

Abstract
Background
The influence of reserve variables and Alzheimer’s disease (AD) biomarkers on cognitive test performance has been fairly well-characterized. However, less is known about the influence of these factors on “non-cognitive” outcomes, including functional abilities and mood.
Objective
We examined whether cognitive and brain reserve variables mediate how AD biomarker levels in cognitively normal persons predict future changes in function, mood, and neuropsychiatric behavior.
Methods
Non-cognitive outcomes were examined in 328 individuals 50 years and older enrolled in ongoing studies of aging and dementia at the Knight Alzheimer Disease Research Center (ADRC). All participants were cognitively normal at baseline (Clinical Dementia Rating [CDR] 0), completed cerebrospinal fluid (CSF) and structural neuroimaging studies within one year of baseline, and were followed for an average of 4.6 annual visits. Linear mixed effects models explored how cognitive reserve and brain reserve variables mediate the relationships between AD biomarker levels and changes in function, mood, and neuropsychiatric behavior in cognitively normal participants.
Results
Education levels did not have a significant effect on predicting non-cognitive decline. However, participants with smaller brain volumes exhibited the worst outcomes on measures of mood, functional abilities, and behavioral disturbance. This effect was most pronounced in individuals who also had abnormal CSF biomarkers.
Conclusions
The findings suggest that brain reserve plays a stronger, or earlier, role than cognitive reserve in protecting against non-cognitive impairment in AD.
Keywords: Alzheimer’s disease, biomarkers, cognitive reserve, dementia
INTRODUCTION
Evidence suggests that the pathophysiological process of AD begins years, possibly decades, before symptoms emerge and a diagnosis of clinical dementia is made [1–4]. Thus a “preclinical” or “asymptomatic” stage of AD exists, where underlying pathology is present in the brain but symptoms have yet to be expressed [5]. The goal of this study was to use biomarkers and variables representing cognitive and brain reserve in the preclinical stage to predict future “non-cognitive” changes. Importantly, the preclinical stage offers a strategic and underutilized opportunity for therapeutic intervention [5]. Such interventions would aim to combat progression of the underlying disease process to slow or halt conversion from asymptomatic to symptomatic AD.
The concept of reserve arose when studies noted a disjunction between the presence of AD pathology in brains examined at autopsy and its clinical expression during life. One of the earliest findings in this area noted advanced AD pathology in the brains of older adults who were clearly cognitively normal during life, particularly those with larger brains [6]. This suggested that individuals with more and larger neurons could harbor advanced AD pathology while remaining cognitively normal. The idea of a buffer or “reserve” between pathology and its clinical expression was hypothesized to explain this disjunction—individuals with greater reserve can withstand more pathology without becoming cognitively impaired than individuals with less reserve. Stern and colleagues proposed two models of reserve, brain reserve and cognitive reserve, to explain how the brain copes with neuropathology [7]. The brain reserve hypothesis is a passive model whereby differences in innate anatomic features, such as larger brain size, allow certain individuals to withstand more damage from AD pathology before dementiarelated cognitive impairment emerges [7, 8]. Under this model, a fixed threshold exists between clinical normality and impairment. Anatomical advantages may represent a physical buffer that must be depleted before the critical threshold for clinical expression is reached. Brain volume is commonly used to assess brain reserve [7]. In contrast to brain reserve, the cognitive reserve hypothesis proposes an active model whereby high reserve individuals actively cope with neuropathology. Cognitive reserve concerns the efficiency of the handling of neural resources; for example, high cognitive reserve individuals may employ alternate cognitive processing paradigms when the primary paradigm becomes impaired. This may take the form of the use of compensatory brain networks [7]. Cognitive reserve is influenced by certain environmental exposures, such as years of education. Other measures, including participation in cognitively stimulating leisure activities, are also believed to reflect cognitive reserve [9].
Advances in AD research have identified biomarkers that allow for the in vivo assessment of AD pathology [5, 10]. These biomarkers can be used to detect evidence of AD pathology in the preclinical stage of the disorder. Cerebrospinal fluid (CSF) biomarkers are ascertained via lumbar puncture [5, 11–15]. Commonly used CSF biomarkers include levels of soluble Aβ (Aβ1–42) as well as tau and phosphorylated tau (ptau181). Additionally, the ratios of tau/Aβ1–42 and ptau181/Aβ1–42 are commonly studied [16]. All of the aforementioned CSF biomarkers have been demonstrated to predict progression to symptomatic AD [16, 17] and are used in this study.
It has also been demonstrated that the inclusion of reserve variables in predictive biomarker models increases the accuracy of predicting future cognitive impairment [17]. The ability to predict future impairment years in advance is of particular interest to medical professionals and healthcare providers, as it would quantify the rate of progression to symptomatic AD and may help in identifying the optimal time of treatment administration.
“Non-cognitive” outcomes of AD, although often overlooked, are important features of the disease [18]. Many studies address AD in the scope of cognitive deficits, but the impact of the disease on functional abilities and other non-cognitive disease features that are important to daily living is addressed much less frequently. Important non-cognitive outcomes include the ability to perform instrumental activities of daily living (IADLs) and mood dysregulation such as anxiety and depression. Although we refer to these non-traditional outcomes as non-cognitive outcomes, we understand that they are not entirely exclusive from cognitive changes, since function is always to some extent dependent on cognition [19, 20]. Researchers are just beginning to examine the relationship between AD biomarkers and non-cognitive outcomes; one longitudinal study found that CSF biomarkers of Aβ1–42, tau, and ptau181 predicted future functional decline among participants who were cognitively normal at baseline [21].
It is unknown how cognitive and brain reserve mediate associations between AD biomarkers and non-cognitive outcomes of the disease. Longitudinal data available in large-scale repositories is supportive of such studies. These repositories allow detailed examination of a large sample of individuals followed over a period of several years. The Washington University Knight ADRC is well-positioned to examine these associations.
This study used longitudinal data from participants enrolled in ongoing studies of normal aging and dementia at the Knight ADRC. Linear mixed effects models explored how cognitive reserve and brain reserve variables mediate the relationships between AD biomarker levels and changes with time in function, mood, and neuropsychiatric behavior in cognitively normal participants. To our knowledge, this study is the first of its kind to examine these temporal outcomes in this manner.
MATERIALS AND METHODS
Participants
Data from participants enrolled in longitudinal studies at the Knight ADRC were used in this study. Specifically, participants were a subset of the participants enrolled in the Knight ADRC whose data were used for a 2013 study by Roe et al. [22], who had completed CSF and structural neuroimaging studies. The Knight ADRC recruits participants for yearly assessments from the greater St. Louis area through word-of-mouth, advertisements, and community events. The recruitment process has been previously described in detail [23]. Unless individuals have health conditions that may interfere with longitudinal follow-up, they are not excluded from participation [24].
Standard protocol approvals, registrations, and patient consents
Study protocols were approved by the Washington University Human Research Protection Office, and written informed consent was obtained for all participants.
Clinical assessments
Accompanied by a collateral source, participants complete a general physical and neurologic examination, psychometric testing, and also provide their health and medication histories at yearly follow-up visits. Experienced clinicians who are blind to each participant’s CSF and neuroimaging results as well as current and prior clinical and psychometric assessments complete a clinical assessment, the results of which yield a Clinical Dementia Rating [25, 26] (CDR). The CDR is derived from the synthesis of information obtained from the neurologic examination and interviews with the participant and separately with the collateral source. The clinician’s judgment about the presence of dementia is based on the principle of intraindividual change where the individual is used as his or her own control. The CDR is derived in accordance with a standard scoring algorithm: 0= no dementia, 0.5 = very mild dementia, 1.5= mild dementia, 2.5= moderate dementia, and 3 = severe dementia. A CDR ≥0.5 indicates clinically significant cognitive impairment [26]. Participants also complete a Mini-Mental State Examination (MMSE) [27] at each visit. Additionally, several tests assess non-cognitive impairment associated with AD.
Non-cognitive outcomes
Functional Activities Questionnaire (FAQ)
The FAQ assesses the ability to perform IADLs [28]. The FAQ asks whether in the last 4 weeks the patient had difficulty or needed help with tasks such as handling financial and business affairs, participating in leisure activities, preparing meals, keeping track of current events, and managing other daily personal affairs. Higher scores indicate greater functional impairment [29].
Neuropsychiatric Inventory Questionnaire (NPI-Q)
The NPI-Q assesses neuropsychiatric symptoms and psychopathology in patients with AD or similar neurodegenerative diseases [30]. It addresses the frequency and severity of several behavioral and psychological symptoms including delusions, hallucinations, aggression, and anxiety. Higher scores indicate greater frequency and severity [31].
Geriatric Depression Scale (GDS)
The GDS is used to identify symptoms of depression specific to older adult populations. Higher scores on the GDS indicate greater depressive symptomatology [32].
Structural MRI
MR acquisition and image processing
All participants underwent baseline magnetic resonance imaging (MRI). Image processing steps have been described in detail in previous publications [33–35] and included averaging across scans, inhomogeneity correction, and inter- and intra-scan motion correction. Intracranial volumes (ICV) were derived using the Atlas Scaling Factor (ASF) method described in detail by Buckner et al. [33]
Regional volumetry
Regional volume estimates are obtained using the FreeSurfer image analysis suite [36–38]. Each voxel in an MR image is assigned a neuroanatomical label based on probabilistic information from a manually labeled training set. This technique generates volumes with a high correspondence to manually generated volumes [38]. The total brain volume estimate was taken as the sum of white and gray matter voxels. Primary analyses did not include the cerebellum in the total brain volume calculation. Whole brain volumes were normalized for differences in ICV using linear regression prior to completion of the main analyses [33].
CSF measurement
Trained neurologists performed lumbar punctures at 8:00 AM following an overnight fast to draw 20–30 mL of CSF from participants using a 22-gauge Sprotte spinal needle. Samples were then gently inverted and centrifuged at low speed to avoid possible gradient effects and frozen at −84°C after aliquotion into propylene tubes [39]. Samples were analyzed using enzyme-linked immunosorbant assay (INNOTEST; Fujirebio, formerly Innogenetics, Ghent, Belgium) to ascertain Aβ1–42, tau, and ptau181 levels. CSF abnormality is indicated by high tau, ptau181, tau/Aβ1–42, and ptau181/Aβ1–42 values, whereas low values of Aβ1–42 are abnormal [10].
Inclusion criteria
Data from participants who (1) donated CSF within one year of a clinical assessment indicating that the participant had normal cognition (CDR 0), (2) had at least one additional assessment following the baseline index assessment, and (3) were age 50 years or older at the time of the index assessment were included in analyses.
Statistical analyses
Analyses examined whether each of the CSF biomarkers in conjunction with reserve variables predicted decline in selected non-cognitive outcomes over time using linear mixed models, which included the intercept and slope terms as random effects. Due to the large number of comparisons, p-values less than 0.01 were considered significant.
Dichotomization of education
Two education groups were defined and each participant was categorized as (1) low education (less than or equal to 16 years; undergraduate studies or less) or (2) high education (greater than 16 years; postgraduate studies).
Dichotomization of intracranial volume and normalized brain volume
Each participant was categorized into one of two groups (high or low) for ICV and total brain volume (normalized by ICV) using a median split.
Dichotomization of CSF biomarker values
Each participant was categorized into one of two groups (normal or abnormal) for each CSF biomarker value based on previously used cutoffs: 500 pg/mL for Aβ1–42, 440 pg/mL for tau, 78 pg/mL for ptau181, 0.94 for tau/Aβ1–42, and 0.15 for ptau181/Aβ1–42 [40].
Creation of multilevel variables
Several four-level variables were then created using the dichotomized variables in order to examine the combined effects of variables of interest. These combinations included (1) each of the biomarkers (normal or abnormal) combined with education (high or low) and (2) each of the biomarkers (normal or abnormal) combined with total brain volume (high or low) or total intracranial volume (high or low).
Linear mixed models
For each of the combination variables created, linear mixed models were used to examine whether there were differences in the slopes of FAQ scores, NPI-Q scores, and GDS scores across follow-up as a function of the combination variables. Models included fixed effects for baseline age, gender, time on study (in years from baseline) and each reserve variable of interest and its interaction with time on study. Random terms were included for participant and time on study. Analyses were conducted in R 3.0.1 using the lme4 package [41].
RESULTS
Demographics
Table 1 provides demographic data for the (n = 328) participants meeting inclusion criteria. Fifty-eight of the participants (17.7%) were no longer classified as cognitively normal by the end of their follow-up. Follow up times ranged from 0.9 years to 14.5 years. Participants had a mean of 4.6 (SD = 2.3) follow-ups each with a total of 1506 person-visits. Abnormal biomarker values were observed in n = 108 (32.9%) individuals for Aβ1–42, n = 51 (15.6%) individuals for tau, n = 52 (15.9%) individuals for ptau181, n = 50 (15.2%) individuals for tau/Aβ1–42, and n = 61 (18.6%) individuals for ptau181/Aβ1–42. The low education group comprised 64.6% (n = 212) of participants, while the high education group comprised 35.4% (n= 116).
Table 1.
Demographics (n = 328)
| Mean (SD) | |
|---|---|
| Age at first visit, y | 68.0 (8.2) |
| Women, N (%) | 206 (62.8) |
| Minority race, N (%) | 20 (6.1) |
| Baseline MMSE | 29.15 (1.1) |
| Baseline CDR-SB | 0.024 (0.11) |
| Progressed to CDR >0, N (%) | 58 (17.7) |
| Education, y | 15.70 (4.9) |
| Time between lumbar puncture and baseline clinical assessment, y | 0.28 (0.14) |
| Time between MRI and baseline clinical assessment, y | 0.84 (1.4) |
| Follow-up visits, N | 4.60 (2.3) |
| Follow-up time, y | 4.90 (2.5) |
MMSE, Mini-Mental State Examination; CDR, Clinical Dementia Rating; CDR-SB, Clinical Dementia Rating Sum of Boxes; MRI, magnetic resonance imaging.
CSF biomarkers and education (Supplementary Table 1; Fig. 1)
Fig. 1.
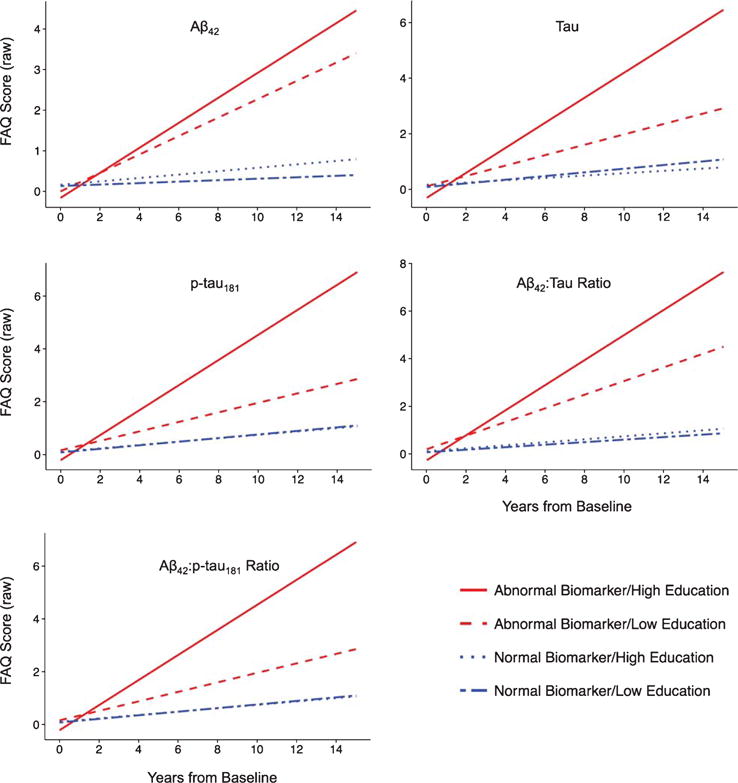
Functional Abilities Questionnaire (FAQ) predicted values adjusted for age and gender and stratified by levels of cerebrospinal fluid biomarkers and education are shown plotted over years from baseline biomarker assessment. Higher scores indicate worse outcomes. Participants with abnormal biomarker levels had the worst outcomes compared to participants with normal biomarker levels (all p’s < 0.05). Plots were similar for other non-cognitive outcomes including the Geriatric Depression Scale (GDS) and the Neuropsychiatric Inventory Questionnaire (NPI-Q).
Abnormal levels of each biomarker were related to higher NPI-Q, GDS, and FAQ scores (poorer outcome) with time. The abnormal biomarker groups tended to show significantly steeper slopes than the normal biomarker groups when biomarker status was combined with education. However, the slopes of the abnormal biomarkers/high education groups and abnormal biomarkers/low education groups did not significantly differ, nor did the slopes between each of the normal biomarkers/high education groups and normal biomarkers/low education groups. This indicates no significant multiplicative effect of education.
CSF biomarkers and brain volume (Supplementary Table 2; Fig. 2)
Fig. 2.
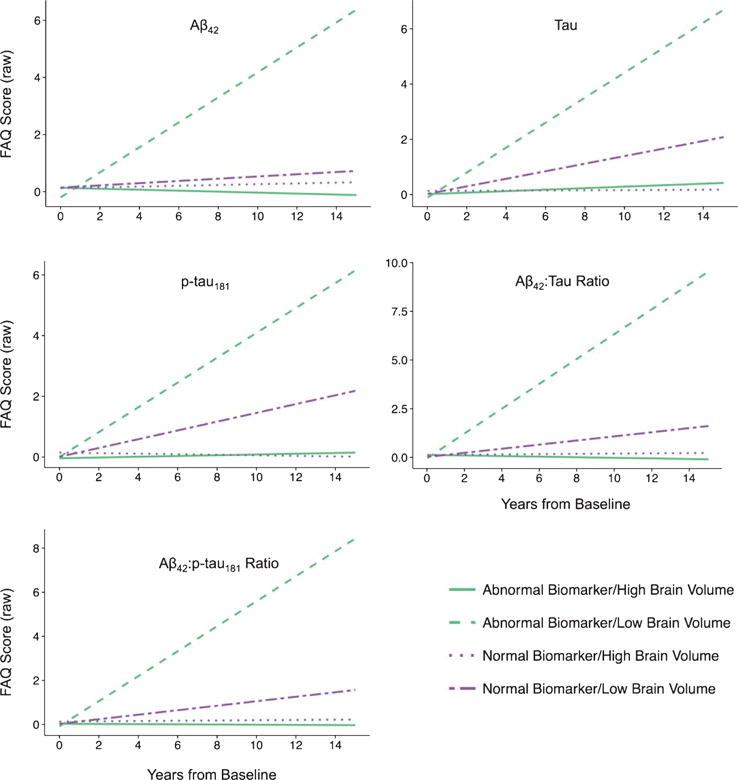
Functional Abilities Questionnaire (FAQ) predicted values adjusted for age and gender and stratified by levels of cerebrospinal fluid biomarkers and brain volume are shown plotted over years from baseline biomarker assessment. Higher scores indicate worse outcomes. Participants with abnormal biomarker levels and low brain volumes had the worst outcomes, compared to all other participants (all p’s < 0.05). Plots were similar for other non-cognitive outcomes including the Geriatric Depression Scale (GDS) and the Neuropsychiatric Inventory Questionnaire (NPI-Q).
Values of each biomarker were associated with change across time in NPI-Q, GDS, and FAQ scores such that abnormal levels of each biomarker and low total brain volume were related to higher scores (poorer outcome) with time. The abnormal biomarkers/low brain volume groups exhibited the worst outcomes. The slopes between each of the abnormal biomarkers/high brain volume groups and abnormal biomarkers/low brain volume groups significantly differed from each other. However, there were no differences in slopes between groups with normal biomarker levels. From most steep decline to least steep decline, the groups ordered: (1) abnormal biomarkers/low brain volume, (2) normal biomarkers/low brain volume, (3) abnormal biomarkers/high brain volume, (4) normal biomarkers/high brain volume.
Our measure of total brain volume did not include the cerebellum. As a sensitivity analysis, models were repeated using the total brain volume estimate including the cerebellum. The results were similar to the models that did not include the cerebellum.
CSF biomarkers and intracranial volume (Supplementary Table 3)
Participants with larger intracranial volumes and abnormal biomarker values had significantly increasing functional impairment on FAQ, neuropsychiatric symptoms on NPI-Q, and depressive symptoms on GDS. Participants with abnormal CSF biomarkers and smaller intracranial volumes also showed increases in FAQ, NPI-Q, and GDS relative to those with normal biomarkers. However, CSF biomarker ratio scores showed little difference in slopes between smaller and larger intracranial volumes suggesting that abnormal CSF biomarkers primarily drove associations in these models, whereas levels of maximal brain growth, as indicated by intracranial volume, had no significant moderating effect on non-cognitive outcomes.
DISCUSSION
The results show that non-cognitive decline was linked to baseline biomarker levels and brain volume. Abnormal biomarker levels and low brain volumes were associated with more difficulties with IADLs and worsening in neuropsychiatric and mood symptoms. Though education or maximal brain growth, as measured by intracranial volumes, did not strongly affect the predictive abilities of CSF biomarkers, brain volume exhibited a pronounced effect when combined with biomarker status to predict changes in function, mood, and behavior.
We have previously explored whether CSF Aβ1–42, tau, ptau181, tau/Aβ1–42, and ptau181/Aβ1–42 predict future decline in the same non-cognitive outcomes we explored [22]. They found that abnormal levels of each biomarker were related to greater impairment over time in all non-cognitive outcomes tested. Our results extend these findings by suggesting that normalized whole brain volume, an easily-acquired individual anatomical characteristic, has a strong mediational effect on this relationship. It is relevant to note that our whole brain volume measure additionally may reflect, at least in part, the AD pathological process through neuronal injury or death.
The ability of biomarkers and reserve variables to predict the onset of recognizable cognitive impairment has been studied [17]. Higher levels of education and greater brain volume seem to delay or slow the onset of cognitive symptomatology associated with AD pathology. According to the reserve theory, higher levels of education and cognitive stimulation may provide resistance to dementia symptoms through promoting the use of alternate or compensatory cognitive processing paradigms in the presence of brain damage [7]. Likewise, larger brain size may confer a greater amount of neuronal resources available for use under pathological insult that would allow for those with larger brain to cope with AD pathology for longer times [7, 8]. Both coping mechanisms, through numerous studies, have been shown to stave off the manifestation of cognitive impairment in AD. The fact that our results did not show a significant effect of education may indicate that although education builds cognitive reserve, it does not protect against non-cognitive impairment. Similarly, we did not find that levels of maximal brain growth had protective effects on non-cognitive outcomes. Thus, cognitive reserve may be specific to cognitive outcomes of AD (e.g., memory, orientation, judgment, executive function). Brain reserve, as quantified by brain volume, was shown to be protective against non-cognitive decline, suggesting that the same anatomical advantages that provide a buffer against cognitive impairment also attenuate non-cognitive impairment.
Growing evidence suggests that the presence of positive AD biomarkers in clinically normal individuals is reflective of preclinical AD and may signal imminent dementia. As noted, approximately 18% of participants developed cognitive problems in our follow-up period. Future research should explore whether the rate of decline in non-cognitive outcomes is linear across the course of AD, from the preclinical stage through the symptomatic stages.
Current efforts are working toward the development of therapies to slow or halt the disease process. It is believed that once these therapies exist, they will likely be most effective if used during the preclinical stage, before symptoms have emerged. It is imperative that we understand the temporal relationship between abnormal biomarker values, the onset of cognitive impairment, and characteristics that affect that relationship to pinpoint the most effective time for treatment. The findings of this study contribute to this cause, elucidating the factors that affect the relationship between biomarkers and future non-cognitive impairment. Recently, Razlighi et al. developed an algorithm that uses data gathered from a single patient visit to predict time from AD onset to disease endpoints including death, institutionalization, and need for full-time care [42]. Our study contributes to the growing body of literature in this field that may lead to the development of individualized risk profiles that can be used by physicians to predict the time course to the emergence of AD symptoms, facilitating the design and use of patient-specific treatment strategies.
There are some important limitations to consider when interpreting these results. Due to the historical legacy of our dataset, our measures of functional ability, neuropsychiatric symptoms, and mood may not be as sensitive or comprehensive as other measures. Also, due to the exploratory nature of these analyses, we did not examine how specific features of non-cognitive outcomes were associated with CSF biomarkers and brain volumes.
In summary, our results indicate that future changes in function, behavior, and mood are linked to baseline biomarker values and total brain volume. Abnormal biomarkers and low brain volume were associated with greater and more rapid non-cognitive impairment as measured by FAQ, NPI-Q, and GDS scores. The lack of an effect of education suggests that cognitive reserve does not serve as buffer against non-cognitive changes with time in AD, at least at its earliest stages. The rather strong effect of brain volume on the other hand suggests that brain reserve acts as a mediating force between increasing AD pathology and non-cognitive decline over time.
Supplementary Material
Acknowledgments
Funding for this study was provided by the Longer Life Foundation; the National Institute on Aging [P50 AG005681, P01 AG003991, and P01 AG026276]; the National Institute of Neurological Disorders and Stroke [P30 NS057105]; the Washington University Institute of Clinical and Translational Sciences grant UL1 TR000448 from the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH); the Neuroimaging Informatics and Analysis Center, supported by NIH grant 5P30NS048056; Fred Simmons and Olga Mohan, the Farrell Family Research Fund, and the Charles and Joanne Knight Alzheimer’s Research Initiative of the Washington University Knight Alzheimer’s Disease Research Center (ADRC). The authors thank the participants, investigators, and staff of the Knight ADRC Clinical (participant assessments) and Genetics Cores (genotyping), the investigators and staff of the Biomarker Core (P01 AG026276) for CSF analytes, and the investigators and staff of the Imaging Core (P01 AG003991) for MRI acquisition and processing. Additionally, Mr. Ingber thanks Mr. David Keith, Mrs. Stephanie Greenwald, and Mr. Ken Kaplan of the Dr. Robert Pavlica Authentic Science Research Program at Byram Hills High School.
Footnotes
Authors’ disclosures available online (http://jalz.com/manuscriptdisclosures/150478r1).
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD150478.
Handling Associate Editor: Natalie Marchant
References
- 1.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC, Network DIA. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morris JC. Early-stage and preclinical Alzheimer disease. Alzheimer Dis Assoc Disord. 2005;19:163–165. doi: 10.1097/01.wad.0000184005.22611.cc. [DOI] [PubMed] [Google Scholar]
- 3.Benzinger TLS, Blazey T, Jack CR, Koeppe RA, Su Y, Xiong C, Raichle ME, Snyder AZ, Ances BM, Bateman RJ, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc Natl Acad Sci U S A. 2013;110:E4502–E4509. doi: 10.1073/pnas.1317918110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fagan AM, Xiong C, Jasielec MS, Bateman RJ, Goate AM, Benzinger TLS, Ghetti B, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Salloway S, Schofield PR, Sperling RA, Marcus D, Cairns NJ, Buckles VD, Ladenson JH, Morris JC, Holtzman DM, Dominantly Inherited Alzheimer Network Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer’s disease. Sci Transl Med. 2014;6:226ra30. doi: 10.1126/scitranslmed.3007901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katzman R, Terry R, DeTeresa R, Brown T, Davies P, Fuld P, Renbing X, Peck A. Clinical, pathological, and neurochemical changes in dementia: A subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol. 1988;23:138–144. doi: 10.1002/ana.410230206. [DOI] [PubMed] [Google Scholar]
- 7.Stern Y. What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc. 2002;8:448–460. [PubMed] [Google Scholar]
- 8.Mortimer JA. Brain reserve and the clinical expression of Alzheimer’s disease. Geriatrics. 1997;52(Suppl 2):S50–S53. [PubMed] [Google Scholar]
- 9.Scarmeas N, Zarahn E, Anderson KE, Habeck CG, Hilton J, Flynn J, Marder KS, Bell KL, Sackeim HA, Van Heertum RL, Moeller JR, Stern Y. Association of life activities with cerebral blood flow in Alzheimer disease: Implications for the cognitive reserve hypothesis. Arch Neurol. 2003;60:359–365. doi: 10.1001/archneur.60.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131–144. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- 11.Blennow K, Vanmechelen E, Hampel H. CSF total tau, Abeta42 and phosphorylated tau protein as biomarkers for Alzheimer’s disease. Mol Neurobiol. 2001;24:87–97. doi: 10.1385/MN:24:1-3:087. [DOI] [PubMed] [Google Scholar]
- 12.Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2003;2:605–613. doi: 10.1016/s1474-4422(03)00530-1. [DOI] [PubMed] [Google Scholar]
- 13.Blennow K. Cerebrospinal fluid protein biomarkers for Alzheimer’s disease. NeuroRx. 2004;1:213–225. doi: 10.1602/neurorx.1.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strozyk D, Blennow K, White LR, Launer LJ. CSF Abeta 42 levels correlate with amyloid-neuropathology in a populationbased autopsy study. Neurology. 2003;60:652–656. doi: 10.1212/01.wnl.0000046581.81650.d0. [DOI] [PubMed] [Google Scholar]
- 15.Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, Pirttilä T. Cerebrospinal fluid beta-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66:382–389. doi: 10.1001/archneurol.2008.596. [DOI] [PubMed] [Google Scholar]
- 16.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–349. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 17.Roe CM, Fagan AM, Williams MM, Ghoshal N, Aeschleman M, Grant EA, Marcus DS, Mintun MA, Holtzman DM, Morris JC. Improving CSF biomarker accuracy in predicting prevalent and incident Alzheimer disease. Neurology. 2011;76:501–510. doi: 10.1212/WNL.0b013e31820af900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zekry D, Graf CE, Giannelli SV, Gold G, Michel JP. Non-cognitive outcomes in trials of disease-modifying drugs for Alzheimer’s disease. Eur Geriatr Med. 2012;3:37–42. [Google Scholar]
- 19.Liu-Seifert H, Siemers E, Sundell K, Price K, Han B, Selzler K, Aisen P, Cummings J, Raskin J, Mohs R. Cognitive and functional decline and their relationship in patients with mild Alzheimer’s dementia. J Alzheimers Dis. 2014;43:949–955. doi: 10.3233/JAD-140792. [DOI] [PubMed] [Google Scholar]
- 20.Zahodne LB, Manly JJ, MacKay-Brandt A, Stern Y. Cognitive declines precede and predict functional declines in aging and Alzheimer’s disease. PLoS One. 2013;8:e73645. doi: 10.1371/journal.pone.0073645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okonkwo OC, Alosco ML, Griffith HR, Mielke MM, Shaw LM, Trojanowski JQ, Tremont G, Initiative ADN. Cerebrospinal fluid abnormalities and rate of decline in everyday function across the dementia spectrum: Normal aging, mild cognitive impairment, and Alzheimer disease. Arch Neurol. 2010;67:688–696. doi: 10.1001/archneurol.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roe CM, Fagan AM, Grant EA, Holtzman DM, Morris JC. CSF biomarkers of Alzheimer disease: “Non-cognitive” outcomes. Neurology. 2013;81:2028–2031. doi: 10.1212/01.wnl.0000436940.78152.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berg L, McKeel DW, Miller JP, Storandt M, Rubin EH, Morris JC, Baty J, Coats M, Norton J, Goate AM, Price JL, Gearing M, Mirra SS, Saunders AM. Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: Relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol. 1998;55:326–335. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- 24.Villareal DT, Grant E, Miller JP, Storandt M, McKeel DW, Morris JC. Clinical outcomes of possible versus probable Alzheimer’s disease. Neurology. 2003;61:661–667. doi: 10.1212/wnl.61.5.661. [DOI] [PubMed] [Google Scholar]
- 25.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566–572. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- 26.Morris JC. The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 27.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 2014;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 28.Pfeffer RI, Kurosaki TT, Harrah CH, Chance JM, Filos S. Measurement of functional activities in older adults in the community. J Gerontol. 1982;37:323–329. doi: 10.1093/geronj/37.3.323. [DOI] [PubMed] [Google Scholar]
- 29.Manly JJ, Schupf N, Tang MX, Stern Y. Cognitive decline and literacy among ethnically diverse elders. J Geriatr Psychiatry Neurol. 2005;18:213–217. doi: 10.1177/0891988705281868. [DOI] [PubMed] [Google Scholar]
- 30.Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: Comprehensive assessment of psychopathology in dementia. Neurology. 1994;44:2308–2314. doi: 10.1212/wnl.44.12.2308. [DOI] [PubMed] [Google Scholar]
- 31.Kaufer DI, Cummings JL, Ketchel P, Smith V, MacMillan A, Shelley T, Lopez OL, DeKosky ST. Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory. J Neuropsychiatry Clin Neurosci. 2000;12:233–239. doi: 10.1176/jnp.12.2.233. [DOI] [PubMed] [Google Scholar]
- 32.Sheikh JI, Yesavage JA. Clinical Gerontology: A Guide to Assessment and Intervention. The Haworth Press; NY: 1986. Geriatric Depression Scale (GDS): Recent evidence and development of a shorter version. [Google Scholar]
- 33.Buckner RL, Head D, Parker J, Fotenos AF, Marcus D, Morris JC, Snyder AZ. A unified approach for morphometric and functional data analysis in young, old, and demented adults using automated atlas-based head size normalization: Reliability and validation against manual measurement of total intracranial volume. Neuroimage. 2004;23:724–738. doi: 10.1016/j.neuroimage.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 34.Fotenos AF, Snyder AZ, Girton LE, Morris JC, Buckner RL. Normative estimates of cross-sectional and longitudinal brain volume decline in aging and AD. Neurology. 2005;64:1032–1039. doi: 10.1212/01.WNL.0000154530.72969.11. [DOI] [PubMed] [Google Scholar]
- 35.Head D, Snyder AZ, Girton LE, Morris JC, Buckner RL. Frontalhippocampal double dissociation between normal aging and Alzheimer’s disease. Cereb Cortex. 2005;15:732–739. doi: 10.1093/cercor/bhh174. [DOI] [PubMed] [Google Scholar]
- 36.Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, Albert MS, Killiany RJ. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 37.Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM. Whole brain segmentation: Automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 38.Fischl B, van der Kouwe A, Destrieux C, Halgren E, Ségonne F, Salat DH, Busa E, Seidman LJ, Goldstein J, Kennedy D, Caviness V, Makris N, Rosen B, Dale AM. Automatically parcellating the human cerebral cortex. Cereb Cortex. 2004;14:11–22. doi: 10.1093/cercor/bhg087. [DOI] [PubMed] [Google Scholar]
- 39.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, De Kosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 40.Tarawneh R, D’Angelo G, Macy E, Xiong C, Carter D, Cairns NJ, Fagan AM, Head D, Mintun MA, Ladenson JH, Lee JM, Morris JC, Holtzman DM. Visininlike protein1: Diagnostic and prognostic biomarker in Alzheimer disease. Ann Neurol. 2011;70:274–285. doi: 10.1002/ana.22448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bates DM. lme4: Mixed-effects modeling with R. 2010 URL http://lme4.r-forge.r-project.org/lMMwR/lrgprt.pdf.
- 42.Razlighi QR, Stallard E, Brandt J, Blacker D, Albert M, Scarmeas N, Kinosian B, Yashin AI, Stern Y. A new algorithm for predicting time to disease endpoints in Alzheimer’s disease patients. J Alzheimers Dis. 2014;38:661–668. doi: 10.3233/JAD-131142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.