

Abstract
Exploiting natural resources for bioactive compounds is an attractive drug discovery strategy in search for new anti-malarial drugs with novel modes of action. Initial screening efforts in our laboratory revealed two preparations of soil-derived actinomycetes (H11809 and FH025) with potent anti-malarial activities. Both crude extracts showed glycogen synthase kinase 3β (GSK3β)-inhibitory activities in a yeast-based kinase assay. We have previously shown that the GSK3 inhibitor, lithium chloride (LiCl), was able to suppress parasitaemia development in a rodent model of malarial infection. The present study aims to evaluate whether anti-malarial activities of H11809 and FH025 involve the inhibition of GSK3β. The acetone crude extracts of H11809 and FH025 each exerted strong inhibition on the growth of Plasmodium falciparum 3D7 in vitro with 50% inhibitory concentration (IC50) values of 0.57 ± 0.09 and 1.28 ± 0.11 µg/mL, respectively. The tested extracts exhibited Selectivity Index (SI) values exceeding 10 for the 3D7 strain. Both H11809 and FH025 showed dosage-dependent chemo-suppressive activities in vivo and improved animal survivability compared to non-treated infected mice. Western analysis revealed increased phosphorylation of serine (Ser 9) GSK3β (by 6.79 to 6.83-fold) in liver samples from infected mice treated with H11809 or FH025 compared to samples from non-infected or non-treated infected mice. A compound already identified in H11809 (data not shown), dibutyl phthalate (DBP) showed active anti-plasmodial activity against 3D7 (IC50 4.87 ± 1.26 µg/mL which is equivalent to 17.50 µM) and good chemo-suppressive activity in vivo (60.80% chemo-suppression at 300 mg/kg body weight [bw] dosage). DBP administration also resulted in increased phosphorylation of Ser 9 GSK3β compared to controls. Findings from the present study demonstrate that the potent anti-malarial activities of H11809 and FH025 were mediated via inhibition of host GSK3β. In addition, our study suggests that DBP is in part the bioactive component contributing to the anti-malarial activity displayed by H11809 acting through the inhibition of GSK3β.
Keywords: Anti-malarial, GSK3β, Actinomycete, Dibutyl Phthalate
Abstract
Pengeksploitasian sebatian bioaktif daripada sumber semula jadi merupakan strategi menarik dalam usaha pencarian drug anti-malaria baru dengan mod tindakan novel. Usaha penyaringan awal di makmal kami telah menemukan dua persediaan aktinomiset tanah (H11809 dan FH025) dengan aktiviti anti-malaria. Kedua-dua ekstrak kasar menunjukkan ciri anti-glikogen sintase kinase 3β (anti-GSK3β) menggunakan sistem pengasaian berasas-yis. Kajian terdahulu telah menunjukkan perencat GSK3, litium klorida (LiCl), berupaya menindas pertumbuhan parasitaemia dalam model infeksi malaria roden. Kajian ini dijalankan untuk menentukan sama ada aktiviti anti-malaria H11809 dan FH025 melibatkan perencatan GSK3β. Kedua-dua ekstrak aseton H11809 dan FH025 menunjukkan perencatan yang baik terhadap pertumbuhan Plasmodium falciparum 3D7 secara in vitro, dengan nilai kepekatan perencatan 50% (IC50) 0.57 ± 0.09 dan 1.28 ± 0.11 µg/mL, masing-masing. Ekstrak yang diuji turut bersifat memilih bagi 3D7 dengan nilai indeks pemilihan melebihi 10. Secara in vivo, ekstrak H11809 dan FH025 menunjukkan aktiviti kemo-penekanan berkadaran-dos dan meningkatkan kemandirian mencit berbanding mencit terinfeksi tanpa perlakuan. Analisis Western menunjukkan peningkatan (6.79 hingga 6.83 kali ganda) terhadap pemfosfatan serin (Ser 9) GSK3β dalam sampel hepar mencit terinfeksi dengan perlakuan ekstrak H11809 atau FH025 berbanding sampel daripada mencit tidak terinfeksi atau mencit terinfeksi tanpa perlakuan. Sebatian bioaktif yang telah dikenal pasti dalam H11809 (data tidak ditunjukkan), iaitu dibutil ftalat (DBP) menunjukkan aktiviti anti-plasmodium yang baik terhadap parasit strain 3D7 (IC50 4.87 ± 1.26 µg/mL bersamaan 17.50 µM) serta aktiviti kemo-penekanan in vivo yang baik (kemo-penekanan 60.80% pada dos 300 mg/kg berat tubuh [bt]). Pemberian DBP turut menyebabkan peningkatan pemfosfatan Ser 9 GSK3β hepar berbanding kawalan. Penemuan daripada kajian ini menunjukkan aktiviti anti-malaria H11809 dan FH025 diperantara melalui perencatan GSK3β hos. Tambahan lagi, kajian ini mencadangkan DBP sebagai satu komponen bioaktif yang menyumbang kepada aktiviti anti-malaria H11809 melalui perencatan GSK3β.
Keywords: Anti-malaria, GSK3β, Aktinomiset, Dibutil Ftalat
INTRODUCTION
Malaria is a highly infectious disease caused by a protozoan parasite of the genus Plasmodium (Karthik et al. 2014). Control and treatment of the malarial disease have been complicated by alarmingly rapid development of the plasmodial parasite’s resistance to existing anti-malarial drugs, as well as the increasing numbers of zoonotic Plasmodium knowlesi infections (Singh et al. 2008). This has necessitated research efforts towards discovery of new anti-malarial drugs with novel modes of action. Actinomycetes, widely-distributed bacteria in terrestrial and aquatic ecosystems, especially soil (Sharma 2014) represent plausible source to be exploited for this purpose. Numerous bioactive secondary metabolites with anti-bacterial, anti-fungal, anti-cancer, anti-oxidant, anti-malarial, and anti-inflammatory activities have been identified from actinomycetes (Deepa et al. 2013).
Protein kinases which regulate parasitic growth and differentiation have emerged as promising new anti-malarial drug targets (Houzé et al. 2014). For example, Plasmodium falciparum GSK3 (PfGSK3) is one of the eukaryotic protein kinases identified essential for the plasmodial growth, thus a novel anti-malaria drug target (Masch & Kunick 2015). GSK3, a serine/threonine protein kinase first identified as one of several protein kinases capable of phosphorylating and inactivating glycogen synthase (Embi et al. 1980), is now known to be associated with many cellular processes and implicated in many human diseases such as cancer, Alzheimer’s disease, diabetes, and pathogen-mediated inflammation (Song et al. 2015). The kinase is activated by phosphorylation at tyrosine (Tyr 216) and conversely, inhibited as a result of serine (Ser 9) phosphorylation (Kockeritz et al. 2006). GSK3β appears to play important roles in a host’s response to viral, fungal, or parasitic infections including malaria (Wang et al. 2014). This has led to various efforts to develop small molecule inhibitors against GSK3 (Kramer et al. 2012). Inhibition of the kinase enzyme by mammalian GSK3 inhibitors were shown to significantly inhibit the activity of recombinant P. falciparum protein PfGSK3 (Droucheau et al. 2004). Masch and Kunick (2015) recently developed selective PfGSK3 inhibitors as potential new anti-malarial agents.
We have previously shown that lithium chloride (LiCl), a known GSK3 inhibitor, suppressed parasitaemia development in Plasmodium berghei-infected mice (Zakaria et al. 2010). Screening of plants and microbes from Sabah for anti-malarial activity conducted in our laboratory revealed potent anti-malarial activities in extracts of two soil actinomycetes, H11809 and FH025. A yeast-based kinase assay showed GSK3β-inhibitory activities in both extracts. The present study aims to evaluate whether anti-malarial activities of H11809 and FH025 involve the inhibition of GSK3β and to identify the component responsible for the inhibition of the protein kinase.
MATERIALS AND METHODS
Preparation of Crude Extracts of H11809 and FH025
The actinomycete strains, H11809 and FH025, were isolated from Imbak Valley and Likas, Sabah, respectively. Both strains were purified using modified humic acid agar with the addition of vitamin B (HVA), pH 5.6 (Hayakawa & Nonomura 1987). The strains were purified using manually-prepared oatmeal agar (OA), pH 7.2, and were incubated at 28°C to induce sporulation for morphological observation. Both strains were cultivated using 10 mL of mannitol peptone liquid medium, and 2% mannitol + 2% peptone + 1% glucose, incubated at 28°C, 210 rpm for 5 days. Absolute acetone at the ratio of 1:1 was then added into the crude extracts to produce acetone crude extracts (Ho et al. 2009).
Fractionation and Identification of Active Compounds from H11809
The acetone crude extract of H11809 was partitioned successively with equal volume of hexane, chloroform, and butanol at the ratio of 1:1 (v/v). The chloroform layer was selected for further fractionation using column chromatography and dibutyl phthalate (DBP, 61.5%) was identified as a major constituent through gas chromatography mass spectrometry (GCMS) (unpublished data).
Yeast-based Assay for GSK3β-inhibitory Activity
The activity of test extracts or compounds against GSK3β was evaluated using an in vivo yeast-based system described by Andoh et al. (2000). Insertion and expression of mammalian GSK3β restored the original phenotype of the temperature-sensitive yeast gsk-3 null mutant in a pKT10-GSK3β yeast strain with genotype of MATa his3 leu2 ura3 trp1 ade2 mck1::TRP1 mds1::HIS3 mrk1 yol128c::LEU2. For screening purposes, a loop-full of a three-day yeast culture was inoculated into 5.0 mL of synthetic complete-uracil (SC-Ura) broth and incubated in a water bath at 37°C, 150 rpm for 48 h. For the assay, 400 μL yeast culture was then added into 100 mL of SC-Ura agar and poured into six plates and left to solidify at room temperature. Disc diffusion agar technique was applied in which 20 μL of the 100 mg/mL test samples were inoculated onto paper discs. The paper discs were then arranged on screening SC-Ura agar plates, incubated at 28°C (permissive temperature) and 37°C (high temperature) for 120 h, and the growth of yeast was observed for 5 days. Screening was carried out in triplicates. GSK3β-inhibitory activity in test samples is indicated by the presence of inhibition zones at 37°C (Cheenpracha et al. 2009).
P. falciparum Culture
Chloroquine-sensitive 3D7 strain of P. falciparum was obtained from Malaria Research and Reference Reagent Resource Centre (MR4, Manassas, Virginia, USA). The parasites were cultivated using a previously described procedure by Trager and Jensen (1976). Non-infected venous human blood group O rhesus-positive samples served as host cells. Complete media consisting of RPMI 1640 media (Gibco, Waltham, Massachusetts, USA) supplemented with 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, HEPES (Gibco, Waltham, Massachusetts, USA), 1.8 mM NaHCO3 (Sigma Aldrich, Missouri, USA), 100 µM hypoxanthine (Sigma Aldrich, Missouri, USA), 12.5 µg/mL gentamicin (Sigma Aldrich, Missouri, USA), and 0.5% Albumax (Gibco, Waltham, Massachusetts, USA) was used to cultivate the parasites. Infected erythrocytes were suspended in culture media at a haematocrit of 1.5% and initial parasitaemia of less than 10% in T25 culture flasks and incubated at 37°C, 3% O2, and 5% CO2. Culture media were changed and thin blood smears prepared every 24 h to monitor parasitaemia levels.
In vitro Anti-plasmodial Assay
Quantitative assessment of parasite (3D7) viability in vitro in the presence of test extracts or compounds was accomplished using parasite lactate dehydrogenase (pLDH) assay method, slightly modified from that described by Makler and Hinrichs (1993). The crude extracts and compound were each dissolved in the culture media and serially-diluted ten-fold to yield eight concentrations ranging from 0.0001 to 1000 µg/mL. Chloroquine diphosphate (Sigma Aldrich, Missouri, USA) (100 µg/mL) was used as the standard reference drug. Normal erythrocytes were added to wells without test samples, while infected erythrocytes were added to wells with extracts, compound, or drug. Asynchronous cultures with parasitaemia of 2%–3% and a final haematocrit of 1.5% were aliquoted into microtiter plates and incubated at 37°C for 48 h for maximum parasite growth. All tests were performed in triplicates. The cultures were then frozen at −20°C overnight, then subjected to three 20-minute freeze-thaw cycles to release cell content and for cultures to resuspend. At the end of the freeze-thaw cycles, 100 μL of Malstat reagent (Sigma Aldrich, Missouri, USA) and 25 μL of nitrobluetetrazolium/phenazine ethosulphate (NBT/PES, Sigma Aldrich, Missouri, USA) solution were added to each well of a fresh microtiter plate. Thereafter, 15 μL aliquots of the resuspended cultures were transferred to the corresponding well of the Malstat plate, thereby initiating colorimetric assessment of the lactate dehydrogenase (LDH) activity. Colour development of the LDH plate was monitored at 650 nm using a microplate reader (Fluostar Optima, Ortenberg, Germany) after 1 h of incubation in the dark. Fifty percent inhibitory concentration (IC50) values were calculated using GraphPad Prism 5 (GraphPad Software Inc., California, USA). Anti-plasmodial activities of the extracts or compounds were expressed as IC50 pLDH (mean ± S.D. of three separate experiments performed in triplicates).
Cytotoxicity Assay
Cytotoxicity of test extracts or compounds were evaluated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay described by Mossman (1983) to assess the effects of these samples on the proliferation of Chang liver cells. Chang liver cells, originally purchased from the American Type Culture Collection (ATCC), USA and were a kind gift from the Institute of Systems Biology (INBIOSIS), Universiti Kebangsaan Malaysia (UKM, Bangi, Selangor, Malaysia). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% foetal bovine serum (FBS, Gibco, Waltham, Massachusetts, USA) in a humidified atmosphere of 5% CO2 at 37°C. Briefly, Chang liver cells were added into 96-well microtiter plates at a density of 5 × 104 cells per well and incubated for 48 h to ensure attachment and confluency. After 48 h incubation, culture media were carefully removed, cells treated with 100 μL of test extracts or compounds at different concentrations ranging from 0.001–10,000 µg/mL and further incubated for 48 h.
Then, the media and test sample mixtures were carefully removed, cells washed with phosphate-buffered saline (PBS, pH 7.2, Sigma Aldrich, Missouri, USA) and the mixture was replaced with 100 μL culture media. Following this, 10 μL of MTT (Sigma Aldrich, Missouri, USA) solution (5 mg MTT/mL in PBS) was added to each well and further incubation was carried out for an additional 3 h. The medium was carefully removed and MTT-formazan products formed were dissolved in 100 μL DMSO (Sigma Aldrich, Missouri, USA). After 30 minutes, the absorbance was measured at 540 nm using a microplate reader (Fluostar Optima, Ortenberg, Germany). IC50 values determined using GraphPad Prism 5 were expressed as IC50 MTT. For each test, growth of cells in media alone was used as positive control. All experiments were performed in triplicates.
Selectivity Index (SI), corresponding to the ratio between cytotoxicity and anti-plasmodial activity, was calculated for test samples according to the following formula (Verma et al. 2011):
In vivo Anti-malarial Test
For the in vivo test, the four-day suppressive test described by Peters et al. (1975) was used. Male ICR mice (6 weeks old, n=6 per group) were obtained from the Animal House Complex, Universiti Kebangsaan Malaysia. The experimental procedures were approved by Universiti Kebangsaan Malaysia Animal Ethics Committee (UKMAEC). Mice were infected by intraperitoneal (i.p.) inoculations with 1 × 107 P. berghei NK65-infected erythrocytes on day 0. The inoculum was prepared by diluting infected stock mice blood at 20%–30% parasitaemia with Alsever’s solution. Within 3 h of inoculations with the parasites (on day 0), experimental animals were given intraperitoneal injections of the test samples; H11809 or FH025 (25, 50, 100, and 250 mg/kg body weight [bw]) or DBP (10, 30, 100, and 300 mg/kg bw). Test samples were administered intraperitoneally for 4 consecutive days on days 0, 1, 2, and 3. For control groups, mice were similarly treated for four consecutive days with chloroquine diphosphate (10 mg/kg bw), lithium chloride (100 mg/kg bw, Sigma Aldrich, Missouri, USA) or saline solution (0.9% NaCl, Sigma Aldrich, Missouri, USA). Parasitaemia was determined by microscopic examination of thin blood smears prepared from blood drawn from the tail. The average percentage of chemo-suppression was calculated using the following formula:
where A is the average percentage of parasitaemia in the non-treated control group and B the average percentage of parasitaemia in the test groups. Survivability of all groups of mice were recorded for 28 days.
The test samples were also evaluated for effects on normal mice survivability at the doses used for the in vivo four-day chemo-suppressive test. Four groups of mice (n=6 per group) were injected intraperitoneally with 25, 50, 100, and 250 mg/kg bw extracts or 10, 30, 100, and 300 mg/kg bw compounds for four consecutive days. Gross signs of toxicity such as death and changes in physical appearance and behaviour were observed. Survivability was observed for 28 days. Mortality occurring before day 5 post-treatment with test samples is an indication of toxicity of the samples towards the experimental animals (Hilou et al. 2006).
Western Analysis
To determine the phosphorylation state of GSK3β in samples, infected mice were administered with effective (chemo-suppressive) dosages of each test sample, lithium chloride or chloroquine for four consecutive days. On day 4 post-infection, liver organs were harvested, homogenised and proteins were extracted using a protein extraction buffer containing 50 mM TrisHCl (Sigma Aldrich, Missouri, USA), 150 mM NaCl, 1% Triton X-100 (Merck, New Jersey, USA), phosphatase and protease inhibitors (1 mM ethylenediaminetetraacetic acid [EDTA], 1 mM ethylene glycol tetraacetic acid [EGTA], 0.5 mM Na3VO4, 0.5 mM phenylmethylsulfonyl fluoride [PMSF], 1 μg/mL aprotinin, 5 μg/mL leupeptin and 1 mM NaF; all procured from Sigma Aldrich, Missouri, USA) (Lee 2007). Samples were then centrifuged at 20,000 g for 20 minutes at 4°C. Protein concentrations were measured using the Bradford method described by Bradford (1976) with bovine serum albumin (BSA, Sigma Aldrich, Missouri, USA) as a standard. Protein samples were separated using 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) (Laemmli 1970). Following gel electrophoresis, proteins were electro-transferred onto nitrocellulose membranes using Novex semi-dry blotter (Invitrogen, California) and the membranes were blocked with 3% BSA for 1 h before overnight incubation with primary monoclonal antibodies; anti-GSK3β (Cell Signaling Technology, Danvers, Massachusetts, USA) or anti-phospho Ser 9-GSK3β (Calbiochem, Seattle, Washington, USA). Blots were then hybridised with horseradish peroxidase-conjugated immunoglobulin G (HRP-conjugated IgG) as secondary antibodies for 2 h at room temperature. Detection of immuno-reactive protein bands was carried out using enhanced chemiluminescence reagent (ECL); western blot detection reagents (Thermo Scientific, Waltham, Massachusetts, USA). Stripping was carried out and membranes were then probed with β-actin antibody (Santa Cruz Biotechnology, Dallas, Texas, USA) to ensure equivalent protein loading. Band area intensity was quantified using a densitometer (Vilbert Lourmat 302, Torcy, France) and scanned blots were processed using the UltraQuant 6.0 software (UltraLum, California, USA) in order to determine the mean intensity of immuno-reactive proteins.
Statistical Analysis
All data obtained were expressed as the mean ± SD and analysed using GraphPad Prism 5 analysis software (GraphPad, USA). A p value of less than 0.05 was considered significant.
RESULTS
H11809 and FH025 Each Exhibited Inhibitory Activity against GSK3β
Actinomycete H11809 and FH025 were isolated from soils in Sabah. Aerial mycelia of H11809 are light brown in colour with no pigmentation (Fig. 1[a]) whereas FH025 produces dark brown aerial mycelia with brown pigmentation (Fig. 1[b]). A yeast-based GSK3 assay was employed to evaluate H11809 and FH025 extracts for GSK3β-inhibitory activities.
Figure 1:


Actinomycetes (a) H11809 and (b) FH025 showing aerial mycelia (light brown and dark brown in colour respectively).
In this assay, insertion of mammalian GSK3β gene into yeast complements the temperature-sensitive phenotype so that the yeast growth is permissible at 37°C. Thus, the presence of GSK3β-inhibitory activity will not allow the growth of the yeast at this temperature. Therefore, larger inhibition zones displayed by the yeast treated with H11809 or FH025 extracts at 37°C (13.8 ± 3.12 and 19.0 ± 1.0 mm, respectively) each compared to those at 25°C (8.88 ± 8.27 and 9.67 ± 0.58 mm, respectively) suggest positive inhibition of mammalian GSK3β (Table 1) by both actinomycetes tested.
Table 1:
GSK3β-inhibitory activities of H11809, FH025, and DBP using a yeast-based GSK3 assay.
| Sample | Inhibitory activity of acetone crude extract against GSK3β (mm) | Remarks | |
|---|---|---|---|
|
| |||
| 25°C | 37°C | ||
| Acetone crude extract of H11809 |
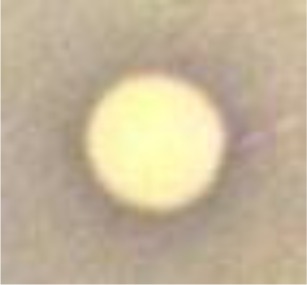 8.88 ± 8.27 (partial inhibition) |
 13.88 ± 3.12 (clear inhibition) |
Inhibition zone is slightly larger at 37°C which indicates the presence of the GSK3β inhibitor. |
| Acetone crude extract of FH025 |
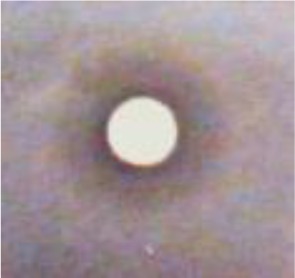 9.67 ± 0.58 (partial inhibition) |
 19.0 ± 1.0 (clear inhibition) |
Inhibition zone is significantly larger and clearer at 37°C which indicates the presence of the GSK3β inhibitor. |
| DBP |
 0.0 ± 0.0 (no inhibition) |
 20.0 ± 0.0 (partial inhibition) |
Inhibition zone was observed only at 37°C which indicates the presence of the GSK3β inhibitor. |
H11809 and FH025 Each Showed Promising Anti-plasmodial Activity
The anti-plasmodial activities of H11809 and FH025 extracts against chloroquine-sensitive P. falciparum 3D7 were determined using pLDH assay. The acetone crude extracts of H11809 and FH025 each displayed active anti-plasmodial activity with IC50 values of 0.57 ± 0.09 and 1.28 ± 0.11 µg/mL, respectively. Extracts with IC50 ≤ 50 μg/mL are regarded as active whereas those with values higher than 50 μg/mL are considered inactive (Ramazani et al. 2010). Therefore, both H11809 and FH025 extracts showed strong anti-plasmodial activities (Table 2).
Table 2:
In vitro anti-plasmodial activities of H11809, FH025, and DBP against P. falciparum 3D7.
| Extracts/drug | Cytotoxic activity of Chang liver cells IC50MTT ± SD (μg/mL) |
Anti-plasmodial activity IC50 pLDH ± SD (μg/mL) |
SI |
|---|---|---|---|
| H11809 (acetone) | 1028.00 ± 1.10 | 0.57 ± 0.09 | 1803.52 |
| FH025 (acetone) | 134.80 ± 1.21 | 1.28 ± 0.11 | 105.31 |
| DBP | 902.90 ± 2.96 | 4.87 ± 1.26 | 185.40 |
| Chloroquine diphosphate | ND | 0.19 ± 0.01 | ND |
Notes: The results are based on average IC50 ± SD (µg/mL) values from three replicates (n = 3) for in vitro cytotoxicity activity and anti-plasmodial activity. ND = not determined.
Extracts were tested for cytotoxic effects against Chang liver cells using MTT assay. The results showed no significant toxicity of H11809 with IC50 of 1028.00 ± 1.10 µg/mL. FH025 however showed mild toxicity towards the mammalian liver cells (IC50 = 134.80 ± 1.21 µg/mL) (Table 2). Further comparison of toxicities of tested extracts towards Chang liver cells with their respective anti-plasmodial (P. falciparum 3D7) activities gave SI values exceeding 10 indicating both crude extracts selectively suppressed development of the human malarial parasite 3D7 strain in vitro with minimal effects on normal mammalian cells (Ramazani et al. 2010). Selectivity of H11809 was determined to be superior (SI = 1803.52) whereas FH025 displayed good selectivity (SI = 105.31) against P. falciparum 3D7.
DBP Displayed Anti-GSK3β Activity and Good Anti-plasmodial Activity
Further fractionation of H11809-chloroform layers yielded one fraction in which DBP was identified as a major constituent using GCMS analysis (data not shown). The yeast-based kinase assay performed showed that DBP displayed a large and clear inhibition zone at 37°C (20.0 ± 0.0mm) (Table 1). Interestingly, DBP was not detected in the acetone crude extract of FH025 (unpublished data).
Determination of the anti-plasmodial activity of DBP showed an IC50 value of 4.87 ± 1.26 µg/mL towards P. falciparum 3D7 and no significant toxicity towards Chang liver cells (IC50 = 902.90 ± 2.96 µg/mL with SI>10) (Table 2). From these findings, DBP showed GSK3β-inhibitory activity and good anti-plasmodial activity.
H11809 and FH025 Suppressed Parasitaemia Development in P. berghei-infected Mice
Administration of the test extracts each at 25, 50, 100, and 250 mg/kg bw for four consecutive days caused dosage-dependent suppression of parasitaemia development in P. berghei NK65-infected mice (Table 3). At the highest dosage tested (250 mg/kg bw), H11809 and FH025 inhibited parasitaemia development in P. berghei-infected mice by 83.02% and 79.36%, respectively. By comparison, the reference drug, chloroquine (10 mg/kg bw) displayed 100% chemo-suppression on day 4. H11809 and FH025 were capable of inhibiting P. berghei parasitaemia development by more than 60% at 100 and 250 mg/kg bw dosages. Thus, these results showed promising chemo-suppressive activities of H11809 and FH025 extracts in P. berghei-infected mice.
Table 3:
In vivo anti-malarial activities of H11809 and FH025 against P. berghei NK65-infected mice.
| Extracts/ compound/drugs | Dosages (mg/kg bw) | Suppression of parasitaemia on day 4 (%) | Median survival time (days) |
|---|---|---|---|
| H11809 | 25 | 48.00* | 15 |
| 50 | 51.90* | 17 | |
| 100 | 80.42* | 18 | |
| 250 | 83.02* | 22 | |
| FH025 | 25 | 44.80* | 17 |
| 50 | 53.71* | 21 | |
| 100 | 65.92* | 22 | |
| 250 | 79.36* | 23 | |
| Chloroquine diphosphate (positive control) | 10 | 100.00* | 28 |
| 0.9% NaCl (negative control) | 0.2 mL | – | 10 |
Notes: The results show chemo-suppression (%) compared with negative control (n = 6).
The symbol * shows significant value (p<0.05).
Infected mice treated with the highest dosage of H11809 or FH025 displayed median survival time of 22 and 23 days, respectively. As comparison, median survival time of 10 days was obtained for non-treated infected mice. Control experimental animals given chloroquine survived throughout the observation period of 28 days (Table 3). These results demonstrated that the administration of H11809 or FH025 improved the median survival time of P. berghei-infected mice.
The crude extract of actinomycetes were also evaluated for effects on normal mice survivability by intraperitoneal injections of (25, 50, 100 or 250 mg/kg bw) for four consecutive days into non-infected mice. All mice survived throughout the 28 days observation period. No signs of toxicity such as diarrhoea, excess urination, and lethargy were observed in the study animals.
DBP Caused Good Inhibition of Parasitaemia Development and Prolonged Survivability of P. berghei-infected Mice
Administration of DBP into P. berghei-infected mice caused good inhibition of P. berghei development. At the highest dosage tested (300 mg/kg bw), DBP suppressed P. berghei NK65 parasitaemia development in mice by 60.80% ± 1.29 (Table 4). The median survival time of infected mice treated with DBP (18 days for highest dosage) was improved compared to non-treated mice. Non-treated mice did not survive beyond day 13 post-infection (Table 4). Reference anti-malarial drug, chloroquine, showed 100% suppression of parasitaemia development on day 4 and all mice survived throughout the observation period of 28 days. The findings showed that DBP suppressed the growth of P. berghei and improved mice median survival time.
Table 4:
In vivo anti-malarial activities of DBP against P. berghei NK65-infected mice.
| Extracts/compound/drugs | Dosages (mg/kg bw) | Suppression of parasitaemia on day 4 (%) | Median survival time (days) |
|---|---|---|---|
| DBP | 10 | 14.56* | 11 |
| 30 | 31.60* | 14 | |
| 100 | 49.60* | 17 | |
| 300 | 60.80* | 18 | |
| LiCl (GSK3 inhibitor) | 100 | 73.20* | 20 |
| Chloroquine diphosphate (positive control) | 10 | 100.00* | 28 |
| 0.9% NaCl (negative control) | 0.2 mL | – | 13 |
Notes: The results show chemo-suppression (%) compared with negative control (n = 6).
The symbol * shows significant value (p<0.05).
DBP (10, 30, 100 or 300 mg/kg bw) was also administered intraperitoneally into normal mice for four consecutive days to evaluate the effects of this compound on the survivability of normal non-infected mice. All mice survived throughout the 28 days observation period with no signs of toxicity observed in the study animals.
Increased Phosphorylation (Ser 9) of GSK3β were Detected in Organ Samples of P. berghei-infected Mice Administered with H11809 or FH025
The phosphorylation state of Ser 9 GSK3β in liver tissues obtained from P. berghei-infected mice administered with H11809 or FH025 crude extracts were determined by western analysis. Western analysis revealed higher levels of Ser 9 GSK3β phosphorylation in samples from infected mice treated with H11809 or FH025 (6.79-fold and 6.97-fold, respectively) compared to samples from their respective controls. Interestingly, fold increases of Ser9 GSK3β in the liver of H11809 or FH025-administered mice were 7.57-fold and 8.16-fold, respectively compared to their own set of controls (Figs. 2–5).
Figure 2:

GSK3β phosphorylation levels in liver. Liver protein samples from P. berghei-infected mice administered with anti-malarial drug, chloroquine diphosphate (CQ), GSK3 inhibitor; LiCl; and acetone crude extract of H11809. Levels of phosphorylated GSK3β (Ser 9) were normalised to total levels of GSK3β. Data represents mean ± SD of treated group compared to non-treated control.
Figure 3:

GSK3β phosphorylation levels in liver. Liver protein samples from normal mice each administered with anti-malarial drug, chloroquine diphosphate (CQ), GSK3 inhibitor; LiCl; and acetone crude extract of H11809. Levels of phosphorylated GSK3β (Ser 9) were normalised to total levels of GSK3β. Data represents mean ± SD of treated group compared to non-treated control.
Figure 4:

GSK3β phosphorylation levels in liver. Liver protein samples from P. berghei-infected mice each administered with anti-malarial drug, chloroquine diphosphate (CQ), GSK3 inhibitor; LiCl; and acetone crude extract of FH025. Levels of phosphorylated GSK3β (Ser 9) were normalised to total levels of GSK3β. Data represents mean ± SD of treated group compared to non-treated control.
Figure 5:

GSK3β phosphorylation levels in liver. Liver protein samples from normal mice each administered with anti-malarial drug, chloroquine diphosphate (CQ), GSK3 inhibitor; LiCl; and acetone crude extract of FH025. Levels of phosphorylated GSK3β (Ser 9) were normalised to total levels of GSK3β. Data represents mean ± SD of treated group compared to non-treated control.
The fold increase observed is comparable to that in LiCl-treated mice (8.16 to 8.39-fold) compared to non-treated infected mice. However, in the presence of chloroquine (10 mg/kg bw), levels of liver pGSK3 were not affected by the P. berghei infection. Both control and normal groups of mice showed lower levels of liver pGSK3β (Ser 9) compared to extracts and LiCl treatments. This finding indicates that anti-malarial activities of extracts prepared from actinomycete H11809 or FH025 resulted in the inhibition of host (liver) GSK3β.
DBP Resulted in Increased Ser 9 Phosphorylation of GSK3β
Similarly, like H11809, administration of DBP in P. berghei-infected mice increased levels of phosphorylated GSK3β (Ser 9) in the liver by 6.85-fold (Fig. 6) compared to non-treated infected mice (Fig. 7). It is noteworthy that increased phosphorylation was also comparable in the liver of LiCl-administered animal (7.95-fold), therefore suggesting the anti-malarial effects of DBP to involve Ser 9 phosphorylation (thus inhibition) of GSK3β.
Figure 6:

GSK3β phosphorylation levels in liver. Liver protein samples from P. berghei-infected mice each administered with anti-malarial drug, chloroquine diphosphate (CQ), GSK3 inhibitor; LiCl; and DBP. Levels of phosphorylated GSK3β (Ser 9) were normalised to total levels of GSK3β. Data represents mean ± SD of treated group compared to non-treated control.
Figure 7:

GSK3β phosphorylation levels in liver. Liver protein samples from normal mice each administered with anti-malarial drug, chloroquine diphosphate (CQ), GSK3 inhibitor; LiCl; and DBP. Levels of phosphorylated GSK3β (Ser 9) were normalised to total levels of GSK3β. Data represents mean ± SD of treated group compared to non-treated control.
DISCUSSION
Actinomycetes are a class of bacteria known to contain diverse chemical compounds which hold promise for the development of novel anti-malarial therapeutics (Sosovele et al. 2012; Xu et al. 2014). Anti-malarial activities have been identified in actinomycetes from various sources. For example, secondary metabolites salinosporamide A (Prudhomme et al. 2008), eponemycin and coronamycin (Rivo et al. 2013) isolated from marine and soil actinomycetes have been shown to inhibit plasmodial growth. Interestingly, another anti-malarial secondary metabolite, manzamine A, produced by sponge-associated bacteria also displayed GSK3β-inhibitory effects (Waters et al. 2014). To our knowledge, anti-GSK3 activity has not been reported in soil actinomycetes.
From our present study, we report that extracts prepared from two soil-derived actinomycetes, H11809 and FH025, with anti-malarial activities also displayed GSK3β-inhibitory properties evaluated via a yeast-based kinase assay. Both strains of Plasmodium used in this study are established models of malarial infection in vitro and in vivo. Both crude extracts were found to be highly selective towards P. falciparum 3D7. Based on the positive in vitro results, in vivo efficacy tests were carried out for H11809 and FH025 using a murine model of malarial infection. Our findings revealed that both extracts showed dosage-dependent chemo-suppressive activities in vivo and improved animal survivability compared to non-treated infected mice. Previously, we reported that LiCl (a GSK3 inhibitor) suppressed P. berghei parasitaemia development and prolonged survivability of malaria-infected mice (Zakaria et al. 2010). Taken together, the above findings suggest that the anti-malarial properties of the extracts could be mediated through inhibition of GSK3β.
Previous studies have shown that GSK3 inhibitors inhibit growth of plasmodial parasite in vitro and in vivo (Masch & Kunick 2015). The P. falciparum homologue of the GSK3 enzyme (PfGSK3) is believed to be essential in the liver and blood stages of the parasite’s development (Droucheau et al. 2004). In addition, Gazarini et al. (2003) demonstrated that protein kinase inhibitors interrupted the blood stage cycle of the rodent malarial parasite, Plasmodium chabaudi, thus clearly suggesting that phosphorylation events are required for normal development of these parasites. In our present study, administration of LiCl into P. berghei-infected mice resulted in phosphorylation of hepatic (Ser 9) GSK3β. It is interesting to note that increased Ser 9 phosphorylation of GSK3β was also detected in the liver of P. berghei-infected mice following administration of H11809 or FH025. The potent anti-malarial activity of H11809 and FH025 can therefore be associated with the inhibition of host GSK3β.
The present study also revealed that DBP, a compound in H11809 (unpublished data), exhibited anti-plasmodial activity in vitro. Similar chemo-suppressive effects were observed with DBP and more importantly, the present study revealed DBP administration also increased the phosphorylation of Ser 9 GSK3β. Therefore, the findings described implicates DBP as plausibly contributing to the anti-malarial effects observed for H11809. These findings corroborate a previous report on disruption of Wnt/β-catenin signalling pathway, in which GSK3 plays a critical regulatory role, by DBP in zebrafish embryos (Fairbairn et al. 2012).
In conclusion, findings from the present study suggest that the potent anti-malarial activities of H11809 and FH025 can therefore be associated with the inhibition of GSK3β in the host implicating immuno-modulatory effects of the two extracts. In addition, our study suggests that DBP is in part the bioactive component contributing to the anti-malarial activity, acting through inhibition of GSK3β displayed by H11809.
Acknowledgments
This research was supported by research grants from Ministry of Science, Technology and Innovation, Malaysia (09-05-IFN-BPH-001), Ministry of Higher Education, Malaysia (FRGS/1/2012/ST04/UKM/02/4) and Universiti Kebangsaan Malaysia (UKM-GUP-2011-212). Chang liver cells, originally purchased from American Type Culture Collection (ATCC), USA were a kind gift from the Institute of Systems Biology (INBIOSIS), Universiti Kebangsaan Malaysia.
REFERENCES
- Andoh T, Hirata Y, Kikuchi A. Yeast glycogen synthase kinase 3 is involved in protein degradation in cooperation with Bul1, Bul2, and Rsp5. Molecular and Cellular Biology. 2000;20(18):6712–6720. doi: 10.1128/mcb.20.18.6712-6720.2000. doi.org/10.1128/MCB.20.18.6712-6720.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72(1–2):248–254. doi: 10.1016/0003-2697(76)90527-3. doi.org/10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cheenpracha S, Zhang H, Mar AM, Foss AP, Foo SH, Lai NS, Jee JM, Seow HF, Ho CC, Chang LC. Yeast glycogen synthase kinase-3beta pathway inhibitors from an organic extract of Streptomyces sp. Journal of Natural Product. 2009;72(8):1520–1523. doi: 10.1021/np900163f. doi.org/10.1021/np900163f. [DOI] [PubMed] [Google Scholar]
- Deepa S, Kanimozhi K, Panneerselvam A. Antimicrobial activity of extracellularly synthesized silver nanoparticles from marine derived actinomycetes. International Journal of Current Microbiology and Applied Science. 2013;2(9):223–230. [Google Scholar]
- Droucheau E, Primot A, Thomas V, Mattei D, Knockaert M, Richardson C, Sallicandro P, et al. Plasmodium falciparum glycogen synthase kinase-3: Molecular model, expression, intracellular localisation and selective inhibitors. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics. 2004;1697(1–2):181–196. doi: 10.1016/j.bbapap.2003.11.023. doi.org/10.1016/j.bbapap.2003.11.023. [DOI] [PubMed] [Google Scholar]
- Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. European Journal of Biochemistry. 1980;107(2):519–527. doi.org/10.1111/j.1432-1033.1980.tb06059.x. [PubMed] [Google Scholar]
- Fairbairn EA, Bonthius J, Cherr GN. Polycylic aromatic hydrocarbons and dibutyl phthalate disrupt dorsal-ventral axis determination via the Wnt/β-catenin signalling pathway in zebrafish embryos. Aquatic Toxicology. 2012;15(124–125):188–196. doi: 10.1016/j.aquatox.2012.08.017. doi.org/10.1016/j.aquatox.2012.08.017. [DOI] [PubMed] [Google Scholar]
- Gazarini ML, Garcia CRS. Interruption of the blood-stage cycle of the malaria parasite, Plasmodium chabaudi, by protein tyrosine kinase inhibitors. Brazilian Journal of Medical Biological Research. 2003;36(11):1465–1469. doi: 10.1590/s0100-879x2003001100003. doi.org/10.1590/S0100-879X2003001100003. [DOI] [PubMed] [Google Scholar]
- Hayakawa M, Nonomura H. Efficacy of artificial humic acid as a selective nutrient in HV agar used for the isolation of soil actinomycetes. Journal of Fermentation Technology. 1987;65(6):609–616. doi.org/10.1016/0385-6380(87)90001-X. [Google Scholar]
- Hilou A, Nacoulma OG, Guiguemde TR. In vivo antimalarial activities of extracts from Amaranthusspinosus L. and Boerhaaviaerecta L. in mice. Journal of Ethnopharmacology. 2006;103(2):236–240. doi: 10.1016/j.jep.2005.08.006. doi.org/10.1016/j.jep.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Ho WL, Chan KW, Bernadr TZV, Lim SH, Ngao WC, Tong ML, Teh SC, et al. Screeming for potential microbial inhibitors against prokaryotic and eukaryotic signal transduction and isocitratelyase in Mycobacterium from Danum Valley, Sabah. Sabah Society Journal. 2009;26(1):67–157. [Google Scholar]
- Houzé S, Hoang N, Lozach O, Bras JL, Meijer L, Galons H, Demange L. Several human cyclin-dependent kinase inhibitors, structurally related to roscovitine, as new anti-malarial agents. Molecules. 2014;19(1):15237–15257. doi: 10.3390/molecules190915237. doi.org/10.3390/molecules190915237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthik L, Kumar G, Keswani T, Bhattacharyya A, Chandar SS, Bhaskara Rao KV. Protease inhibitors from marine actinobacteria as a potential source of antimalarial compound. PLoS ONE. 2014;9(3):e90972. doi: 10.1371/journal.pone.0090972. doi.org/10.1371/journal.pone.0090972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockeritz L, Doble B, Patel S, Woodgett JR. Glycogen synthase kinase-3-an overview of an over-achieving protein kinase. Current Drug Targets. 2006;27(11):1377–1388. doi: 10.2174/1389450110607011377. doi.org/10.2174/1389450110607011377. [DOI] [PubMed] [Google Scholar]
- Kramer T, Schmidt B, Monte FL. Small-molecule inhibitors of GSK-3: Structural insights and their application to Alzheimer’s disease models. International Journal of Alzheimer’s Disease. 2012;2012(6):381029–381061. doi: 10.1155/2012/381029. doi.org/10.1155/2012/381029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. doi.org/10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lee C. Protein extraction from mammalian tissues. Methods in Molecular Biology. 2007;362(1):385–389. doi: 10.1007/978-1-59745-257-1_29. doi.org/10.1007/978-1-59745-257-1_29. [DOI] [PubMed] [Google Scholar]
- Makler MT, Hinrichs DJ. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assesment of parasitemia. The American Journal of Tropical Medicine and Hygiene. 1993;48(2):205–210. doi: 10.4269/ajtmh.1993.48.205. [DOI] [PubMed] [Google Scholar]
- Masch A, Kunick C. Selective inhibitors of Plasmodium falciparum glycogen synthase-3 (PfGSK3): New anti-malarial agents? Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics. 2015;1854(10):1644–1649. doi: 10.1016/j.bbapap.2015.03.013. doi.org/10.1016/j.bbapap.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Mossman T. Rapid colorimetric assay for cellulase growth and survival: Application to proliferation and cytotoxicity assays. Journal of Immunology. 1983;65(1):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Peters W. The chemotheraphy of rodent malaria, XXII. The value of drug-resistant strains of P. berghei in screening for blood schizontisidal activity. Annals of Tropical Medicine and Parasitology. 1975;69(2):155–171. doi.org/10.1080/00034983.1975.11686997. [PubMed] [Google Scholar]
- Prudhomme J, McDaniel E, Ponts N, Bertani S, Fenical W, Jensen P, Le Roch K. Marine actinomycetes: A new source compounds against the human malaria parasite. PLoS One. 2008;3:e2335. doi: 10.1371/journal.pone.0002335. doi.org/10.1371/journal.pone.0002335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramazani A, Zakeri S, Sardari S, Khodakarim N, Djadidt ND. In vitro and in vivo anti-malarial activity of Boerhavia elegans and Solanum surattense. Malaria Journal. 2010;9(1):124–131. doi: 10.1186/1475-2875-9-124. doi.org/10.1186/1475-2875-9-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivo YB, Alkarimah A, Ramadhani NN, Cahyono AW, Laksmi DA, Winarsih S, Fitri LE. Metabolite extract of Streptomyces hygroscopicus Hygroscopicus inhibit the growth of Plasmodium berghei through inhibiton of ubiquitinproteasome system. Tropical Biomedicine. 2013;30(2):291–300. [PubMed] [Google Scholar]
- Sharma M. Actinomycetes: Source, identification, and their applications. International Journal of Current Microbiology and Applied Sciences. 2014;3(2):801–832. [Google Scholar]
- Singh JC, Davis TME, Lee KS, Shamsul SSG, Matusop A, Ratnam HA, Conway DJ, Singh B. Plasmodium knowlesi malaria in human is widely distributed and potentially life threatening. Clinical Infectious Disease. 2008;46(2):165–171. doi: 10.1086/524888. doi.org/10.1086/524888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, Song EAC, Jung MS, Choi SH, Baik HH, Jin BK, Kim JH, Chung SH. Phosphorylation and Inactivation of GSK3β by dual-specificity tyrosine (y)-phosphorylation-regulated kinase 1A. The Journal of Biological Chemistry. 2015;290(4):2321–2333. doi: 10.1074/jbc.M114.594952. doi.org/10.1074/jbc.M114.594952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosovele ME, Bergmann B, Lyimo TJ, Hosea KM, Mueller BI. Antimalarial activity of marine actinomycetes isolated from Dar Es Salaam mangrove sediments. International Journal of Research in Biological Sciences. 2012;2(4):177–181. [Google Scholar]
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193(4254):673–675. doi: 10.1126/science.781840. doi.org/10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Verma G, Dua VK, Agarwal DD, Atul PK. Anti-malarial activity of Holarrhena antidysenterica and Viola canescens, plants traditionally used against malaria in the Garhwal region of north-west Himalaya. Malaria Journal. 2011;10(1):20–24. doi: 10.1186/1475-2875-10-20. doi.org/10.1186/1475-2875-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu DB, Ye WW, Han Y, Deng ZX, Hong K. Natural products from mangrove actinomycetes. Marine Drugs. 2014;12(1):2590–2613. doi: 10.3390/md12052590. doi.org/10.3390/md12052590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HH, Lamont RJ, Kumar A, Scott DA. GSK3β and the control of infectious bacterial diseases. Trends in Microbiology. 2014;22(4):208–217. doi: 10.1016/j.tim.2014.01.009. doi.org/10.1016/j.tim.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AL, Peraud O, Kasanah N, Sims JW, Kothalawala N, Anderson MA, Abbas SH, et al. An analysis of the sponge Acanthostrongylophora igens’ microbiome yields an actinomycete that produces the natural product Manzamine A. Marine Science. 2014;1(54):1–15. doi: 10.3389/fmars.2014.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakaria NA, Embi N, Sidek H. Suppression of Plasmodium berghei parasitemia by LiCl in an animal infection model. Tropical Biomedicine. 2010;27(3):624–631. [PubMed] [Google Scholar]