lane.
Functional metagenomic assembly and annotation
Illumina paired-end sequence reads were binned by barcode (exact match required), such that independent selections were assembled and annotated in parallel. Assembly of the resistance-conferring DNA fragments from each selection was achieved using PARFuMS17 (Parallel Annotation and Re-assembly of Functional Metagenomic Selections), a tool developed specifically for the high-throughput assembly and annotation of functional metagenomic selections. Assembly with PARFuMS consists of: (1) three iterations of variable job size with the short-read assembler Velvet, (2) two iterations of assembly with Phrap, and (3) custom scripts to clean sequence reads, remove chimeric assemblies, and link contigs by coverage and common annotation, as previously described. Of the 336 antibiotic selections performed, 183 yielded antibiotic-resistant E. coli transformants (Supplementary Fig. 4) and from each selection we assembled fragments larger than 500 bp and annotated AR genes.
Open reading frames (ORFs) were predicted in assembled contigs using the gene finding algorithm MetaGeneMark41 and annotated by searching amino acid sequences against an AR gene specific profile hidden Markov model (pHMM) database, Resfams26 (http://www.dantaslab.org/resfams), with HMMER342. MetaGeneMark was run using default gene-finding parameters while hmmscan (HMMER3) was run with the option –cut_ga as implemented in the script annotate_ functional_selections.py. Proteins were classified as AR genes if they had a significant hit to a Resfams pHMM using profile-specific gathering thresholds. Of the 3,506 unique predicted AR genes, 784 (23%) were classified as ARGs.
Percentage identity of selected ORFs against NCBI and ARG-specific databases
Percentage identity of all ARGs were examined via a BlastP query against both the NCBI protein Non-Redundant (NR) database (retrieved 20 August 2013) or a combined database of all proteins from CARD27 and a curated list of β-lactamases from Lahey Clinic (http://www.lahey.org/Studies/) to identify the top local alignment. Once the top local alignment was identified using BlastP, it was the used for a global alignment using the EMBOSS v.6.3.1 implementation of the Needleman-Wunsch global alignment algorithm using the needle program with the following non-default parameters: -gapopen-10 –gapextend=0.5. This analysis was implemented with calculate_percent_identity_to_db.py.
Taxonomic classification of functionally selected fragments
All bacterial genomes (ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/all.fna.tar.gz) and bacterial plasmids (ftp://ftp.ncbi.nlm.nih.gov/genomes/Plasmids/plasmids.all.fna.tar.gz) were downloaded from NCBI (retrieved 26 November 2014). Blastn was used to align all contigs assembled from functional metagenomic selections to bacterial genomes and plasmids. Fragments were assigned to a bacterial genome or plasmid if they had greater than 95% nucleotide identity over 95% of the fragment.
Multidrug resistance in preterm infant gut microbiota
In order to identify AR genes which were selected on multiple antibiotics (as in Fig. 2d), all identified AR genes were clustered at 100% identity using cd-hit43 with the following non-default parameters: -aS 1.0 -g 1 -d 0 -bak 1. Protein clusters that included antibiotic-resistance proteins identified on multiple antibiotics were classified as MDR and could arise in one of two ways: (1) the protein itself could be bifunctional (across antibiotic classes) or have a broad-spectrum of activity (within antibiotic classes) or (2) the antibiotic resistance protein could be co-localized on the same 1–5 kb fragment with another protein that provides resistance to an independent antibiotic. Multidrug resistance was visualized in Fig. 2d using Cytoscape. Percentage sharing of antibiotic resistance genes across antibiotics (as in Fig. 2d inset) was calculated as ((2×number of shared AR genes between antibiotic 1 and antibiotic 2)/(number of AR genes selected on antibiotic 1×number of AR genes selected on antibiotic 2))×100.
Metagenome assembly
VelvetOptimiser was used to assemble metagenomes using quality filtered reads with all human reads removed (methods described above) with the following parameters: -t 6 –s 81 –e 131. A summary of all metagenome assemblies can be found in Supplementary Table 3.
Clinical metadata associated with subjects and samples
All clinical metadata associated with samples and subjects are listed in Supplementary Table 1a–c. Postmenstrual age (PostMenst_Age: reported in weeks) is calculated as gestational age (Gestational_Age: reported in weeks) plus day of life (DOL: reported in days). Birthweight is reported in grams. Experimental details include the number of raw 150 bp paired-end reads (Shotgun:NumSeq_Total) for each sample, total number of reads that passed quality filtering (Shotgun:NumSeq_QualityFiltered) and removal of human reads (Shotgun:NumSeq_HumanRemoved) as well as method of total metagenomic DNA extraction (Extraction_Method). Antibiotic treatments for which samples were collected directly before initiation and directly after termination of treatment are recorded in Antibiotic_Treatment and the clinical indication for antibiotic used is listed in Indication. The total number of cumulative days of intravenous antibiotic exposure prior to sample collection is listed under Infant Cumulative Antibiotics for nine antibiotics plus total antibiotic exposure (Total_abx). Total_abx does not sum across all nine antibiotics as some antibiotics were given in combination and therefore are not counted as independent days. Location and room-type (private or public) of infant in Saint Louis Children’s Hospital NICU are listed under Bed Space. Total cumulative days of enteral feeding prior to sample collection including maternal human milk, pasteurized donor milk and formula each with and without cows-milk-based fortification is listed under Diet. Total cumulative days of exposure prior to sample collection to other medications commonly used in the NICU is listed under Other Medications. Maternal intrapartum antibiotics indicate if the mother received antibiotics within 24 h before birth for 13 antibiotics, including just β-lactam or any antibiotic. Maternal health includes a diagnosis for chorioamnionitis, Group-B Streptococcal colonization status (if known), pre-eclampsia status, antenatal steroids within 24 h of birth, and duration of membrane rupture in hours prior to delivery. Infant infections are listed as days since last recorded infection for either body site or type of species.
Chorioamnionitis was an intrapartum clinical diagnosis by the attending obstetrician based on key clinical findings of fever, uterine tenderness, maternal tachycardia, fetal tachycardia and foul amniotic fluid44. We found the rate of chorioamnionitis to be 7% in our cohort compared to 9% in the general study population similar to other reported rates for clinical definitions45. When considering the effect of this covariate, it is important to note that the median day of life (DOL) for our samples is 30.3, well after the day of birth which could influence our results and chorioamnionitis diagnosis.
Statistical modelling of preterm infant gut microbiota species richness and AR gene abundance
In order to formally model the trends in preterm infant gut microbiota species richness and AR gene abundance, a generalized linear mixed model (GLMM) with a Poisson family was fit by maximum likelihood (Laplace Approximation) using the lme4 package in R. Individual preterm infant was defined as the random effect to control for longitudinal sampling. As the scales for predictor variables were diverse, all predictors were centered and scaled using the generic function, scale, in R with default parameters. Marginal and conditional R2 values were calculated using an implementation of Schielzeth and Nakagwa’s R2 for generalized linear mixed effects models in R.
Random forests classification of species richness response for vancomycin and gentamicin treated individuals
Random forests classification was used to predict the species richness response following treatment with vancomycin and gentamicin based on the species (based on MetaPhlAn 2.0 relative abundance) and AR gene composition (based on ShortBRED relative abundance) prior to treatment. The default parameters of the R implementation of the algorithm (R package ‘randomForest’) were used with the following non-default parameters: ntree=10,000, importance=T. The full model with all species and all AR genes known to provide resistance to gentamicin (no relevant vancomycin resistance was identified) was used to identify the most informative predictors and then predictors were removed in order to minimize both out-of-box error rate and the number of predictors.
Correlation of species and antibiotic resistance genes across all 401 preterm infant metagenomes
Pearson correlation coefficients reported in Fig. 3b were calculated for all 546 antibiotic resistance genes identified in the preterm infant gut resistome by all samples (n = 401) analysed using the corrcoef function from the Numpy package in Python. Only those with r > 0.2 are depicted in Fig. 3.
Determination of appropriate sequencing depth
In order to determine the appropriate sequencing depth necessary to fully characterize the composition and function of the preterm infant gut microbiota, we rarefied all samples and identified the sequencing depth for which we did not identify any new species using clade-specific markers in the majority of samples. First all quality-filtered samples with human reads removed were rarefied ten times using the random_subsample.py script from the QIIME package including all sampling depths included in the following range that were less than the total sequences in the sample of interest: 10,000; 100,000; 1,000,000; 2,000,000; 3,000,000; 5,000,000; 10,000,000 and 15,000,000. For each subsample replicated and depth, the species richness was calculated from MetaPhlAn 2.0 output and averaged across all replicates for a particular sample and sequencing depth. Using this analysis, we show that the average sequencing depth ± s.d. for all of our samples occurs well into the plateaued portion of the rarefaction curve (Supplementary Fig. 9a) and the majority of samples (77%) do not identify any additional species beyond the average sequencing depth (Supplementary Fig. 9b).
Supplementary Material
Acknowledgments
We thank A. Moore for initial study conception and clinical insight, C. Hall-Moore for assistance in access to cohort samples, M. Wallace for cultured strain characterization, and members of the Dantas lab for discussions of the results and analyses. We also thank families and clinical staff in the Saint Louis Children’s Hospital Neonatal Intensive Care Unit for their cooperation with study, and Laura Linneman and Julie Hoffmann for their efforts in enrolment and data accrual. This work is supported in part by awards to G.D. through the Children’s Discovery Institute (MD-II-2011-117 and 127), the March of Dimes Foundation (6-FY12-394), and the National Institute of General Medical Sciences of the National Institutes of Health (R01-GM099538). This work is also supported in part by an award to Washington University School of Medicine through a Clinical and Translational Science Award (CTSA) Grant (UL1 TR000448). This collection was also supported by the US National Institutes of Health Grants UH3AI083265 and P30DK052574 (Biobank Core), along with funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, and Foundation for the National Institutes of Health (made possible by support from the Gerber Foundation). M.K.G. is a Mr. and Mrs. Spencer T. Olin Fellow at Washington University and a National Science Foundation (NSF) graduate research fellow (DGE-1143954). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Author contributions
P.I.T. and B.B.W. assembled the cohort and developed protocols and infrastructure to obtain the biologics and clinical metadata from the cohort. M.K.G. and G.D. designed and conceived the study. M.K.G. performed 16S rRNA gene sequencing for comparison to term infants. B.W. prepared shotgun metagenomic sequencing libraries. S.A. created metagenomic libraries, performed functional selections, and prepared sequencing libraries. M.K.G. performed functional metagenomic data analysis, shotgun metagenomic data analysis, statistical modelling. M.K.G. wrote the manuscript with contributions from C.A. B., P.I.T., B.B.W., and G.D.
Supplementary information is available online.
Competing interests
The authors declare no competing financial interests.
References
- 1.Groer MW et al. Development of the preterm infant gut microbiome: a research priority. Microbiome. 2014;2:38. doi: 10.1186/2049-2618-2-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark RH, Bloom BT, Spitzer AR, Gerstmann DR. Reported medication use in the neonatal intensive care unit: data from a large national data set. Pediatrics. 2006;117:1979–1987. doi: 10.1542/peds.2005-1707. [DOI] [PubMed] [Google Scholar]
- 3.La Rosa PS, et al. Patterned progression of bacterial populations in the premature infant gut. Proc Natl Acad Sci USA. 2014;111:12522–12527. doi: 10.1073/pnas.1409497111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vangay P, Ward T, Gerber JS, Knights D. Antibiotics, pediatric dysbiosis, and disease. Cell Host Microbe. 2015;17:553–564. doi: 10.1016/j.chom.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenwood C, et al. Early empiric antibiotic use in preterm infants is associated with lower bacterial diversity and higher relative abundance of Enterobacter. J Pediatr. 2014;165:23–29. doi: 10.1016/j.jpeds.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arboleya S, et al. Intestinal microbiota development in preterm neonates and effect of perinatal antibiotics. J Pediatr. 2015;166:538–544. doi: 10.1016/j.jpeds.2014.09.041. [DOI] [PubMed] [Google Scholar]
- 7.Yatsunenko T, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azad MB, et al. Gut microbiota of healthy Canadian infants: profiles by mode of delivery and infant diet at 4 months. Can Med Assoc J. 2013;185:385–394. doi: 10.1503/cmaj.121189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gronlund MM, Lehtonen OP, Eerola E, Kero P. Fecal microflora in healthy infants born by different methods of delivery: permanent changes in intestinal flora after cesarean delivery. J Pediatr Gastroenterol Nutr. 1999;28:19–25. doi: 10.1097/00005176-199901000-00007. [DOI] [PubMed] [Google Scholar]
- 10.Backhed F, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17:690–703. doi: 10.1016/j.chom.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 11.Rosenthal VD, et al. International Nosocomial Infection Control Consortium (INICC) report, data summary of 36 countries, for 2004–2009. Am J Infect Control. 2012;40:396–407. doi: 10.1016/j.ajic.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 12.Richards MJ, Edwards JR, Culver DH, Gaynes RP. Nosocomial infections in pediatric intensive care units in the United States. National Nosocomial Infections Surveillance System. Pediatrics. 1999;103:e39. doi: 10.1542/peds.103.4.e39. [DOI] [PubMed] [Google Scholar]
- 13.Dicksved J, et al. Molecular analysis of the gut microbiota of identical twins with Crohn’s disease. ISME J. 2008;2:716–727. doi: 10.1038/ismej.2008.37. [DOI] [PubMed] [Google Scholar]
- 14.Abrahamsson TR et al. Low gut microbiota diversity in early infancy precedes asthma at school age. Clin Exp Allergy. 2014;44:842–850. doi: 10.1111/cea.12253. [DOI] [PubMed] [Google Scholar]
- 15.Fujimura KE, Slusher NA, Cabana MD, Lynch SV. Role of the gut microbiota in defining human health. Expert Rev Anti-infect Ther. 2010;8:435–454. doi: 10.1586/eri.10.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, et al. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 2009;3:944–954. doi: 10.1038/ismej.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forsberg KJ, et al. The shared antibiotic resistome of soil bacteria and human pathogens. Science. 2012;337:1107–1111. doi: 10.1126/science.1220761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pehrsson EC, Forsberg KJ, Gibson MK, Ahmadi S, Dantas G. Novel resistance functions uncovered using functional metagenomic investigations of resistance reservoirs. Front Microbiol. 2013;4:145. doi: 10.3389/fmicb.2013.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forsberg KJ, et al. Bacterial phylogeny structures soil resistomes across habitats. Nature. 2014;509:612–616. doi: 10.1038/nature13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore AM, et al. Gut resistome development in healthy twin pairs in the first year of life. Microbiome. 2015;3:27. doi: 10.1186/s40168-015-0090-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore AM, et al. Pediatric fecal microbiota harbor diverse and novel antibiotic resistance genes. PLoS ONE. 2013;8:e78822. doi: 10.1371/journal.pone.0078822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sommer MO, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science. 2009;325:1128–1131. doi: 10.1126/science.1176950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riesenfeld CS, Goodman RM, Handelsman J. Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. Environ Microbiol. 2004;6:981–989. doi: 10.1111/j.1462-2920.2004.00664.x. [DOI] [PubMed] [Google Scholar]
- 24.Handelsman J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev. 2004;68:669–685. doi: 10.1128/MMBR.68.4.669-685.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chai G, et al. Trends of outpatient prescription drug utilization in US children 2002–2010. Pediatrics. 2012;130:23–31. doi: 10.1542/peds.2011-2879. [DOI] [PubMed] [Google Scholar]
- 26.Gibson MK, Forsberg KJ, Dantas G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 2015;9:207–216. doi: 10.1038/ismej.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McArthur AG, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. 2013;57:3348–3357. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boucher HW, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 29.Peterson LR. Bad bugs, no drugs: no ESCAPE revisited. Clin Infect Dis. 2009;49:992–993. doi: 10.1086/605539. [DOI] [PubMed] [Google Scholar]
- 30.Brooks B, et al. Microbes in the neonatal intensive care unit resemble those found in the gut of premature infants. Microbiome. 2014;2:1. doi: 10.1186/2049-2618-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaminski J, et al. High-specificity targeted functional profiling in microbial communities with ShortBRED. PLoS Comput Biol. 2015;11:e1004557. doi: 10.1371/journal.pcbi.1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiener M, L A. Classification and Regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- 33.Mahoney TF, Silhavy TJ. The Cpx stress response confers resistance to some, but not all, bactericidal antibiotics. J Bacteriol. 2013;195:1869–1874. doi: 10.1128/JB.02197-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caporaso JG, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 36.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmieder R, Edwards R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE. 2011;6:e17288. doi: 10.1371/journal.pone.0017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Segata N, et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nature Methods. 2012;9:811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markowitz VM, et al. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 2012;40:D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010;38:e132. doi: 10.1093/nar/gkq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 44.Tita AT, Andrews WW. Diagnosis and management of clinical chorioamnionitis. Clin Perinatol. 2010;37:339–354. doi: 10.1016/j.clp.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Avila C, et al. Usefulness of two clinical chorioamnionitis definitions in predicting neonatal infectious outcomes: a systematic review. Am J Perinatol. 2015;32:1001–1009. doi: 10.1055/s-0035-1547325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.