

Abstract
New Findings
-
What is the topic of this review?
In this review, we discuss recent findings that provide a novel insight into the mechanisms that link glial cell function with the pathogenesis of cardiovascular disease, including systemic arterial hypertension and chronic heart failure.
-
What advances does it highlight?
We discuss how glial cells may influence central presympathetic circuits, leading to maladaptive and detrimental increases in sympathetic activity and contributing to the development and progression of cardiovascular disease.
Increased activity of the sympathetic nervous system is associated with the development of cardiovascular disease and may contribute to its progression. Vasomotor and cardiac sympathetic activities are generated by the neuronal circuits located in the hypothalamus and the brainstem. These neuronal networks receive multiple inputs from the periphery and other parts of the CNS and, at a local level, may be influenced by their non‐neuronal neighbours, in particular glial cells. In this review, we discuss recent experimental evidence suggesting that astrocytes and microglial cells are able to modulate the activity of sympathoexcitatory neural networks in disparate physiological and pathophysiological conditions. We focus on the chemosensory properties of astrocytes residing in the rostral ventrolateral medulla oblongata and discuss signalling mechanisms leading to glial activation during brain hypoxia and inflammation. Alterations in these mechanisms may lead to heightened activity of sympathoexcitatory CNS circuits and contribute to maladaptive and detrimental increases in sympathetic tone associated with systemic arterial hypertension and chronic heart failure.
Introduction
Cardiovascular disease remains the most common cause of death in Western societies, although the relative mortality attributable to it has decreased significantly in the last decade (Go et al. 2013). Hypertension is an important risk factor for the development of cardiovascular disease, and recent statistics from the American Heart Association suggest that approximately one‐third of adults in the USA are hypertensive (Go et al. 2013). Despite significant progress in the prevention, diagnosis and treatment of hypertension, only half of patients show satisfactory response to treatment (Go et al. 2013). Inadequate adherence to antihypertensive medication is an important factor that affects disease management. However, relatively poor efficacy of the existing antihypertensive therapies might also be due to the fact that conventional treatments target the peripheral mechanisms that maintain high systemic blood pressure, whereas primary CNS factors are responsible for the development and progression of the disease and remain untreated.
The pathophysiology of cardiovascular disease is complex, multifactorial and, in many respects, poorly understood. In this review, we focus on essential hypertension and chronic heart failure, which are conditions known to be associated with significant changes in the central nervous mechanisms of cardiovascular control. Indeed, recent advances in autonomic nervous system monitoring techniques in humans (including microneurography, measurement of noradrenaline spillover, assessment of heart rate variability and baroreflex sensitivity) have shown that increased activity of the sympathetic nervous system is intimately linked with the development and progression of both essential hypertension and chronic heart failure (Hasking et al. 1986; Leimbach et al. 1986; Esler et al. 1989; Kaye et al. 1995; Grassi et al. 1998; La Rovere et al. 1998, 2003; Esler & Kaye, 2000; Esler et al. 2001). In these conditions, complex interactions between behavioural, nutritional and humoral factors, altered activity of cardiovascular afferents (baro‐ and chemoreceptors) and modifications in the central nervous control mechanisms all lead to increased sympathetic efferent activity (Vallbo et al. 1979; Trzebski et al. 1982; Grassi et al. 1988, 2002; Mark, 1990; van de Borne et al. 1997; Schultz & Sun, 2000; Carlyle et al. 2002; Schultz et al. 2007; Grassi, 2009; Siński et al. 2012; May et al. 2013). In hypertension, enhanced sympathetic tone, along with humoral and vascular factors, is believed to play a significant role in the development and maintenance of high arterial blood pressure. In heart failure, sympathetic drive increases in parallel with the progression of the disease as a compensatory measure aimed to preserve ventricular contractile function. However, in the long‐term, sustained elevation of sympathetic tone becomes maladaptive and detrimental, leading to morphological and functional changes in the peripheral vasculature and the myocardium, including hypertrophy and proliferation of smooth muscle cells (Bevan, 1984), increased arterial stiffness (Boutouyrie et al. 1994), endothelial dysfunction (Pettersson et al. 1990), atherosclerosis (Kaplan et al. 1987), increased left ventricular mass (Simpson, 1983) and increased arrhythmogenesis (Lown & Verrier, 1976). Understanding the CNS mechanisms that underlie the increases in activity of the sympathetic nervous system is, therefore, important for the development of novel therapeutic strategies to treat hypertension and heart failure, and may ultimately help to reduce the clinical, social and economic burden of cardiovascular disease.
Vasomotor and cardiac neural activities of spinal sympathetic preganglionic neurones depend on tonic descending excitatory drive generated by sympathoexcitatory (presympathetic) neuronal circuits residing in the brainstem and the hypothalamus (Dampney, 1994; Spyer, 1994; Guyenet, 2000; Dampney et al. 2003; Madden & Sved, 2003). These circuits include the rostral ventrolateral medulla (RVLM), rostral ventromedial and mid‐line medulla, the A5 cell group of the pons and the hypothalamic paraventricular nucleus (PVN; Strack et al. 1989; Dampney et al. 2003; Madden & Sved, 2003; Kanbar et al. 2011; Marina et al. 2011). Although RVLM neurones responsible for sympathetic control of cardiovascular activities have been studied extensively (see Guyenet, 2013), little or no attention has been paid to their non‐neuronal neighbours, astrocytes and microglia. Here, we review recent evidence suggesting that changes in astroglial and microglial activities contribute to modifications in central nervous autonomic control and, therefore, may play an important role in the development and progression of cardiovascular disease associated with increased activity of the sympathetic nervous system (Marina, 2013; Marina, 2015; Rana et al. 2010; Shi et al. 2010; Zubcevic et al. 2011).
Glial cells
Astrocytes are the most abundant CNS glial cells and occupy non‐overlapping territories that define their functional domains (Halassa et al. 2007). Astrocytes were traditionally considered as a rather passive CNS cellular component that assisted neuronal circuits to maintain their function by providing nutritional and structural support. However, significant evidence accumulated in the last two decades suggests that astrocytes may play an active role in the regulation of synaptic strength and information processing (Halassa et al. 2009). In pathological conditions, astrocytes respond to CNS injury with significant changes in gene expression, morphology and function. This process, called astrogliosis, might have both beneficial and detrimental effects on tissue repair and might potentially influence the activity of neuronal networks (Sofroniew, 2015).
Microglial cells are resident immune cells in the CNS involved in the detection of invading organisms and brain injury. Microglial cells are less abundant than astrocytes, making up ∼20% of all brain glial cells. In resting conditions, microglial cells present a ramified morphology characterized by a small soma and extensive branched and long processes, which might make direct contact with neurones, astrocytes and cerebral vasculature. In response to brain injury, microglial cells become activated, undergo significant morphological changes and release various substances that may exert either neuroprotective or neurotoxic effects (Badoer, 2010).
Gliotransmission
Significant experimental evidence indicates that communication between glial cells and CNS neurones is bidirectional in nature. Astrocytes are capable of controlling synaptic communication via complex interactions with presynaptic and postsynaptic elements (Perea et al. 2009). Astrocytes release (glio)transmitters in response to activation of G‐protein‐coupled receptors and, when activated, generate intracellular Ca2+ responses. Several molecules identified as gliotransmitters can be released by activated astrocytes, including ATP/adenosine, d‐serine, glutamate, GABA, l‐lactate and possibly some others (Volterra & Meldolesi, 2005; Holmström et al. 2013; Tang et al. 2014; Marina et al. 2015).
One well‐documented astroglial signalling mechanism involves the release of ATP. The ATP released by activated astrocytes into the extracellular space acts on purinergic (P2) metabotropic and ionotropic receptors expressed by adjacent astrocytes, neurones and other glial cells (Gourine et al. 2009). Ectonucleotidases break down ATP to ADP, AMP and adenosine, which in turn can activate a different group of G‐protein‐coupled adenosine (P1) receptors (Burnstock, 2007).
Although release of (glio)transmitter(s) can be triggered by many stimuli, the physiological significance of gliotransmission has been debated. According to some estimates, the amount of glutamate contained within astroglial ‘vesicles’ is not sufficiently high to trigger changes in the neuronal activity following its release (Bramham et al. 1990). Indeed, the functional significance of gliotransmission is difficult to demonstrate because the approaches commonly used to stimulate astrocytes and to interfere with gliotransmitter release are generally non‐specific (Hamilton & Attwell, 2010). When genetic methods of selective activation and blockade of astrocytic Gq G‐protein‐coupled receptor‐mediated Ca2+ signalling mechanisms were applied in animal models, neuronal excitatory synaptic transmission in the hippocampus, as well as short‐ and long‐term plasticity were found to be unaffected (Fiacco et al. 2007; Agulhon et al. 2010). This may be explained by the fact that mature astrocytes do not express metabotropic glutamate receptor 5, thus casting further doubt on the role of glutamate in mediating communication between neurones and astrocytes (Sun et al. 2013). These results emphasize the importance of ATP‐mediated signalling by astrocytes, which is further supported by the recent studies demonstrating that ATP release by brainstem astrocytes plays an important role in the CNS mechanisms that maintain cardiovascular and respiratory homeostasis.
Glial cells and central chemosensitivity
There is significant evidence that astrocytes are involved in central chemosensory mechanisms that maintain cardiorespiratory homeostasis (Gourine et al. 2010; Marina et al. 2013). In mammals, these mechanisms are vital in producing adaptive changes in breathing in response to changes in /pH and in the arterial blood and brain parenchyma. Systemic hypercapnia, leading to decreases in blood and brain pH, is associated with a rapid release of ATP within the chemosensory areas of the brainstem (Gourine et al. 2005 b). Interestingly, these chemosensitive areas are located immediately underneath and in close proximity to the populations of RVLM presympathetic and ventral respiratory column neurones, which generate co‐ordinated sympathetic and respiratory rhythms (Gourine et al. 2005 a,b; 2010). Hypercapnia‐induced ATP release is independent of the peripheral chemoreceptor inputs (Gourine et al. 2005 b) and occurs within the appropriate time frame and in sufficient quantities to be responsible for the subsequent increases in breathing. Consistent with this, blockade of ATP receptors at the sites of release reduces the CO2‐induced increase in breathing, whereas application of ATP evokes an increase in the respiratory activity, mimicking the effect of CO2 (Gourine et al. 2005 b). Recent studies have also provided evidence that brainstem astrocytes are exquisitely sensitive to changes in pH within the physiological range and may represent the main source of ATP released in the brainstem during hypercapnia (Gourine et al. 2010). In contrast to cortical astrocytes, ventral brainstem astrocytes respond to acidification with elevations in intracellular Ca2+ and an increased rate of exocytosis of ATP‐containing vesicular compartments (Kasymov et al. 2013). This suggests that ventral brainstem astrocytes are functionally specialized for monitoring physiological increases in /[H+] and responding to these increases with the release of ATP (Kasymov et al. 2013). ATP propagates Ca2+ excitation among neighbouring astrocytes, activates neurones of the ventral respiratory column and, thus, contributes to the adaptive increases in the respiratory activity (Gourine et al. 2010).
Astrocytes also appear to be sensitive to physiological changes in brain O2 content. They respond to decreases in a few millimetres of mercury below normal brain oxygenation with robust increases in intracellular [Ca2+]. Hypoxia is detected in mitochondria, where O2 is consumed (Angelova et al. 2015). Decreases in inhibit mitochondrial respiration, leading to mitochondrial depolarization, production of free radicals, lipid peroxidation, activation of phospholipase C and Inositol triphosphate (IP3) receptors, resulting in recruitment of Ca2+ from the intracellular stores and vesicular release of ATP (Angelova et al. 2015; Fig. 1). This hypoxia‐induced astroglial ATP release has been demonstrated to maintain enhanced respiratory activity even in the absence of oxygen sensing by the peripheral chemoreceptors (Angelova et al. 2015).
Figure 1. Neuroglial interactions in the rostral ventrolateral medulla oblongata hypothesized to underlie pathological increases in sympathetic nerve activity in chronic heart failure and essential hypertension .
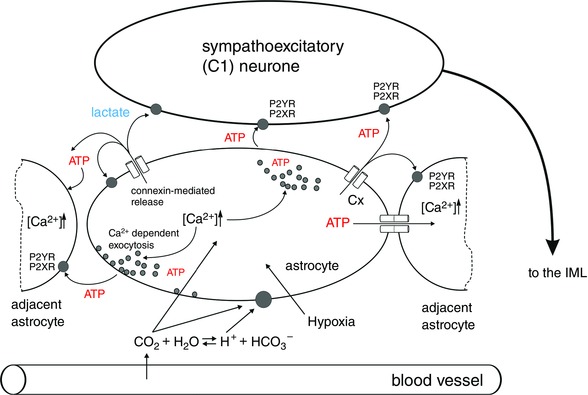
Tissue hypoxia and increased concentration of CO2/H+ is detected by astrocytes, leading to the release of ATP and lactate, which in turn increase the excitability of bulbospinal sympathoexcitatory (C1) neurones. Abbreviations: Cx, connexin; and IML, intermediolateral cell column.
Role of brainstem glia in the pathogenesis of cardiovascular disease
New evidence suggests that glial dysfunction may be responsible for the pathogenesis of certain neurodevelopmental disorders previously attributed to primary neuronal abnormalities. For example, recent studies demonstrated that altered astroglial and microglial functions contribute to the pathogenesis of Rett syndrome (Lioy et al. 2011; Derecki et al. 2012; Okabe et al. 2012), a prototypical neurological disorder associated with mutations of the methyl‐CpG‐binding protein 2 (MECP2) gene, which is manifested by neurological, respiratory and autonomic deficiencies. In this section, we discuss putative mechanisms that link altered glial cell activity with maladaptive increases in excitability of CNS presympathetic circuits, which may contribute to the detrimental sympathetic activation in cardiovascular disease.
Hypoxia
The (patho)physiological conditions associated with activation of RVLM astrocytes and downstream responses of presympathetic circuits are still unclear, although this mechanism may become significant in conditions of central hypoxia, which is a powerful stimulus for the release of ATP and lactate in the RVLM (Gourine et al. 2005 b; Karagiannis et al. 2015). ATP was previously shown to be released from within the RVLM in response to hypoxia (10% inspired O2, 5 min). This release was only slightly attenuated in animals with bilateral sectioning of vagi, aortic and carotid sinus nerves, suggesting that most of the hypoxia‐induced ATP release originates from astrocytes. It was also found that the amount of hypoxia‐induced ATP released from the ventral medullary structures is similar to that released in response to CO2 (Gourine et al. 2005 a). Activation of ATP receptors in the RVLM by microinjections of ATP or stable ATP analogues has been shown to activate bulbospinal presympathetic neurones, leading to marked increases in the arterial blood pressure, heart rate and renal sympathetic nerve activity (Sun et al. 1992; Horiuchi et al. 1999; Ralevic, 2000). These data are consistent with the hypothesis that increases in the level of ‘ambient’ ATP within the RVLM as a result of hypercapnia and/or hypoxia‐induced activation of brainstem astrocytes may contribute to the increases in central sympathetic drive (Fig. 1).
Factors that trigger and maintain astrocytic activation during the development and progression of heart failure remain unknown. Both obstructive sleep apnoea and central sleep apnoea are common clinical features in heart failure patients (Bradley & Floras, 2003 a,b). These conditions are associated with recurrent episodes of systemic hypoxia and hyper/hypocapnia. Brain tissue oxygen content in many heart failure patients was found to be significantly decreased despite nearly normal oxygen levels measured in the arterial blood (Rifai et al. 2012). Therefore, a hypoxic environment is likely to result in higher extracellular concentrations of ATP within the brainstem and this, as discussed above, may increase the activity of presympathetic neurones and result in higher sympathetic tone (Fig. 1). Indeed, sympathetic tone appears to be significantly higher in heart failure patients with sleep apnoea in comparison to heart failure patients with normal breathing patterns (Naughton et al. 1995; Mansfield et al. 2003).
Similar mechanisms may also underlie sympathoexcitation associated with the development of systemic arterial hypertension (Marina et al. 2015). Earlier studies demonstrated that basilar artery diameter in animal models of hypertension (spontaneously hypertensive rats, SHRs) is reduced, resulting in increased vascular resistance (Cates et al. 2011, 2012). Structural changes of brain vasculature are unlikely to be a consequence of hypertension because they are evident in prehypertensive animals (Cates et al. 2011, 2012). It was reported recently that tissue in the RVLM of the SHR is lower than in normotensive counterparts (despite normal levels of arterial ), suggesting that in the SHR the sympathoexcitatory networks are exposed to a hypoxic environment (Marina et al. 2015). Thus, in neurogenic hypertension, the Cushing mechanism is hypothesized to maintain brain perfusion by increasing systemic arterial blood pressure. Increased sympathetic activity and systemic hypertension can be viewed as a compensatory mechanism activated to preserve oxygen delivery and maintain brain oxygenation at the expense of systemic hypertension (Rodbard & Stone, 1955; Cates et al. 2011, 2012).
Brain hypoxia is known to trigger robust activation of the RVLM presympathetic neurones (Sun & Reis, 1994). There is evidence suggesting that the sensitivity of the RVLM presympathetic neurones to decreases in is mostly indirect and mediated by prior release and actions of both ATP and lactate, released by astrocytes (Fig. 2; Marina et al. 2015).
Figure 2. Release and actions of ATP and lactate mediate activation of rostral ventrolateral medulla (RVLM) neurons during hypoxia .
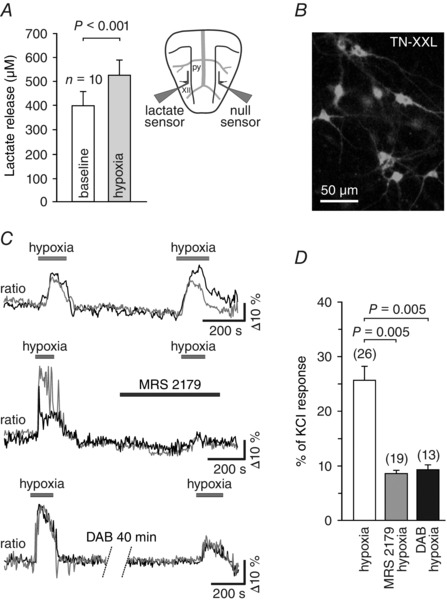
A, summary data obtained in vitro using horizontal slices of the rat brainstem, showing tonic release of lactate from the ventral surface of the medulla oblongata and peak lactate release during hypoxia. Inset, schematic drawing of a horizontal brainstem slice, illustrating dual recording configuration of lactate and null (control) biosensors placed on the ventral medullary surface. The difference in current between lactate and null biosensors was used to determine the amount of lactate release. Abbreviations: py, pyramidal tract; and XII hypoglossal rootlets. B, putative presympathetic C1 RVLM neurons visualized in organotypic brainstem slices after transduction with an adenoviral vector to express genetically encoded Ca2+ indicator TN‐XXL under the control of PRSx8 promoter. C, raw traces (changes in intracellular [Ca2+] of two individual neurons are shown on each plot), illustrating robust and reproducible responses of the RVLM neurons to hypoxia (top traces) as well as the effects of the ATP receptor antagonist MRS2179 (30 μmol l−1; middle traces) and the glycogenolysis inhibitor 1,4‐dideoxy‐1,4‐imino‐d‐arabinitol (DAB; 500 μmol l−1; bottom traces) on hypoxia‐induced [Ca2+]i responses of these neurons (ratiometric imaging using TN‐XXL). D, summary data illustrating the effects of MRS2179 and DAB on hypoxia‐induced [Ca2+]i responses of putative C1 neurons. Data are presented as means ± SEM. Reproduced from Marina et al. (2015).
In a similar manner to ATP, lactate, which is also released by activated astrocytes (Tang et al. 2014), produces strong excitation of C1 neurons in vitro and increases in sympathetic nerve activity and the arterial blood pressure when applied on the brainstem surface (Marina et al. 2015). Together, these lines of evidence suggest that both heart failure and hypertension are associated with a decrease in brain parenchymal oxygen content, accumulation of ATP and lactate within the presympathetic brainstem areas, leading to increased activity of sympathoexcitatory neurons and concomitant sustained elevation of sympathetic drive (Fig. 1). It remains to be determined whether in the human brain, tissue extracellular ATP and lactate concentrations are correlated with the severity of the sympathetically mediated cardiovascular diseases.
Increased purinergic (glio)transmission in the brainstem
Activated brainstem astrocytes signal to bulbospinal sympathoexcitatory neurones. It was shown that optogenetic stimulation of RVLM astrocytes (transduced with viral vectors to express light‐sensitive channels, e.g. channelrhodopsin 2) excites presympathetic RVLM neurones belonging to the C1 catecholaminergic group (Marina et al. 2013). These responses were attenuated in the presence of an ATP‐degrading enzyme, apyrase, demonstrating a key role for ATP in mediating communication between activated astrocytes and presympathetic neurones (Marina et al. 2013). Furthermore, in vivo experiments conducted in anaesthetized and artificially ventilated rats showed that optogenetic activation of channelrhodopsin 2‐expressing astrocytes within the RVLM produces robust elevations in renal sympathetic nerve activity, heart rate and arterial blood pressure (Marina et al. 2013; Fig. 3). Thus, selective recruitment of [Ca2+]i in RVLM astrocytes using an optogenetic approach is sufficient to trigger activation of sympathoexcitatory CNS circuits.
Figure 3. Optogenetic stimulation of RVLM astrocytes evokes sympathoexcitation in vivo .
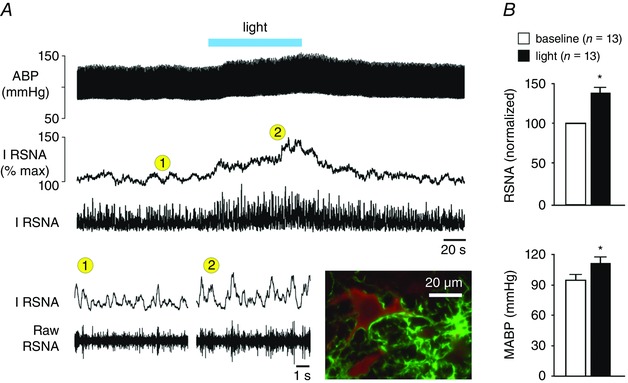
A, unilateral optogenetic stimulation of RVLM astrocytes expressing channelrhodopsin 2 increases sympathetic nerve activity and arterial blood pressure in an anaesthetized and artificially ventilated rat. Abbreviations: ABP, arterial blood pressure; I RSNA, integrated renal sympathetic nerve activity; and RSNA, renal sympathetic nerve activity. Inset microphotograph depicts an example of a tyrosine hydroxylase (red immunofluorescence)‐expressing C1 neurone embraced by astrocytic processes expressing channelrhodopsin 2‐Venus (green fluorescence). B, summary data illustrating the effect of optogenetic stimulation of RVLM astrocytes on mean arterial blood pressure (MABP) and RSNA. Group data are shown as means + SEM. *P < 0.05 (paired t‐test). Reproduced from Marina et al. (2013) with permission from Springer.
It was also shown in a rat model of heart failure that ATP‐mediated purinergic signalling in the RVLM plays an important role in sympathoexcitation, which is associated with, and may contribute to, the progression of left ventricular remodelling after a myocardial infarction. Given that specific ‘inhibition’ of astroglial activity/function is difficult to achieve (and it is not always easy to choose which of their functions has to be targeted), we reasoned that the role of astrocytes in the control of sympathetic activity in pathological conditions could be investigated by using experimental tools that prevent communication between these cells. To interfere with ATP‐mediated signalling, a lentiviral vector was developed in order to drive expression of a potent ectonucleotidase, transmembrane prostatic acid phosphatase (TMPAP), the effect of which is to facilitate rapid breakdown of extracellular as well as vesicular ATP (Wells et al. 2015). The TMPAP was tagged with green fluorescent protein, anchored to the plasma membrane and had a catalytic domain facing the extracellular space. Bilateral overexpression of TMPAP within the RVLM presympathetic circuits reduced sympathetic activity in developing heart failure (evident from a lower plasma concentration of noradrenaline) and slowed the progression of left ventricular remodelling and dysfunction (Marina et al. 2013). These data provided the first experimental evidence suggesting that altered glial activity leading to a higher level of ‘ambient’ ATP in the brainstem might be responsible for the increases in sympathetic tone and, by doing so, contribute to progression of heart failure (Fig 1).
Recent studies have demonstrated that ATP‐mediated purinergic signalling in the RVLM may also play a role in the development of neurogenic hypertension. Facilitated breakdown of ATP with targeted overexpression of virally driven TMPAP in the RVLM resulted in a significant reduction of the systemic arterial blood pressure in the SHR (Marina et al. 2015). There is also recent evidence to suggest that ATP‐mediated signalling may contribute to alterations in the central nervous mechanisms of autonomic control and development of hypertension in a rat model of chronic intermittent hypoxia. Increases in sympathetic nerve discharge evoked by microinjections of ATP into the RVLM were found to be significantly higher in rats exposed to chronic intermittent hypoxia (Zoccal et al. 2011). Chronic intermittent hypoxia was also associated with a significant upregulation of ATP receptor (P2X3 and P2X4 subunits in particular) expression in the RVLM (Zoccal et al. 2011). It remains to be determined whether increased ATP‐mediated signalling in the RVLM in a rat model of chronic intermittent hypoxia originates from activation of astrocytes in response to recurrent changes in the brainstem tissue (Angelova et al. 2015).
Microglial cell activation, astrogliosis and inflammation
Subtle chronic inflammation is a hallmark feature in the pathogenesis of chronic heart failure and essential hypertension. Indeed, in an animal model of heart failure, the expression of the pro‐inflammatory cytokines interleukin‐1β (IL‐1β) and tumour necrosis factor‐α (TNF‐α) was found to be significantly increased in at least one brain site that controls sympathetic outflow, namely the hypothalamic paraventricular nucleus (Kang et al. 2008, 2009). Clear evidence of microglial activation within the PVN after myocardial infarction was also reported (Rana et al. 2010).
Experimental models of hypertension (rats receiving chronic infusion of angiotensin II) provided further evidence of microglial activation and enhanced production of pro‐inflammatory cytokines in the presympathetic regions of the brain (PVN in particular; Shi et al. 2010). Activated microglial cells and inflammatory cytokines can evoke and amplify gliotransmitter release by astrocytes (Cotrina et al. 2000; Bezzi et al. 2001; Domercq et al. 2006; Pascual et al. 2012). Interestingly, in the model of hypertension induced by angiotensin II infusion, intracerebroventricular application of an anti‐inflammatory antibiotic, minocyclin, decreased the number of activated microglial cells and reduced the expression of pro‐inflammatory cytokines in the PVN. Suppression of microglial activity and PVN inflammation was associated with lower systemic arterial blood pressure, attenuated ventricular hypertrophy and lower plasma noradrenaline, all of which are indicative of a reduced sympathetic activity (Shi et al. 2010).
Immunohistochemical studies have also revealed microglial activation in the PVN of SHRs (Shi et al. 2010; Zubcevic et al. 2011). Recent data demonstrated that cultured hypothalamic microglial cells are directly activated by prorenin, a component of the renin–angiotensin system, resulting in induction of pro‐inflammatory mechanisms in these cells via activation of nuclear factor‐κB (Shi et al. 2014). It remains to be determined whether heart failure and hypertension are associated with similar changes in microglial function in other CNS areas involved in cardiovascular control.
There is also significant evidence to suggest that in pathological conditions, astrogliosis may lead to altered gliotransmission, which may affect neuronal excitability and result in neurotoxicity. Chronic astroglial activation occurs in response to an increase in extracellular ATP as well as in response to the actions of inflammatory cytokines, including IL‐1β and TNF‐α (Bezzi et al. 2001; Liu et al. 2011; Pascual et al. 2012), released locally by activated microglia. For example, in acute hippocampal slices, activated microglia release small amounts of ATP, which triggers waves of Ca2+ excitation in astrocytic networks (via stimulation of P2Y1 receptors), leading to the release of glutamate and an increased frequency of excitatory postsynaptic events in adjacent neurones (Pascual et al. 2012). Furthermore, TNF‐α released by activated microglia induces further release of TNF‐α by local astrocytes. These interactions between activated micro‐ and astroglia result in a significant production of TNF‐α, followed by a release of glutamate (Bezzi et al. 2001). All these factors are capable of initiating propagating waves of Ca2+ excitation within astroglial networks and modulating the excitability of local neuronal circuits (Cotrina et al. 2000; Bezzi et al. 2001; Pascual et al. 2012). Thus, in pathological conditions, the activity of CNS neuronal networks may be affected by altered gliotransmission initiated and maintained by tissue hypoxia and by the actions of inflammatory cytokines released by activated microglia.
There is morphological evidence of glial abnormalities in the brains of hypertensive subjects (Tomassoni et al. 2004). In an attempt to ‘inhibit’ astrocytic activity in SHRs, non‐specific pharmacological agents, such as arundic acid, have been used. Arundic acid suppresses synthesis of S100B (Asano et al. 2005), a glia‐specific Ca2+‐binding protein important for the expression of pro‐inflammatory cytokines and control of apoptosis (Marenholz et al. 2004). Interestingly, treatment of SHRs with arundic acid resulted in a significant reduction in the arterial blood pressure and prevented hypertension‐related strokes (Higashino et al. 2009).
Previous studies have also revealed an association of events that may link microvascular inflammation and brain hypoxia with astroglial activation of presympathetic networks. Experiments using RT‐PCR showed increased expression of a pro‐inflammatory molecule, junctional adhesion molecule‐A (JAM‐A), in the endothelial cells of arterioles supplying the dorsal brainstem structures of both young (pre‐hypertensive) and adult SHRs (Waki et al. 2007). Overexpression of JAM‐A in the dorsal medullary regions using viral vectors led to a significant increase in systolic blood pressure in control (Wistar) rats, and this was accompanied by leucocyte adhesion to the brainstem microvasculature (Waki et al. 2007, 2008). Moreover, body‐wide downregulation of JAM‐A using a specially formulated ‘vivo’ morpholino delayed development of hypertension in the SHR (Xu et al. 2012). There is also evidence that infiltration of monocytes in the brain microvasculature triggers the synthesis of cytokines, including monocyte chemoattractant protein‐1, by astrocytes (Andjelkovic et al. 2000). Therefore, leucocyte adhesion to the brainstem microvasculature may result in astroglial activation and upregulation of cytokine expression (Waki et al. 2008). In addition, increased leucocyte adhesion may produce partial blood flow obstruction, resulting in increased vascular resistance, reduced tissue perfusion and hypoxia (Paton & Waki, 2009). This may be sufficient to trigger astroglial activation, inflammation and gliotransmitter (ATP) release, leading to increased excitability of presympathetic circuits and enhanced central sympathetic drive in neurogenic hypertension.
Conclusion
The active role of glial cells in the central nervous mechanisms that maintain cardiovascular homeostasis in physiological and pathological conditions is a novel concept, and the intimate details of neuroglial interactions in the brainstem are only starting to be explored. The experimental evidence reviewed here identifies two potential mechanisms of how glial cells may impart their activity on presympathetic CNS networks (both hypothalamic and brainstem), leading to maladaptive and detrimental increases in sympathetic activity associated with the development and progression of heart failure and hypertension, as follows: (i) a decrease in cerebral O2 content that causes hypoxia‐induced astroglial activation (Fig. 1); and (ii) increased production of ATP (Fig. 1) and pro‐inflammatory cytokines by activated astrocytes and microglia. Further advances in our understanding of micro‐ and astroglial function will not only reveal the role of these cells in modulating the activities of vital presympathetic networks that control the heart and the vasculature, but may also help to identify novel therapeutic modalities for the treatment of cardiovascular disease.
Additional information
Competing interests
None declared.
Author contributions
A.V.G. and N.M. drafted the manuscript. A.G.T. and S.K. revised the manuscript critically for intellectual content. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The research in our laboratories referred to in this report was funded by The Wellcome Trust and the British Heart Foundation. N.M. is a British Heart Foundation Intermediate Basic Research Science Fellow FS/13/5/29927. A.V.G. is a Wellcome Trust Senior Research Fellow. S.K. is funded by MRC MR/L020661/1 and BBSRC BB/L019396/1.
[The copyright line for this article was changed on 13th September 2016 after original online publication]
References
- Agulhon C, Fiacco TA & McCarthy KD (2010). Hippocampal short‐ and long‐term plasticity are not modulated by astrocyte Ca2+ signaling. Science 327, 1250–1254. [DOI] [PubMed] [Google Scholar]
- Andjelkovic AV, Kerkovich D & Pachter JS (2000). Monocyte:astrocyte interactions regulate MCP‐1 expression in both cell types. J Leukoc Biol 68, 545–552. [PubMed] [Google Scholar]
- Angelova PR, Kasymov V, Christie I, Sheikhbahaei S, Turovsky E, Marina N, Korsak A, Zwicker J, Teschemacher AG, Ackland GL, Funk GD, Kasparov S, Abramov AY & Gourine AV (2015). Functional oxygen sensitivity of astrocytes. J Neurosci 35, 10460–10473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano T, Mori T, Shimoda T, Shinagawa R, Satoh S, Yada N, Katsumata S, Matsuda S, Kagamiishi Y & Tateishi N (2005). Arundic acid (ONO‐2506) ameliorates delayed ischemic brain damage by preventing astrocytic overproduction of S100B. Curr Drug Targets CNS Neurol Disord 4, 127–142. [DOI] [PubMed] [Google Scholar]
- Badoer E (2010). Microglia: activation in acute and chronic inflammatory states and in response to cardiovascular dysfunction. Int J Biochem Cell Biol 42, 1580–1585. [DOI] [PubMed] [Google Scholar]
- Bevan RD (1984). Trophic effects of peripheral adrenergic nerves on vascular structure. Hypertension 6, III19–III26. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J & Volterra A (2001). CXCR4‐activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat Neurosci 4, 702–710. [DOI] [PubMed] [Google Scholar]
- Boutouyrie P, Lacolley P, Girerd X, Beck L, Safar M & Laurent S (1994). Sympathetic activation decreases medium‐sized arterial compliance in humans. Am J Physiol Heart Circ Physiol 267, H1368–H1376. [DOI] [PubMed] [Google Scholar]
- Bradley TD & Floras JS (2003. a). Sleep apnea and heart failure: Part I: obstructive sleep apnea. Circulation 107, 1671–1678. [DOI] [PubMed] [Google Scholar]
- Bradley TD & Floras JS (2003. b). Sleep apnea and heart failure: Part II: central sleep apnea. Circulation 107, 1822–1826. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Torp R, Zhang N, Storm‐Mathisen J & Ottersen OP (1990). Distribution of glutamate‐like immunoreactivity in excitatory hippocampal pathways: a semiquantitative electron microscopic study in rats. Neuroscience 39, 405–417. [DOI] [PubMed] [Google Scholar]
- Burnstock G (2007). Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev 87, 659–797. [DOI] [PubMed] [Google Scholar]
- Carlyle M, Jones OB, Kuo JJ & Hall JE (2002). Chronic cardiovascular and renal actions of leptin: role of adrenergic activity. Hypertension 39, 496–501. [DOI] [PubMed] [Google Scholar]
- Cates MJ, Dickinson CJ, Hart ECJ & Paton JFR (2012). Neurogenic hypertension and elevated vertebrobasilar arterial resistance: is there a causative link? Curr Hypertens Rep 14, 261–269. [DOI] [PubMed] [Google Scholar]
- Cates MJ, Steed PW, Abdala AP, Langton PD & Paton JFR (2011). Elevated vertebrobasilar artery resistance in neonatal spontaneously hypertensive rats. J Appl Physiol 111, 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Lin JH, Lopez‐Garcia JC, Naus CC & Nedergaard M (2000). ATP‐mediated glia signaling. J Neurosci 20, 2835–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampney RA (1994). The subretrofacial vasomotor nucleus: anatomical, chemical and pharmacological properties and role in cardiovascular regulation. Prog Neurobiol 42, 197–227. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Horiuchi J, Tagawa T, Fontes MA, Potts PD & Polson JW (2003). Medullary and supramedullary mechanisms regulating sympathetic vasomotor tone. Acta Physiol Scand 177, 209–218. [DOI] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG & Kipnis J (2012). Wild‐type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domercq M, Brambilla L, Pilati E, Marchaland J, Volterra A & Bezzi P (2006). P2Y1 receptor‐evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor‐α and prostaglandins. J Biol Chem 281, 30684–30696. [DOI] [PubMed] [Google Scholar]
- Esler M & Kaye D (2000). Measurement of sympathetic nervous system activity in heart failure: the role of norepinephrine kinetics. Heart Fail Rev 5, 17–25. [DOI] [PubMed] [Google Scholar]
- Esler M, Lambert G & Jennings G (1989). Regional norepinephrine turnover in human hypertension. Clin Exp Hypertens A 11 Suppl 1, 75–89. [DOI] [PubMed] [Google Scholar]
- Esler M, Rumantir M, Kaye D, Jennings G, Hastings J, Socratous F & Lambert G (2001). Sympathetic nerve biology in essential hypertension. Clin Exp Pharmacol Physiol 28, 986–989. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, Taves SR, Petravicz J, Casper KB, Dong X, Chen J & McCarthy KD (2007). Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron 54, 611–626. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D & Turner MB (2013). Executive summary: heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 127, 143–152. [DOI] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K & Kasparov S (2010). Astrocytes control breathing through pH‐dependent release of ATP. Science 329, 571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Llaudet E, Dale N & Spyer KM (2005. a). ATP is a mediator of chemosensory transduction in the central nervous system. Nature 436, 108–111. [DOI] [PubMed] [Google Scholar]
- Gourine AV, Llaudet E, Dale N & Spyer KM (2005. b). Release of ATP in the ventral medulla during hypoxia in rats: role in hypoxic ventilatory response. J Neurosci 25, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Wood JD & Burnstock G (2009). Purinergic signalling in autonomic control. Trends Neurosci 32, 241–248. [DOI] [PubMed] [Google Scholar]
- Grassi G (2009). Assessment of sympathetic cardiovascular drive in human hypertension: achievements and perspectives. Hypertension 54, 690–697. [DOI] [PubMed] [Google Scholar]
- Grassi G, Colombo M, Seravalle G, Spaziani D & Mancia G (1998). Dissociation between muscle and skin sympathetic nerve activity in essential hypertension, obesity, and congestive heart failure. Hypertension 31, 64–67. [DOI] [PubMed] [Google Scholar]
- Grassi G, Dell'Oro R, Seravalle G, Foglia G, Trevano FQ & Mancia G (2002). Short‐ and long‐term neuroadrenergic effects of moderate dietary sodium restriction in essential hypertension. Circulation 106, 1957–1961. [DOI] [PubMed] [Google Scholar]
- Grassi G, Giannattasio C, Cleroux J, Cuspidi C, Sampieri L, Bolla GB & Mancia G (1988). Cardiopulmonary reflex before and after regression of left ventricular hypertrophy in essential hypertension. Hypertension 12, 227–237. [DOI] [PubMed] [Google Scholar]
- Guyenet PG (2000). Neural structures that mediate sympathoexcitation during hypoxia. Respir Physiol 121, 147–162. [DOI] [PubMed] [Google Scholar]
- Guyenet PG, Stornetta RL, Bochorishvili G, Depuy SD, Burke PG & Abbott SB (2013). C1 neurons: the body's EMTs (2013). Am J Physiol Regul Integr Comp Physiol 305, 187–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Fellin T & Haydon PG (2009). Tripartite synapses: roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology 57, 343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Takano H, Dong JH & Haydon PG (2007). Synaptic islands defined by the territory of a single astrocyte. J Neurosci 27, 6473–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton NB & Attwell D (2010). Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci 11, 227–238. [DOI] [PubMed] [Google Scholar]
- Hasking GJ, Esler MD, Jennings GL, Burton D, Johns JA & Korner PI (1986). Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation 73, 615–621. [DOI] [PubMed] [Google Scholar]
- Higashino H, Niwa A, Satou T, Ohta Y, Hashimoto S, Tabuchi M & Ooshima K (2009). Immunohistochemical analysis of brain lesions using S100B and glial fibrillary acidic protein antibodies in arundic acid‐ (ONO‐2506) treated stroke‐prone spontaneously hypertensive rats. J Neural Transm 116, 1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmström KM, Marina N, Baev AY, Wood NW, Gourine AV & Abramov AY (2013). Signalling properties of inorganic polyphosphate in the mammalian brain. Nat Commun 4, 1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi J, Potts PD, Tagawa T & Dampney RA (1999). Effects of activation and blockade of P2x receptors in the ventrolateral medulla on arterial pressure and sympathetic activity. J Auton Nerv Syst 76, 118–126. [DOI] [PubMed] [Google Scholar]
- Kanbar R, DePuy SD, West GH, Stornetta RL & Guyenet PG (2011). Regulation of visceral sympathetic tone by A5 noradrenergic neurons in rodents. J Physiol 589, 903–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YM, He RL, Yang LM, Qin DN, Guggilam A, Elks C, Yan N, Guo Z & Francis J (2009). Brain tumour necrosis factor‐α modulates neurotransmitters in hypothalamic paraventricular nucleus in heart failure. Cardiovasc Res 83, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YM, Zhang ZH, Xue B, Weiss RM & Felder RB (2008). Inhibition of brain proinflammatory cytokine synthesis reduces hypothalamic excitation in rats with ischemia‐induced heart failure. Am J Physiol Heart Circ Physiol 295, H227–H236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JR, Manuck SB, Adams MR, Weingand KW & Clarkson TB (1987). Inhibition of coronary atherosclerosis by propranolol in behaviorally predisposed monkeys fed an atherogenic diet. Circulation 76, 1364–1372. [DOI] [PubMed] [Google Scholar]
- Karagiannis A, Sylantyev S, Hadjihambi A, Hosford PS, Kasparov S & Gourine AV (2015). Hemichannel‐mediated release of lactate. J Cereb Blood Flow Metab DOI: 10.1177/0271678X15611912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasymov V, Larina O, Castaldo C, Marina N, Patrushev M, Kasparov S & Gourine AV (2013). Differential sensitivity of brainstem versus cortical astrocytes to changes in pH reveals functional regional specialization of astroglia. J Neurosci 33, 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye DM, Lefkovits J, Jennings GL, Bergin P, Broughton A & Esler MD (1995). Adverse consequences of high sympathetic nervous activity in the failing human heart. J Am Coll Cardiol 26, 1257–1263. [DOI] [PubMed] [Google Scholar]
- La Rovere MT, Bigger JT Jr, Marcus FI, Mortara A & Schwartz PJ (1998). Baroreflex sensitivity and heart‐rate variability in prediction of total cardiac mortality after myocardial infarction. ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) Investigators. Lancet 351, 478–484. [DOI] [PubMed] [Google Scholar]
- La Rovere MT, Pinna GD, Maestri R, Mortara A, Capomolla S, Febo O, Ferrari R, Franchini M, Gnemmi M, Opasich C, Riccardi PG, Traversi E & Cobelli F (2003). Short‐term heart rate variability strongly predicts sudden cardiac death in chronic heart failure patients. Circulation 107, 565–570. [DOI] [PubMed] [Google Scholar]
- Leimbach WN Jr, Wallin BG, Victor RG, Aylward PE, Sundlof G & Mark AL (1986). Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation 73, 913–919. [DOI] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N & Mandel G (2011). A role for glia in the progression of Rett's syndrome. Nature 475, 497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Tang Y & Feng J (2011). Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci 89, 141–146. [DOI] [PubMed] [Google Scholar]
- Lown B & Verrier RL (1976). Neural activity and ventricular fibrillation. N Engl J Med 294, 1165–1170. [DOI] [PubMed] [Google Scholar]
- Madden CJ & Sved AF (2003). Cardiovascular regulation after destruction of the C1 cell group of the rostral ventrolateral medulla in rats. Am J Physiol Heart Circ Physiol 285, H2734–H2748. [DOI] [PubMed] [Google Scholar]
- Mansfield D, Kaye DM, Brunner La Rocca H, Solin P, Esler MD & Naughton MT (2003). Raised sympathetic nerve activity in heart failure and central sleep apnea is due to heart failure severity. Circulation 107, 1396–1400. [DOI] [PubMed] [Google Scholar]
- Marenholz I, Heizmann CW & Fritz G (2004). S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem Biophys Res Commun 322, 1111–1122. [DOI] [PubMed] [Google Scholar]
- Marina N, Abdala AP, Korsak A, Simms AE, Allen AM, Paton JFR & Gourine AV (2011). Control of sympathetic vasomotor tone by catecholaminergic C1 neurones of the rostral ventrolateral medulla oblongata. Cardiovasc Res 91, 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Ang R, Machhada A, Kasymov V, Karagiannis A, Hosford PS, Mosienko V, Teschemacher AG, Vihko P, Paton JFR, Kasparov S & Gourine AV (2015). Brainstem hypoxia contributes to the development of hypertension in the spontaneously hypertensive rat. Hypertension 65, 775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Tang F, Figueiredo M, Mastitskaya S, Kasimov V, Mohamed‐Ali V, Roloff E, Teschemacher AG, Gourine AV & Kasparov S (2013). Purinergic signalling in the rostral ventro‐lateral medulla controls sympathetic drive and contributes to the progression of heart failure following myocardial infarction in rats. Basic Res Cardiol 108, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark AL (1990). Regulation of sympathetic nerve activity in mild human hypertension. J Hypertens 8 Suppl, S67–S75. [PubMed] [Google Scholar]
- May CN, Yao ST, Booth LC & Ramchandra R (2013). Cardiac sympathoexcitation in heart failure. Auton Neurosci 175, 76–84. [DOI] [PubMed] [Google Scholar]
- Naughton MT, Benard DC, Liu PP, Rutherford R, Rankin F & Bradley TD (1995). Effects of nasal CPAP on sympathetic activity in patients with heart failure and central sleep apnea. Am J Respir Crit Care Med 152, 473–479. [DOI] [PubMed] [Google Scholar]
- Okabe Y, Takahashi T, Mitsumasu C, Kosai K, Tanaka E & Matsuishi T (2012). Alterations of gene expression and glutamate clearance in astrocytes derived from an MeCP2‐null mouse model of Rett syndrome. PLoS One 7, e35354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual O, Ben AS, Rostaing P, Triller A & Bessis A (2012). Microglia activation triggers astrocyte‐mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci USA 109, E197–E205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton JFR & Waki H (2009). Is neurogenic hypertension related to vascular inflammation of the brainstem? Neurosci Biobehav Rev 33, 89–94. [DOI] [PubMed] [Google Scholar]
- Perea G, Navarrete M & Araque A (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32, 421–431. [DOI] [PubMed] [Google Scholar]
- Pettersson K, Bejne B, Björk H, Strawn WB & Bondjers G (1990). Experimental sympathetic activation causes endothelial injury in the rabbit thoracic aorta via β1‐adrenoceptor activation. Circ Res 67, 1027–1034. [DOI] [PubMed] [Google Scholar]
- Potapenko ES, Biancardi VC, Zhou Y & Stern JE (2012). Altered astrocyte glutamate transporter regulation of hypothalamic neurosecretory neurons in heart failure rats. Am J Physiol Regul Integr Comp Physiol 303, R291–R300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapenko ES, Biancardi VC, Zhou Y & Stern JE (2013). Astrocytes modulate a postsynaptic NMDA‐GABAA‐receptor crosstalk in hypothalamic neurosecretory neurons. J Neurosci 33, 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralevic V (2000). P2 receptors in the central and peripheral nervous systems modulating sympathetic vasomotor tone. J Auton Nerv Syst 81, 205–211. [DOI] [PubMed] [Google Scholar]
- Rana I, Stebbing M, Kompa A, Kelly DJ, Krum H & Badoer E (2010). Microglia activation in the hypothalamic PVN following myocardial infarction. Brain Res 1326, 96–104. [DOI] [PubMed] [Google Scholar]
- Rifai L, Winters J, Friedman E & Silver MA (2012). Initial description of cerebral oximetry measurement in heart failure patients. Congest Heart Fail 18, 85–90. [DOI] [PubMed] [Google Scholar]
- Rodbard S & Stone W (1955). Pressor mechanisms induced by intracranial compression. Circulation 12, 883–890. [DOI] [PubMed] [Google Scholar]
- Schultz HD, Li YL & Ding Y (2007). Arterial chemoreceptors and sympathetic nerve activity: implications for hypertension and heart failure. Hypertension 50, 6–13. [DOI] [PubMed] [Google Scholar]
- Schultz HD & Sun SY (2000). Chemoreflex function in heart failure. Heart Fail Rev 5, 45–56. [DOI] [PubMed] [Google Scholar]
- Shi P, Diez‐Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C & Raizada MK (2010). Brain microglial cytokines in neurogenic hypertension. Hypertension 56, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Grobe JL, Desland FA, Zhou G, Shen XZ, Shan Z, Liu M, Raizada MK & Sumners C (2014). Direct pro‐inflammatory effects of prorenin on microglia. PLoS One 9, e92937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson P (1983). Norepinephrine‐stimulated hypertrophy of cultured rat myocardial cells is an alpha1 adrenergic response. J Clin Invest 72, 732–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siński M, Lewandowski J, Przybylski J, Bidiuk J, Abramczyk P, Ciarka A & Gaciong Z (2012). Tonic activity of carotid body chemoreceptors contributes to the increased sympathetic drive in essential hypertension. Hypertens Res 35, 487–491. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV (2015). Astrogliosis. Cold Spring Harb Perspect Biol 7, a020420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyer KM (1994). Annual review prize lecture. Central nervous mechanisms contributing to cardiovascular control. J Physiol 474, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack AM, Sawyer WB, Hughes JH, Platt KB & Loewy AD (1989). A general pattern of CNS innervation of the sympathetic outflow demonstrated by transneuronal pseudorabies viral infections. Brain Res 491, 156–162. [DOI] [PubMed] [Google Scholar]
- Sun MK & Reis DJ (1994). Hypoxia selectively excites vasomotor neurons of rostral ventrolateral medulla in rats. Am J Physiol Regul Integr Comp Physiol 266, R245–R256. [DOI] [PubMed] [Google Scholar]
- Sun MK, Wahlestedt C & Reis DJ (1992). Action of externally applied ATP on rat reticulospinal vasomotor neurons. Eur J Pharmacol 224, 93–96. [DOI] [PubMed] [Google Scholar]
- Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, Lovatt D, Han X, Smith Y & Nedergaard M (2013). Glutamate‐dependent neuroglial calcium signaling differs between young and adult brain. Science 339, 197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Lane S, Korsak A, Paton JFR, Gourine AV, Kasparov S & Teschemacher AG (2014). Lactate‐mediated glia‐neuronal signalling in the mammalian brain. Nat Commun 5, 3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassoni D, Avola R, Di Tullio MA, Sabbatini M, Vitaioli L & Amenta F (2004). Increased expression of glial fibrillary acidic protein in the brain of spontaneously hypertensive rats. Clin Exp Hypertens 26, 335–350. [DOI] [PubMed] [Google Scholar]
- Trzebski A, Tafil M, Zoltowski M & Przybylski J (1982). Increased sensitivity of the arterial chemoreceptor drive in young men with mild hypertension. Cardiovasc Res 16, 163–172. [DOI] [PubMed] [Google Scholar]
- Vallbo AB, Hagbarth KE, Torebjörk HE & Wallin BG (1979). Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev 59, 919–957. [DOI] [PubMed] [Google Scholar]
- van de Borne P, Mark AL, Montano N, Mion D & Somers VK (1997). Effects of alcohol on sympathetic activity, hemodynamics, and chemoreflex sensitivity. Hypertension 29, 1278–1283. [DOI] [PubMed] [Google Scholar]
- Volterra A & Meldolesi J (2005). Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 6, 626–640. [DOI] [PubMed] [Google Scholar]
- Waki H, Gouraud SS, Maeda M & Paton JFR (2008). Gene expression profiles of major cytokines in the nucleus tractus solitarii of the spontaneously hypertensive rat. Auton Neurosci 142, 40–44. [DOI] [PubMed] [Google Scholar]
- Waki H, Liu B, Miyake M, Katahira K, Murphy D, Kasparov S & Paton JFR (2007). Junctional adhesion molecule‐1 is upregulated in spontaneously hypertensive rats: evidence for a prohypertensive role within the brain stem. Hypertension 49, 1321–1327. [DOI] [PubMed] [Google Scholar]
- Wells JA, Christie IN, Hosford PS, Huckstepp RT, Angelova PR, Vihko P, Cork SC, Abramov AY, Teschemacher AG, Kasparov S, Lythgoe MF & Gourine AV (2015). A critical role for purinergic signalling in the mechanisms underlying generation of BOLD fMRI responses. J Neurosci 35, 5284–5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Oliveira‐Sales EB, McBride F, Liu B, Hewinson J, Toward M, Hendy EB, Graham D, Dominiczak AF, Giannotta M, Waki H, Ascione R, Paton JFR & Kasparov S (2012). Upregulation of junctional adhesion molecule‐A is a putative prognostic marker of hypertension. Cardiovasc Res 96, 552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoccal DB, Huidobro‐Toro JP & Machado BH (2011). Chronic intermittent hypoxia augments sympatho‐excitatory response to ATP but not to l‐glutamate in the RVLM of rats. Auton Neurosci 165, 156–162. [DOI] [PubMed] [Google Scholar]
- Zubcevic J, Waki H, Raizada MK & Paton JFR (2011). Autonomic‐immune‐vascular interaction: an emerging concept for neurogenic hypertension. Hypertension 57, 1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]