

Abstract
Background
Larvae of the Holarctic mayfly genus Rhithrogena Eaton, 1881 (Ephemeroptera, Heptageniidae) are a diverse and abundant member of stream and river communities and are routinely used as bio-indicators of water quality. Rhithrogena is well diversified in the European Alps, with a number of locally endemic species, and several cryptic species have been recently detected. While several informal species groups are morphologically well defined, a lack of reliable characters for species identification considerably hampers their study. Their relationships, origin, timing of speciation and mechanisms promoting their diversification in the Alps are unknown.
Results
Here we present a species-level phylogeny of Rhithrogena in Europe using two mitochondrial and three nuclear gene regions. To improve sampling in a genus with many cryptic species, individuals were selected for analysis according to a recent DNA-based taxonomy rather than traditional nomenclature. A coalescent-based species tree and a reconstruction based on a supermatrix approach supported five of the species groups as monophyletic. A molecular clock, mapped on the most resolved phylogeny and calibrated using published mitochondrial evolution rates for insects, suggested an origin of Alpine Rhithrogena in the Oligocene/Miocene boundary. A diversification analysis that included simulation of missing species indicated a constant speciation rate over time, rather than any pronounced periods of rapid speciation. Ancestral state reconstructions provided evidence for downstream diversification in at least two species groups.
Conclusions
Our species-level analyses of five gene regions provide clearer definitions of species groups within European Rhithrogena. A constant speciation rate over time suggests that the paleoclimatic fluctuations, including the Pleistocene glaciations, did not significantly influence the tempo of diversification of Alpine species. A downstream diversification trend in the hybrida and alpestris species groups supports a previously proposed headwater origin hypothesis for aquatic insects.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-016-0758-1) contains supplementary material, which is available to authorized users.
Keywords: Dated phylogeny, Speciation, Diversification, Lineage-through-time, Pleistocene glaciations, Freshwater insects, Headwater, Mayfly, Ephemeroptera
Background
Mountainous areas are hotspots of biodiversity and endemism (e.g. [1, 2]), and headwater streams are increasingly recognized for their contribution to freshwater biodiversity [3–5]. In Europe, the Alps have strongly influenced the evolution of many taxa, acting as a barrier or refuge for populations during climatic fluctuations [6, 7]. Pleistocene glaciations are recognized as important promoters of population divergence and ultimately speciation through repeated isolation in allopatric refugia, increasing speciation rates in a variety of organisms (e.g. [6, 8–11]). However, the role of recent glaciations in speciation has also been largely debated [12–17], and a number of studies favor a model of constant diversification rate during the Quaternary ice ages (e.g. [18–22]).
The processes that drive speciation in aquatic insects remain poorly understood, despite the significant contribution of aquatic insects to global insect diversity [23]. Several studies of aquatic insects have uncovered evidence for the important influence of glaciations on divergence patterns at the level of both populations (e.g. [24–27]) and species (e.g. [28–30]). In mayflies (Ephemeroptera), population divergence within particular species has often been interpreted to result from their separation into distinct refugia during the Pleistocene (e.g. [31, 32]). Although some authors have suggested that Pleistocene glaciations promoted mayfly speciation in the Palearctic [33] and Australasia [34], extensive analyses of speciation rates during the Quaternary are lacking. Earlier workers proposed that the ancestral position of cold-adapted aquatic insect lineages was in headwaters, with subsequent downstream diversification into warmer waters (e.g. [35–37]). This headwater origin hypothesis has been supported by more recent studies of European caddisflies (Trichoptera, Hydropsychidae) [38, 39], and in some, but not all, lineages of tropical diving beetles (Coleoptera, Dytiscidae) in New Guinea [40]. In contrast, there was no support for the hypothesis in a study of tropical heptageniid mayflies (Heptageniidae) in Madagascar [41]. Findings to date suggest that the headwater origin hypothesis requires further testing and refinement with additional phylogenetic studies.
Rhithrogena Eaton, 1881 (Ephemeroptera, Heptageniidae) is a predominantly Holarctic mayfly genus [42]. Highest Rhithrogena species diversity is found in the Palaearctic [43], with a few species in northern Neotropical and Oriental regions [33, 44, 45]. It is the second-most species-rich mayfly genus, with 148 recognized species to date [46], 74 of which have been recorded in Europe [47, 48]. European species are particularly diverse in the Alpine region, with more than 30 described species, half of which are thought to be local endemics. Rhithrogena immature stages are often an abundant and diverse component of the benthic invertebrate community, and are routinely used as bio-indicators of water quality [49]. They inhabit most types of running waters, from large floodplain rivers to small and mountainous headwater streams. However, many species are cold-tolerant, with larvae living in fast-flowing, well-oxygenated streams and rivers [43], suggesting that adaptation to such environmental conditions may have played an important role in their diversification in the Alps.
The identification of many European Rhithrogena species remains challenging because of a lack of reliable morphological characters in both flying adults and aquatic immature stages (e.g. [43, 50]). A recent study of European species membership using one mitochondrial (mt hereafter) and one nuclear (nuc hereafter) gene fragment highlighted the occurrence of several cryptic species within the genus [51]. Phenotypic plasticity, convergence, or local adaptation could explain such ambiguities (e.g. [52–55]), as could a recent origin of species and incomplete morphological differentiation. Rhithrogena species are arranged into several “species groups” [56–58] that are themselves easily identifiable. Although Vuataz et al. [51] recovered five of six studied species groups to be monophyletic, relationships within and among species groups remained unclear.
Here we used two mitochondrial and three nuclear gene regions to reconstruct a species-level phylogeny of European Alpine Rhithrogena. Our first aim was to resolve the relationships among Rhithrogena species and species groups, where a previous phylogeny showed insufficient resolution. We then determined whether their origin, diversification and changes in speciation rates could be linked to paleoclimatic events, and whether a phylogeny of Rhithrogena provides support for a headwater origin hypothesis in the European Alps.
Methods
Sampling and sequencing
Larval Rhithrogena were collected from streams between March 2006 and May 2010 at 35 localities in the European Alps (France, Switzerland, Austria, Italy) and from additional localities in the Pyrenees and Corsica (France), the Jura Mountains (Switzerland), the Apennine Mountains and Sardinia (Italy), the Bohemian Forest and the Sudetes Mountains (Czech Republic), the Apuseni Mountains (Romania), the Occidental Carpathians (Poland), the Dinarides (Montenegro), the Cambrian (Wales), and the Kroumir Mountains (Tunisia; Fig. 1). Individuals were preserved in pure ethanol in the field, and stored in the laboratory at -20 °C in fresh pure ethanol. Ventral-, dorsal- and lateral-view photographs of each individual were taken prior to DNA extraction as described in Vuataz et al. [51]. For phylogenetic analysis, we first chose one representative from each of the 31 GMYC species reported in Vuataz et al. [51]. Of these, 27 were vouchers from Vuataz et al. [51], and four were newly sequenced for this study. Seven additional specimens were then included, based on morphological and mt evidence (data not shown) that they were distinct species not included in Vuataz et al. [51]. In each case, we selected individuals 1) with the most available sequence data in order to obtain a data matrix that was as complete as possible, and 2) sampled from type localities (topotypes) when available (see Additional file 1: Table S1, for a more detailed description of the sampling).
Fig. 1.
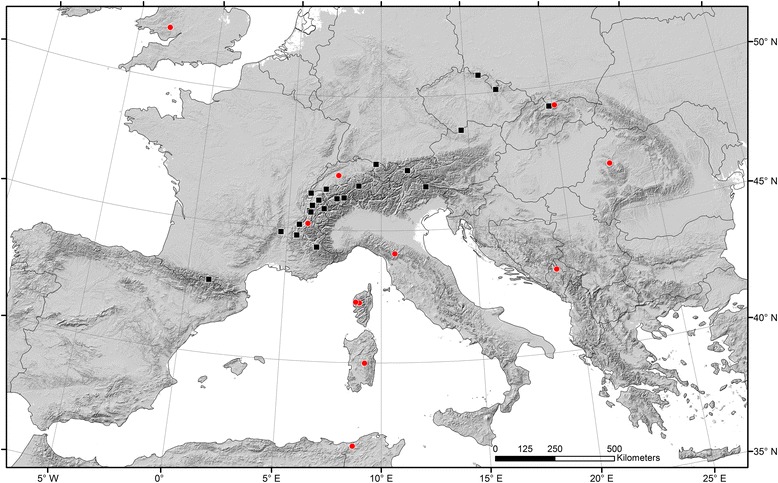
Map of sampling localities from which Rhithrogena individuals were collected. Red circles indicate newly sampled localities; black squares indicate data from Vuataz et al. [51]
For all individuals, five gene fragments were sequenced for phylogenetic analyses: mt cytochrome c oxidase subunit I (cox1) and the large ribosomal subunit 16S rRNA; nuc phosphoenolpyruvate carboxykinase (PEPCK), elongation factor 1-alpha (EF-1α) and wingless (wg). Cox1 and PEPCK were amplified and sequenced as described in Vuataz et al. [51]. The other genes were amplified and sequenced as described in Vuataz et al. [41].
Sequence alignment and models of sequence evolution
Forward and reverse sequencing reads were assembled and edited using CodonCode Aligner v. 3.7.1.1 (CodonCode Corporation, Dedham, MA). Heterozygous sites (in nuc genes) were identified as double peaks within the chromatograms and were scored according to the IUPAC code. Sequence alignment was performed using MAFFT [59] as implemented in Jalview v. 2.6.1 [60]. Sequence alignment was straightforward for cox1, 16S, EF-1α, wg and the coding regions of PEPCK because there were no insertions or deletions other than a 1-bp indel in the 16S sequences. A length-variable non-coding (intron) PEPCK section was detected using amino acid translation, with boundaries identified following the GT-AG rule (see [51]). A total of 201 bp within the intron could be unambiguously aligned (123 bp at the 5’ end, 14 bp internal and 64 bp at the 3’ end) and were retained for analyses. The remaining intron region was removed from the PEPCK alignment.
Identical haplotypes (cox1, 16S) and genotypes (PEPCK, EF-1α, wg) were removed from each alignment using Collapse v. 1.2 [61], and the number of polymorphic and parsimony-informative sites were calculated in MEGA v. 5.03 [62]. The best evolutionary model was selected for each gene fragment (Table 1) following the second-order Akaike information criterion (AICc) implemented in MrAIC v. 1.4.4 [63] under the option using the models implemented in MrBayes. In order to accommodate different substitution rates among codon positions, we used partitioned models of evolution for cox1 and PEPCK (e.g. [64, 65]). Following the partition scheme from Vuataz et al. [51], we examined cox1 in two partitions: one with first and second codon positions and one with third positions (1 + 2, 3); for PEPCK, we used one partition with first and second codon positions and a second with third positions and the intron (1 + 2, 3 + intron).
Table 1.
Sequence variation for each gene region and for the three concatenated matrices (mitochondrial: cox1 + 16S; nuclear: PEPCK + EF-1α + wg; combined: all five gene fragments combined)
| Data set (n = 38) | bp | K | S | S i | %S i | Model |
|---|---|---|---|---|---|---|
| cox1 | 658 | 38 | 237 | 217 | 33 | GTR + Γ + I |
| 16S | 511 | 36 | 67 | 45 | 9 | GTR + Γ + I |
| PEPCK | 557 | 37 | 154 | 94 | 17 | HKY + Γ |
| EF-1α | 139 | 24 | 27 | 12 | 9 | K2P + Γ |
| wg | 463 | 27 | 31 | 11 | 2 | GTR + I |
| mitochondrial | 1169 | 38 | 304 | 262 | 22 | |
| nuclear | 1147 | 37 | 212 | 120 | 10 | |
| combined | 2316 | 38 | 516 | 383 | 17 |
n = number of sequences, bp = size of aligned matrices, K = number of haplotypes (cox1, 16S) or genotypes (PEPCK, EF-1α, wg) or a combination of both (combined), S = number of polymorphic sites, S i = number of parsimony-informative sites, Model = best evolutionary model selected following the second-order Akaike information criterion (AICc). Concatenated matrices were examined with partitioned analysis using the best model for each gene fragment (see Methods)
Tree reconstruction
Our alignments were examined under both coalescent-based species tree and supermatrix (concatenated) approaches. Species tree reconstructions were carried out under a multispecies coalescent framework [66, 67] as implemented in *BEAST v. 2.3.2 [68]. As outgroup for all reconstructions, we used Cinygmula McDunnough, 1933, which belongs to the same subfamily as Rhithrogena (Rhithrogeninae; see [44]). The species tree reconstructions were performed using the haplotype alignments. For this, the genotypes of the nuclear gene alignments (PEPCK, EF-1α, wg) were phased using the probabilistic Bayesian algorithm implemented in PHASE v. 2.1.1 [69, 70] with a cutoff value of 0.6 [71, 72]. One site originally coded as ‘D’ was implemented as ‘N’. Input and output files were formatted using the scripts from SeqPHASE [73]. Heterozygous sites that could not be resolved were coded as ambiguity codes and remained in the data set for subsequent sequence analyses. All matrices were re-aligned after phasing with MAFFT. The individuals were a priori assigned to GMYC species (see Additional file 1: Table S1). As models of sequence evolution, we initially implemented the model combination selected by AICc (see Table 1). Since this specification resulted in poor sampling of several parameters (ESS < 80), even when running up to two billion generations, we used for each partition the model HKY + Γ (sensu [65]). The maximum clade credibility trees produced under both model combinations resulted in very similar topologies, which only silghtly differed in poorly supported terminal nodes (posterior probabilities < 0.4) within the semicolorata group (data not shown). We used a relaxed uncorrelated lognormal clock for gene tree estimation at each locus and a Yule speciation-process prior with two independent runs of 900 million generations with sampling for each every 90,000 generations. The two runs were combined in LogCombiner v. 2.3.2 [68] with a burnin of 10 %, whereby all parameters reached ESS > 200. The maximum clade credibility trees were obtained using TreeAnnotator v. 2.4.1 [68] applying a burnin of 40 %.
For the supermatrix-based approach, Bayesian inference (BI) and maximum likelihood (ML) tree reconstructions were conducted using MrBayes v. 3.1.2 [74] and Garli v. 2.01 [75, 76], respectively. We first performed separate tree searches on mtDNA (cox1, 16S) and nDNA (PEPCK, EF-1α, wg). After detecting no major incongruence (see Additional file 2: Figure S1 and Additional file 3: Figure S2), we ran a concatenated analysis using all five gene fragments. In all analyses, each gene fragment was used as a partition in addition to the cox1 and PEPCK partition scheme defined in the previous section. For each BI analysis, two independent analyses of four MCMC chains were run for 20 million generations sampling trees every 1000 generations. The stationary nucleotide frequencies, alpha shape parameter of the gamma distribution, relative rate of substitution and proportion of invariant sites were unlinked across partitions, and the ratepr command was set to variable (see [77]). Two million generations were discarded as a burnin after visually verifying that likelihood curves had flattened-out and that the independent runs converged using Tracer v. 1.6 [78]. Maximum likelihood phylogenetic inference was performed with 1000 bootstrap replicates, which were summarized in SumTrees v. 4.0.0 [79]. Heuristic searches were used to find the topology with the best likelihood score. Thereby, the searches were conducted using automatic termination, after a maximum of five million generations, or, alternatively, after 10,000 generations without significant (p < 0.01) improvement of scoring topology.
Molecular dating and diversification
We chose a commonly used mt mutation rate of 0.0115 substitutions per site per million years (My hereafter), equivalent to 2.3 % divergence per My [80], to date diversification events. This is because no fossils, geological/paleoclimatic events or independent studies could be used to calibrate tree nodes for Rhithrogena. This widely used rate (see [81]) was calculated from studies of arthropods and is based on both protein-coding and ribosomal mt genes. Our concatenated mt data set (cox1 + 16S) was used for branch-length estimation using a Bayesian relaxed (uncorrelated lognormal) molecular clock approach implemented in BEAST v. 1.8.3 [82]. We initially used a GTR + Γ + I model of evolution for each partition, as selected by AICc (see Table 1). Since this specification resulted in poor sampling of several parameters (ESS < 50), we used a HKY + Γ + I model for each partition, which was the second-best model according to AICc (data not shown). A Yule process was preferred because it provides useful tree priors for species-level studies [83]. The clock models and the trees were linked for the concatenated mt data set. The substitution models of evolution were implemented separately (i.e. unlinked) for each of the three partitions (cox1 codon position 1 + 2; cox1 codon position 3; 16S), and substitution rate parameters and base frequencies were unlinked across cox1 partitions. All base frequencies were estimated from the data, the number of gamma categories was set to six, and a random starting tree was used. Well-supported nodes (BI posterior probabilitiy (PP) > 0.95 and ML bootstrap support (BS) > 80) of the concatenated mt phylogeny were constrained as monophyletic (18 constrained nodes; see Fig. 3 and Additional file 4: Figure S3). For node constraints, we favored the concatenated over the coalescent-base phylogeny, because it was the best supported phylogeny overall (see Results and Fig. 2). All other options and priors were set to default. Two independent MCMC chains were run for 100 million generations and sampled every 1000 generations, resulting in 100,000 trees for each run. Run convergence was verified using Tracer (as above) and the first 10,000 trees were then discarded from each run as a burnin, whereby all parameters reached ESS > 2000. Log and tree files were combined using LogCombiner v. 1.8.3 [82], resulting in 180,000 trees in the combined analyses. The maximum clade credibility tree was built with TreeAnnotator v. 1.8.3 [82] with all options set to default.
Fig. 3.
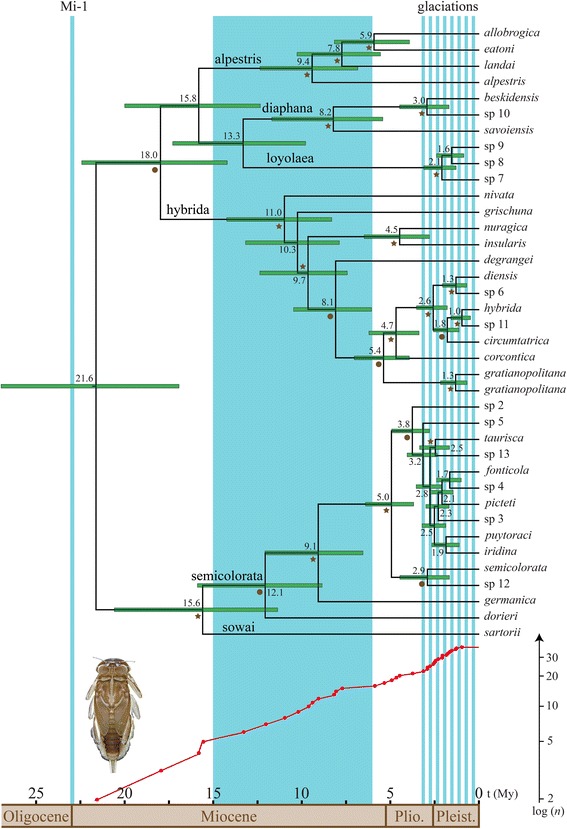
Ultrametric mitochondrial concatenated (cox1 + 16S) phylogeny (top) obtained using a relaxed lognormal molecular clock and a standard insect mutation rate of 0.0115 substitution/site/My in BEAST, and the corresponding LTT plot of the empirical data (bottom), with vertical axis (number of GMYC species) in logarithmic scale. For each node, estimated ages (horizontal axis, time before present in My) are given. Horizontal green bars indicate the 95 % highest posterior density interval. Stars indicate lineages that were constrained to be monophyletic according to the mt + nuc concatenated phylogeny (i.e. PP > 0.95 and BS > 80; see Fig. 2 and Additional file 4: Figure S3); circles indicate PP > 0.9. The blue zone represents a period of global climatic cooling trend, empty zones warming trends, following Zachos et al. [109]. Blue vertical bars symbolize the Oligocene/Miocene boundary glacial maximum (Mi-1; 23 Mya) and the Quaternary glacial cycles (glaciations; 3.2 Mya to present). Species groups are specified above corresponding branches
Fig. 2.
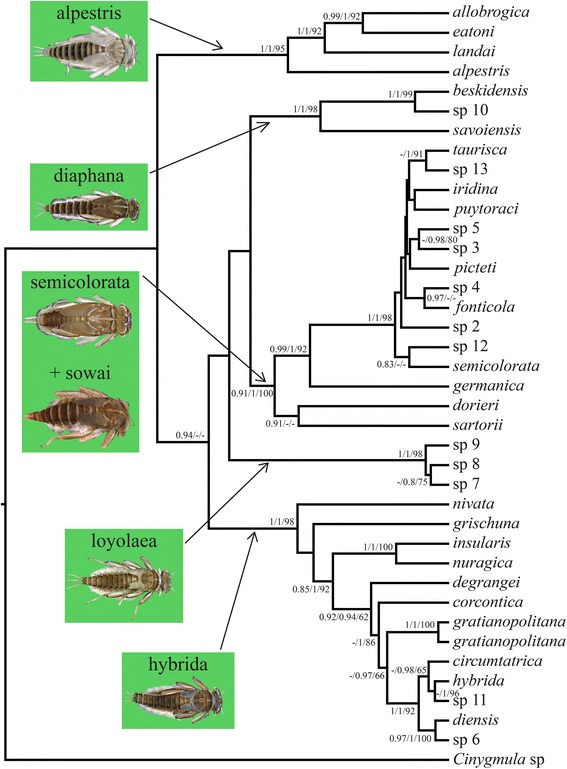
Coalescent-based chronogram (*BEAST maximum clade credibility tree) reconstructed from the five-gene data set. Species groups are indicated with green shading, including a photograph of a species group member. For each node, *BEAST Bayesian posterior probability (*PP), MrBayes Bayesian posterior probability (PP), and maximum likelihood bootstrap support (BS) values are given (*PP/PP/BS) only if posterior probabilities > 0.8 and bootstrap support > 60
The diversification rate of Rhithrogena lineages was examined using γ-statistics [84] and a birth-death likelihood (BDL) analysis [85]. An important problem with empirical diversification analyses is incomplete taxon sampling, which could lead to a variety of misinterpretations of phylogenies (e.g. [86–88]). One recent advance for handling missing species is the CorSiM approach [89, 90], which is based on the simulation of missing splits under speciation/extinction models. To correct for incomplete sampling in our empirical Rhithrogena phylogeny, we used the CorSiM approach to simulate 1000 trees with 36 additional (i.e., missing) species. This brought the total to 74 species, which corresponds to the currently accepted level of species diversity of the genus in Europe [47, 48]. We assumed a random sampling (sensu [90]) because we focused on Alpine species rather than selected species from each species group. The γ-statistic was calculated using the function gamStat in the laser v. 2.4-0 [85] package for R v. 3 [91]. Values of γ > 0 indicate an increasing number of nodes toward the tips of the phylogenetic tree. The BDL analysis with two constant-rate models (Yule, birth-death) and three variable-rate models (logistic density-dependence, exponential density-dependence, two-rate variant of the Yule model) was performed using the function fitdAICrc in laser.
Ancestral state reconstruction
To test whether European Rhithrogena fit the headwater origin hypothesis with subsequent downstream diversification, we reconstructed the ancestral altitude range of the two species groups hybrida and alpestris using the current species distributions as raw data. The other species groups were not included in the ancestral state analyses for specific reasons: the semicolorata group phylogeny was poorly resolved (see Results), the diaphana group is mainly Mediterranean and thus most species were not included in our Alpine-oriented sampling, and all members of the loyolaea group live exclusively at high elevation. In the absence of published altitude ranges of Rhithrogena species at the European scale, we used our own extensive sampling (including unpublished data) to assign elevational distributions. For each species, we considered our minimum and maximum altitude records as a proxy for actual altitude ranges (Additional file 1: Table S3). Subtrees of the hybrida and alpestris species groups were extracted from both our species tree chronogram (coalescent-based approach), and concatenated ML phylogram (supermatrix approach). We then performed residual maximum likelihood (REML) ancestral state reconstructions [92] of elevation on both topologies using the ace function of the ape [93] package for R, which assumes a Brownian motion model of character evolution implemented for continuous variables.
Results
Data set characteristics
The concatenated data matrix of five gene fragments was > 99 % complete. There were no missing data in the cox1, 16S and wg matrices. Missing PEPCK data resulted from seven sequences that lacked 3’ and/or 5’ ends or a small intron section (261 missing characters in the matrix). There was a single EF-1α sequence for which a small internal section of 11 bp was ambiguous. A total of 103 nucleotides in the nuc data set were heterozygous. The concatenated mt data set contained 59 % of the total variation, primarily found in the third codon positions of the cox1 fragment (68 % of the mt variation). Most of the nuc variation (68 %) was found in the third codon positions and in the intron section of PEPCK. The percentage of parsimony-informative sites within each gene fragment ranged from 33 % (cox1) to 2 % (wg; Table 1). Two individuals had identical nuc genotypes (voucher 110FRSAGT and 111FRSAGT; Additional file 1: Table S1), whereas all individuals had distinct mt haplotypes.
Phylogenetic reconstruction
All four reconstructions, namely the mt + nuc coalescent-based (Fig. 2), the mt concatenated (Additional file 2: Figure S1), the nuc concatenated (Additional file 3: Figure S2), and the mt + nuc concatenated (Additional file 4: Figure S3), produced five well supported monophyletic species groups: (1) alpestris, (2) diaphana, (3) semicolorata + Rh. germanica + Rh. sartorii, (4) loyolaea, and (5) hybrida + hercynia + Rh. insularis + Rh. nuragica. The only variation occurred in the placement of Rh. sartorii from Tunisia, the sole representative of the sowai species group included here (see Discussion). In the coalescent-based tree, Rh. sartorii + Rh. dorieri formed a sister clade to the semicolorata species group. In the mt concatenated tree, Rh. sartorii was sister to the semicolorata species group. In the other reconstructions, its placement was equivocal. The alpestris species group was sister clade to the others in the mt + nuc coalescent-based tree, but all other reconstructions did not resolve relationships among the species groups.
Species-level relationships within the alpestris, diaphana and loyolaea species groups were fully congruent in all reconstructions, with increased node support in the mt + nuc coalescent-based and mt + nuc concatenated reconstructions compared to the mt and nuc concatenated reconstructions. Species-level relationships within the semicolorata species group were generally poorly supported in all reconstructions, but with Rh. germanica, Rh. dorieri and Rh. sartorii well supported as sister taxa to the other species group members in all reconstructions. Species-level relationships within the hybrida species group were weak overall in the mt + nuc coalescent-based, in the mt concatenated and in the nuc concatenated reconstructions, but were fully resolved and well supported in the mt + nuc concatenated reconstruction; the only exception was the equivocal relationships between Rh. nivata and Rh. dorieri, well supported as sister taxa to the other species group members.
Dating and diversification
The root of our phylogenetic tree was dated at 21.6 million years ago (Mya hereafter), with a 95 % highest posterior density interval (95 % HPD hereafter) of 27.0 – 16.9 Mya. This node age roughly corresponds to the Oligocene-Miocene boundary (Fig. 3). Most species group diversifications occurred in the middle and late Miocene: semicolorata (12.1 Mya; 95 % HPD 15.9 – 8.9 Mya), hybrida (11.0 Mya; 95 % HPD 14.3 – 8.3 Mya), alpestris (9.4 Mya; 95 % HPD 12.4 – 6.9 Mya), and diaphana (8.2 Mya; 95 % HPD 11.7 – 5.4 Mya). The loyolaea species group diversification appeared notably younger, dated at 2.1 Mya (95 % HPD 3.1 – 1.3 Mya), in the Pleistocene. Examined globally, 17 of 37 (46 %) speciation events (i.e., nodes) occurred between 3.1 Mya (late Pliocene) and 1.0 Mya (early Pleistocene), broadly overlapping the Quaternary glaciations (Fig. 3). Terminal nodes from this recent diversification include Rh. diensis, Rh. hybrida, Rh. circumtatrica, Rh. gratianopolitana, Rh. beskidensis, Rh. semicolorata, Rh. taurisca, Rh. puytoraci, Rh. iridina, Rh. picteti, and Rh. fonticola.
Speciation and extinction rates calculated using TreePar (bd.shifts.optim() function) and used for tree simulations with CorSiM were λ = 0.24 and μ = 0.08. The data set completed by simulation (Fig. 4) was characterised by γ = 1.07 (SD 0.44, one-tailed p = 0.82), indicating that rate constancy was not rejected. The BDL analyses indicated a preference for a Yule-2-rates model in 62 % of data sets (AIC -113.21; SD 7.92; Additional file 1: Table S4), with a decrease in the diversification rate at 0.22 Mya, corresponding to the most recent branching events in the clock-constrained tree (Fig. 4). A Yule (pure birth) model was preferred in 33 % of data sets (AIC -112.18; SD 6.73) and a birth-death model in 6 % of data sets (AIC -111.84; SD 7.78). The empirical LTT plot of the mt ultrametric tree suggested an increase in speciation rate curve occurring from ca. 3.0 to 1.0 Mya in the late Pliocene and early Pleistocene (Figs. 5, bottom and 6). The pattern departure of simulated (74-taxon) trees from the empirical (38-taxon) tree suggested that deeper nodes were oversampled in our empirical tree (see Fig. 1 in [90]).
Fig. 4.
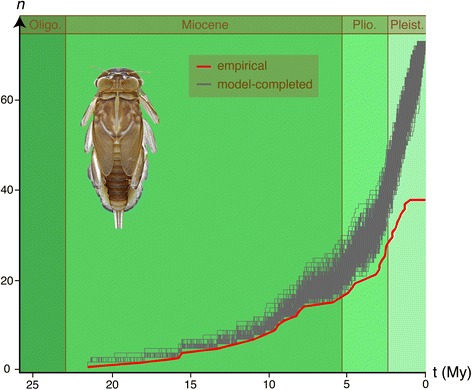
LTT plots of the European Rhithrogena, showing the number of GMYC species (n) versus time before present (t, in My). Red line corresponds to the empirical data (BEAST mitochondrial concatenated (cox1 + 16S) incomplete phylogeny), grey lines to the 1000 simulated complete phylogenies
Fig. 5.
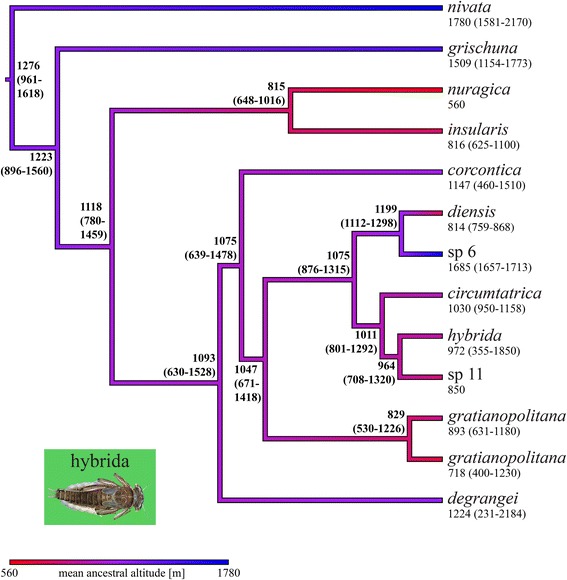
Coalescent-based chronogram (*BEAST maximum clade credibility tree) of the hybrida species group extracted from the global five-gene species tree, with ancestral altitudes reconstructed using the REML method. Mean ancestral altitudes (with minimum and maximum ancestral altitudes between brackets) are indicated for each node. Mean altitudes (with altitude ranges between brackets) extracted from authors’ own records (see Additional file 1: Table S3), and used as raw data for the ancestral altitude reconstruction, are given below each terminal GMYC species. The color gradient symbolizing the mean altitude gradient along the tree branches was obtained using the contMap function of the phytools package [129] for R
Fig. 6.
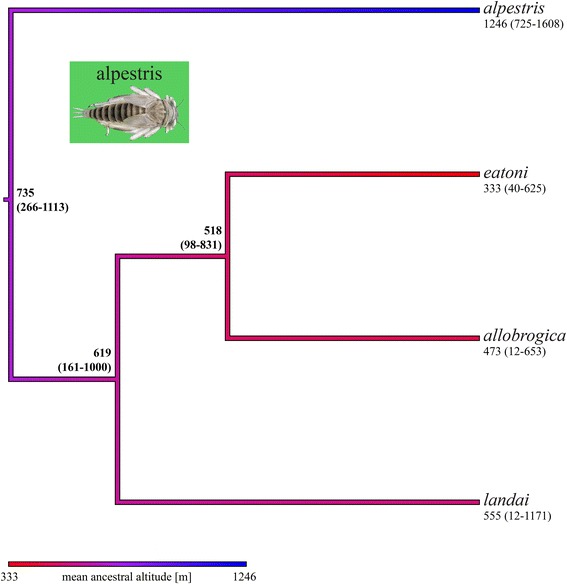
Coalescent-based chronogram (*BEAST maximum clade credibility tree) of the alpestris species group extracted from the global five-gene species tree, with ancestral altitudes reconstructed using the REML method. The altitudes and the color gradient are indicated as in Fig. 5
Ancestral state reconstruction
The ancestral state reconstructions of elevation indicated a trend of decreasing altitude ranges toward the present for the species tree chronograms and for the concatenated ML phylograms in the hybrida and alpestris species groups. For the ancestral species of the hybrida group, the species tree chronogram (Fig. 5) indicated a mean altitude of 1276 m (range 961–1618 m), and the concatenated ML phylogram (Additional file 5: Figure S4) a mean altitude of 1343 m (range 966–1732 m), suggesting an origin of the group in mid to high-elevation streams. For the ancestral species of the alpestris group (Fig. 6), the species tree chronogram indicated a mean altitude of 735 m (range 266–1113 m), and the concatenated ML phylogram (Additional file 6: Figure S5) a mean altitude of 846 m (range 365–1233 m), suggesting an origin of the group in low to mid-elevation streams.
Discussion
Species groups
Several species groups within European Rhithrogena have been proposed based on larval and adult morphology (e.g. [56, 57, 94]). The alpestris, diaphana, loyolaea, semicolorata (including germanica) and hybrida (including hercynia) groups were recently recovered as monophyletic entities in independent mt (cox1) and nuc (PEPCK) gene tree reconstructions [51]. Based on additional gene fragments, increased taxon sampling (Additional file 1: Table S1), and a coalescent-based approach applied here, these findings were confirmed. We also included Rh. insularis from Corsica and Rh. nuragica from Sardinia, both members of an additional putative species group, insularis [95], but both were recovered here within the hybrida species group. The only other genetic analysis to date [58] used allozymes and a genetic-distance tree to divide Rhithrogena into only two species groups. Our study included most species from the Zurwerra et al. [58] study and while broadly congruent, notably within hybrida and semicolorata species groups, there were important differences. Specifically, Zurwerra et al. [58] grouped members of alpestris and hybrida with the loyolaea species group, as well as semicolorata with diaphana, whereas we found clear evidence for their separation.
Rhithrogena sartorii, recently described from Tunisia [96], was thought to share morphological characteristics with the insularis species group [96], although a more recent examination of mounted specimens and different characters, including the shape of the first gill plica and the spines on the upper face of the femora, related this species to members of the sowai species group (M. Sartori, unpublished data). This group is well diversified in the Mediterranean area, particularly the Iberian and Balkan peninsulas [97, 98], indicating Rh. sartorii is a marginal representative. Our mt concatenated phylogenies recovered Rh. sartorii as sister taxon to the semicolorata species group, whereas the other reconstructions included it in the semicolorata group or were inconclusive. A phylogenetic analysis including more members of the sowai species group would help to determine whether the group is monophyletic, and whether it should be fused with semicolorata.
Timing of diversification
For molecular dating, we used a previously estimated evolution rate of mtDNA nucleotide substitution [80] because reliable estimates of node ages are difficult to obtain for mayflies. The only known fossils of Rhithrogena were described by Demoulin [99] from Baltic Amber (40 – 50 Mya; e.g. [100]); however, their attribution to Rhithrogena was contested by Kluge [101], who noted that Heptageniidae genera are based on larval characteristics rather than the adult features used by Demoulin [99]. Geological events, such as island formation, are also problematic. Our analysis revealed sister taxa on neighboring islands (Rh. insularis on Corsica and Rh. nuragica on Sardinia), but using the separation of the Corso-Sardinian microplate from the continent or the separation of Sardinia from Corsica as calibration points is potentially problematic. There is strong evidence for overseas dispersal in the two mayfly families examined to date: Heptageniidae [41] and Baetidae [102, 103]. Although there is evidence for diverging rates between lineages (e.g. [104–106]), the commonly used mtDNA rate we selected was broadly supported by Papadopoulou et al. [81] in a study of tenebrionid beetles using a number of geological calibration points from the mid-Aegean trench.
Our empirical tree indicated an increase in Rhithrogena speciation rate from ca. 3.0 to 1.0 Mya, corresponding to the late Pliocene and early Pleistocene. This period of time largely overlaps the Quaternary glacial cycles, which are well known for having promoted lineage divergence and speciation in a variety of organisms (see [6] for a review). In contrast, our analysis using simulated trees to account for incomplete sampling of Rhithrogena species detected a significant reduction in diversification rate at 0.22 Mya (Additional file 1: Table S4, and Fig. 4). This shift potentially results from the absence of intraspecific variation in our sampling (e.g. [86, 107]), which included a single individual per GMYC species. Alternatively, this shift could be due to a lack of recent species in our sampling, either because they occur in regions poorly coverred by our sampling like the Iberian and Italian peninsulas, or because the cox1 failed to detect the most recent speciation events. Rather than any increase in diversification, the simulation approach indicated a relatively steady increase in the number of lineages over time, suggesting that speciation is relatively constant and ongoing, and that glacial cycles have not been disproportionately important. Nonetheless, more than 50 % of the sampled species arose during the 3.0 – 1.5 Mya period in the empirical tree, regardless of whether speciation rate varied significantly. Species that recently arose include all members of the loyolaea species group, most of the semicolorata species group, and some members of the hybrida and diaphana species groups. A recent divergence between these species may partly explain the difficulty of finding reliable morphological characters for Rhithrogena species identification [43, 50].
Interestingly, the origin of European Rhithrogena roughly corresponded to the Oligocene/Miocene boundary (ca. 23 Mya), which coincided with one of the three major climatic aberrations of the Cenozoic: the Oligocene/Miocene glacial maximum [108, 109]. This deep glaciation spanning 0.2 My, followed by a series of smaller glaciations, which happened in a global context of climate warming, promoted rates of turnover and speciation in many groups, including mammals, birds, ostracods, and planktonic organisms [110–112]. Similar turnovers in a number of groups (e.g. [113–116]) were also linked to another major climatic aberration, the older Eocene/Oligocene glacial period [108, 109].
The Messinian Salinity Crisis (MSC, 5.96 – 5.33 Mya), a geological event that had considerable impact on the Mediterranean flora and fauna (e.g. [117, 118]), potentially promoted freshwater invertebrate dispersal between Mediterranean islands, with subsequent population isolation after the rapid refilling of the Mediterranean Sea [119]. A recent cox1-based study [120] found that genetic differentiation of several continental and insular mayfly populations coincided with the MSC. The origin of the three insular endemic species included in our sampling also largely overlapped the MSC period: Rh. eatoni from Corsica (5.9 Mya; 95 % HPD 8.2 – 3.9), Rh. insularis from Corsica and Rh. nuragica from Sardinia (4.5 Mya; 95 % HPD 6.5 – 2.8), thus supporting the important role of this geological event in the speciation of Corso-Sardinian mayflies.
Using the currently recognized taxonomy would have resulted in species over-splitting because single GMYC species have up to four names (see [51]). Our use of GMYC species (see Material and Methods) was intended to minimize this problem. However, Vuataz et al. [51] defined four cases of potential over-splitting in the delimited GMYC species (involving Rh. gratianopolitana, sp 4, sp 6 and sp 10; Additional file 1: Table S1), which if correct would still leave ca. 50 % of species having diversified 3.0 – 1.0 Mya. Although several low elevation species from the Iberian and Italian peninsulas were not sampled here, most central European species are represented. Our Alpine sampling was extensive and included all described species, most of them from their type localities. We can thus be confident that our sampling provides a very good representation of the Alpine diversity.
Additional evidence for the accuracy of our GMYC species comes from the present-day distribution of sampled species. Based on our sampling in the Alps and other mountain chains in Europe (data not shown), several closely related Rhithrogena species have distinct or adjacent (i.e., allopatric or parapatric) distribution ranges. Based on our dating analysis, these species diverged from one another during the Pleistocene. Within the hybrida species group, the individuals from one well-supported clade (Rh. circumtatrica + Rh. hybrida + sp 11 + Rh. diensis + sp 6; Fig. 2) are distributed in the Apennines (sp 11), in a small area in the western Alps (Rh. diensis, sp 6), in the Carpathians (Rh. circumtatrica) and widely in the Alps (Rh. hybrida). Within the well-supported loyolaea species group (Fig. 2), sp 7 occured in the Carpathians and the eastern Alps, sp 8 in the central Alps, and sp 9 in the western Alps and the Pyrenees. Such present-day allopatric or parapatric distribution ranges are typically observed in closely related species or populations that diverged in separate refugia during the Quaternary glacial periods (e.g. [7, 121]).
Ancestral habitat
The headwater origin hypothesis proposes an ancestral position of cold-adapted aquatic insect lineages in headwaters and subsequent diversification into warmer, larger rivers downstream [35–37]. Recently, Statzner et al. [38] and Statzner & Doledec [39] found evidence to support this hypothesis using an analysis of mtDNA and morphological characters from nine Hydropsyche (Trichoptera) species along the Loire River (ca. 0 – 1300 m), although node support was partly inconclusive. In contrast, Vuataz et al. [41] found evidence that lowland mayflies (Heptageniidae) diversified to more upstream (colder) areas in Madagascar, while Toussaint et al. [40] reported a complex combination of upstream and downstream diversification events in Exocelina diving beetles (Coleoptera, Dytiscidae) in Papua New Guinea.
Our ancestral state reconstructions of elevation constitute the first attempt for mayflies of which we are aware, and provide additional corroboration for the original hypothesis of headwater origins. However, our findings constitute only a preliminary estimate of ancestral aquatic habitats, being derived from a phylogeny of ca. 50 % of the European diversity, and because our own sampling sites served as a proxy of current altitude ranges of Rhithrogena species. Two future improvements would verify our findings. First, including more species, particularly low-elevation species from the Iberian and Italian peninsulas that are lacking in our Alpine-oriented sampling, would produce more complete phylogenies. Second, collecting more altitude data at a broader scale, particularly for rare species that we could sample from only one or a few localities, would improve the accuracy of the altitude ranges.
Interestingly, Rh. nivata and Rh. grischuna were the sister species to all other hybrida group members (Fig. 2), and live at higher elevations than all of them. The distribution of Rh. nivata ranges from 1100 to 2400 m in Switzerland [122] and 1160 to 1650 m in France [123]. Rh. grischuna, which is thought to inhabit small to medium rivers, was only found above 1150 m in our extensive sampling. In contrast, most other hybrida species group members exploit a wide altitudinal range (Rh. degrangei: 200 – 2200 m; Rh. gratianopolitana: 400 – 1200 m; Rh. hybrida: 400 – 1900 m; Rh. corcontica: 500 – 1500 m), or were sampled from lower elevations than Rh. grischuna. Similarly, the sister species to all other alpestris group members, Rh. alpestris, occurs at a higher elevation than any other member of the species group, and immature stages inhabit headwater streams and small to medium rivers between 500 and 2300 m (greatest occurrence between 1000 and 1800 m) in Switzerland [122] and from 1715 to 2020 m in France [123]. In contrast, Rh. landai (including Rh. vaillanti; see [51]) larvae inhabit only medium to large rivers between 400 and 700 m in Switzerland [122], although our sampling included three occurences between 1000 and 1200 m; Rh. allobrogica lives between 210 and 715 m [124] and Rh. eatoni lives exclusively in Corsica from 18 to 1100 m [123]. The loyolaea species group is restricted to cold-adapted species living in high elevation streams (mostly between 1700 and 2200 m; [122]), thus their ancestor was also probably cold adapted. It appears that the diversification of this species group is more recent (Fig. 3) and that there has been no diversification into lower elevations. The semicolorata species group members were sampled at wide altitudinal ranges, with the notable exception of Rh. germanica, an inhabitant of large floodplain rivers (low elevations) that has become very rare [125]. The diaphana species group is mainly composed of Mediterranean species [94, 126, 127], the Alpine ones included in our study being marginal representatives.
Conclusions
Our analysis of two mitochondrial and three nuclear gene regions, combined with an increased taxon sampling compared to previous studies, provided greater resolution and clearer definitions of species groups within European Rhithrogena. While our coalescent-based and concatenated phylogenies produced very similar topologies, node supports from the concatenated approach were repeatedly higher, particularly within the hybrida species group. The simulation approach of estimating diversification rates indicated that European Rhithrogena speciation is constant and ongoing, with the more recent half of the speciation events overlapping the Quaternary glaciations (<3 Mya). The recent origin and ongoing diversification potentially explain the morphologically cryptic characteristics of many European Rhithrogena species. The previously proposed headwater origin hypothesis with subsequent downstream diversification was supported by ancestral state reconstructions in the hybrida and alpestris species groups, indicating speciation has proceeded in part via diversification into warmer and larger rivers downstream. Testing whether this is a consistent pattern in Alpine aquatic insects requires additional analyses of a broader range of lineages.
Acknowledgments
We are grateful to André Wagner who collected and identified most of the Rhithrogena individuals of this study, and to Romain Studer for his precious assistance with data analyses. We thank Arnaud Maeder, Pascal Stucki, Jindřiška Bojková, Sonia Zrelli, Éva Váncsa and Anca Avram for providing additional samples, and Emilie Cavallo, Camille Marshall, Daniel Vuataz, Micaël Vuataz and Marina Vilenica for their help in the field. The laboratory work would not have been possible without Anne-Lyse Ducrest Roulin, Roberto Sermier, Nadège Remollino-Duvoisin, Christine La Mendola, Catherine Berney, Françoise Dolivo and Consolée Aletti from University of Lausanne. Finally, we are grateful to Olivier Broennimann for the beautiful sampling map, to Daniel Cherix and Luca Fumagalli at the University of Lausanne, to the team at the Museum of Zoology in Lausanne and to Denise Vuataz for their kind support. This is publication number 43 of the Berlin Center for Genomics in Biodiversity Research. We thank handling editor Matthew Symonds and three anonymous referees from Axios Review for their useful comments on the initial version of this manuscript.
Funding
Research was funded by the Swiss National Science Foundation (FNS nr. 31003A-116049; www.snf.ch) and the Leibniz Association PAKT für Forschung und Innovation (‘FREDIE’ project: SAW-2011-ZFMK-3). Individual support was provided by the Japan Society for the Promotion of Science (Long-Term Research Fellowship L-15543 to MTM) and the Swiss National Science Foundation (Early PostDoc.Mobility fellowship P2SKP3_158698 to SR).
Availability of data and materials
Extracted DNA, individuals and photographs are deposited at the Museum of Zoology, Lausanne, Switzerland (MZL). All sequences are available from Genbank/ENA database (see Additional file 1: Table S2, for accession numbers). The matrices (fasta files) corresponding to the five gene fragments, the *BEAST input (xml) and tree files, the MrBayes input and tree files (nexus) including the five gene trees, the Garli tree files, the BEAST input (xml) and tree files, and the R script of the CorSiM analyses, are available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.q0j90 [128].
Authors’ contributions
LV, MTM, and MS designed the study. LV performed field collecting and laboratory analyses. LV and SR carried out data analyses. LV, SR, MTM, and MS wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- AIC
Akaike information criterion
- AICc
Second-order Akaike information criterion
- BDL
Birth-death likelihood
- BI
Bayesian inference
- bp
Base pair
- BS
Bootstrap support
- cox1
Cytochrome c oxidase subunit I
- EF-1α
Elongation factor 1-alpha
- ESS
Effective sample size
- GMYC species
Species delimited using the general mixed Yule-coalescent model
- HPD
Highest posterior density interval
- LTT
Lineage-through-time
- Mi-1
Oligocene/Miocene boundary glacial maximum
- ML
Maximum likelihood
- MSC
Messinian Salinity Crisis
- mt
Mitochondrial
- My
Million years
- Mya
Million years ago
- nuc
Nuclear
- PEPCK
Phosphoenolpyruvate carboxykinase
- PP
Posterior probability
- REML
Residual maximum likelihood
- rRNA
Ribosomal ribonucleic acid
- SD
Standard deviation
- wg
Wingless
Additional files
(PDF 133 kb)
Bayesian majority-rule consensus tree reconstructed from the concatenated mitochondrial (cox1 + 16S; 1169 bp) data set. Species groups are indicated with green shading, including a photograph of a species group member. For each node, Bayesian posterior probability (PP) and maximum likelihood bootstrap support (BS) values are given (PP/BS) if PP > 0.8 and BS > 60. (TIF 7398 kb)
Bayesian majority-rule consensus tree reconstructed from the concatenated nuclear (PEPCK + EF-1α + wg; 1147 bp) data set. Species groups and node support are indicated as in Additional file 2: Figure S1. (TIF 7255 kb)
Bayesian majority-rule consensus tree of the mitochondrial + nuclear concatenated (2316 bp) data set. Species groups and node support are indicated as in Additional file 2: Figure S1. (TIF 7566 kb)
ML tree of the hybrida species group extracted from the concatenated (five-gene supermatrix) ML phylogram, with ancestral altitudes reconstructed using the REML method. Mean ancestral altitudes (with minimum and maximum ancestral altitudes between brackets) are indicated for each node. Mean altitudes (with altitude ranges between brackets) extracted from authors’ own records (see Additional file 1: Table S3), and used as raw data for the ancestral altitude reconstruction, are given below each terminal GMYC species. The color gradient symbolizing the mean altitude gradient along the tree branches was obtained using the contMap function of the phytools package [129] for R. (TIF 3301 kb)
ML tree of the alpestris species group extracted from the concatenated (five-gene supermatrix) ML phylogram, with ancestral altitudes reconstructed using the REML method. The altitudes and the color gradient are indicated as in Additional file 5: Figure S4. (TIF 2765 kb)
Footnotes
Michael T. Monaghan and Michel Sartori are the senior authors of the paper.
References
- 1.Diaz HF, Grosjean M, Graumlich L. Climate variability and change in high elevation regions: Past, present and future. Clim Change. 2003;59(1-2):1–4. doi: 10.1023/A:1024416227887. [DOI] [Google Scholar]
- 2.Körner C. Mountain biodiversity, its causes and function. Ambio Spec Rep. 2004;13:11–17. [PubMed] [Google Scholar]
- 3.Meyer JL, Strayer DL, Wallace JB, Eggert SL, Helfman GS, Leonard NE. The contribution of headwater streams to biodiversity in river networks. J Am Water Resour As. 2007;43:86–103. doi: 10.1111/j.1752-1688.2007.00008.x. [DOI] [Google Scholar]
- 4.Clarke A, Mac Nally R, Bond NR, Lake PS. Conserving macroinvertebrate diversity in headwater streams: the importance of knowing the relative contributions of alpha and beta diversity. Divers Distrib. 2010;16(5):725–736. doi: 10.1111/j.1472-4642.2010.00692.x. [DOI] [Google Scholar]
- 5.Finn DS, Bonada N, Murria C, Hughes JM. Small but mighty: headwaters are vital to stream network biodiversity at two levels of organization. J N Am Benthol Soc. 2011;30(4):963–980. doi: 10.1899/11-012.1. [DOI] [Google Scholar]
- 6.Hewitt GM. Genetic consequences of climatic oscillations in the Quaternary. Philos T Roy Soc B. 2004;359(1442):183–195. doi: 10.1098/rstb.2003.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitt T. Biogeographical and evolutionary importance of the European high mountain systems. Front Zool. 2009;6:9. doi: 10.1186/1742-9994-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hewitt GM. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc. 1996;58(3):247–276. doi: 10.1111/j.1095-8312.1996.tb01434.x. [DOI] [Google Scholar]
- 9.Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF. Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol. 1998;7(4):453–464. doi: 10.1046/j.1365-294x.1998.00289.x. [DOI] [PubMed] [Google Scholar]
- 10.Bennett KD. Continuing the debate on the role of Quaternary environmental change for macroevolution. Philos T Roy Soc B. 2004;359(1442):295–303. doi: 10.1098/rstb.2003.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stewart JR, Lister AM, Barnes I, Dalen L. Refugia revisited: individualistic responses of species in space and time. P Roy Soc B-Biol Sci. 2010;277(1682):661–671. doi: 10.1098/rspb.2009.1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zink RM, Slowinski JB. Evidence from molecular systematics for decreased avian diversification in the Pleistocene epoch. Proc Natl Acad Sci U S A. 1995;92(13):5832–5835. doi: 10.1073/pnas.92.13.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klicka J, Zink RM. The importance of recent ice ages in speciation: a failed paradigm. Science. 1997;277(5332):1666–1669. doi: 10.1126/science.277.5332.1666. [DOI] [Google Scholar]
- 14.Klicka J, Zink R. Pleistocene phylogeographic effects on avian evolution. Proc R Soc Lond B. 1999;266:695–700. doi: 10.1098/rspb.1999.0691. [DOI] [Google Scholar]
- 15.Arbogast BS, Slowinski JB. Pleistocene speciation and the mitochondrial DNA clock. Science. 1998;282(5396):1955. doi: 10.1126/science.282.5396.1955a. [DOI] [Google Scholar]
- 16.Avise JC, Walker D. Pleistocene phylogeographic effects on avian populations and the speciation process. P Roy Soc B-Biol Sci. 1998;265(1395):457–463. doi: 10.1098/rspb.1998.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lovette IJ. Glacial cycles and the tempo of avian speciation. Trends Ecol Evol. 2005;20(2):57–59. doi: 10.1016/j.tree.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 18.Zink RM, Klicka J, Barber BR. The tempo of avian diversification during the Quaternary. Philos T Roy Soc B. 2004;359(1442):215–220. doi: 10.1098/rstb.2003.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zink RM, Klicka J. The tempo of avian diversification: a comment on Johnson and Cicero. Evolution. 2006;60(2):411–412. doi: 10.1111/j.0014-3820.2006.tb01118.x. [DOI] [PubMed] [Google Scholar]
- 20.Burbrink FT, Pyron RA. How does ecological opportunity influence rates of speciation, extinction, and morphological diversification in New World ratsnakes (tribe Lampropeltini)? Evolution. 2010;64(4):934–943. doi: 10.1111/j.1558-5646.2009.00888.x. [DOI] [PubMed] [Google Scholar]
- 21.Harmon LJ, Schulte JA, Larson A, Losos JB. Tempo and mode of evolutionary radiation in iguanian lizards. Science. 2003;301(5635):961–964. doi: 10.1126/science.1084786. [DOI] [PubMed] [Google Scholar]
- 22.Barber BR, Jensen G. Quaternary climate change was not an engine of diversification in New World bats (Chiroptera) J Mamm Evol. 2012;19(2):129–133. doi: 10.1007/s10914-011-9180-z. [DOI] [Google Scholar]
- 23.Dijkstra K-DB, Monaghan MT, Pauls SU. Freshwater Biodiversity and Insect Diversification. Annu Rev Entomol. 2014;59:143. doi: 10.1146/annurev-ento-011613-161958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pauls SU, Lumbsch HT, Haase P. Phylogeography of the montane caddisfly Drusus discolor: evidence for multiple refugia and periglacial survival. Mol Ecol. 2006;15(8):2153–2169. doi: 10.1111/j.1365-294X.2006.02916.x. [DOI] [PubMed] [Google Scholar]
- 25.Balint M, Barnard PC, Schmitt T, Ujvarosi L, Popescu O. Differentiation and speciation in mountain streams: a case study in the caddisfly Rhyacophila aquitanica (Trichoptera) J Zool Syst Evol Res. 2008;46(4):340–345. doi: 10.1111/j.1439-0469.2008.00480.x. [DOI] [Google Scholar]
- 26.Kubow KB, Robinson CT, Shama LNS, Jokela J. Spatial scaling in the phylogeography of an alpine caddisfly, Allogamus uncatus, within the central European Alps. J N Am Benthol Soc. 2010;29(3):1089–1099. doi: 10.1899/09-084.1. [DOI] [Google Scholar]
- 27.Lehrian S, Balint M, Haase P, Pauls SU. Genetic population structure of an autumn-emerging caddisfly with inherently low dispersal capacity and insights into its phylogeography. J N Am Benthol Soc. 2010;29(3):1100–1118. doi: 10.1899/09-100.1. [DOI] [Google Scholar]
- 28.Ribera I, Vogler AP. Speciation of Iberian diving beetles in Pleistocene refugia (Coleoptera, Dytiscidae) Mol Ecol. 2004;13(1):179–193. doi: 10.1046/j.1365-294X.2003.02035.x. [DOI] [PubMed] [Google Scholar]
- 29.Previsic A, Walton C, Kucinic M, Mitrikeski PT, Kerovec M. Pleistocene divergence of Dinaric Drusus endemics (Trichoptera, Limnephilidae) in multiple microrefugia within the Balkan Peninsula. Mol Ecol. 2009;18(4):634–647. doi: 10.1111/j.1365-294X.2008.04046.x. [DOI] [PubMed] [Google Scholar]
- 30.Weiss S, Stradner D, Graf W. Molecular systematics, evolution and zoogeography of the stonefly genus Siphonoperla (Insecta: Plecoptera, Chloroperlidae) J Zool Syst Evol Res. 2012;50(1):19–29. doi: 10.1111/j.1439-0469.2011.00639.x. [DOI] [Google Scholar]
- 31.Smith PJ, McVeagh SM, Collier KJ. Genetic diversity and historical population structure in the New Zealand mayfly Acanthophlebia cruentata. Freshwater Biol. 2006;51(1):12–24. doi: 10.1111/j.1365-2427.2005.01462.x. [DOI] [Google Scholar]
- 32.Theissinger K, Balint M, Haase P, Johannesen J, Laube I, Pauls SU. Molecular data and species distribution models reveal the Pleistocene history of the mayfly Ameletus inopinatus (Ephemeroptera: Siphlonuridae) Freshwater Biol. 2011;56(12):2554–2566. doi: 10.1111/j.1365-2427.2011.02681.x. [DOI] [Google Scholar]
- 33.Barber-James HM, Gattolliat JL, Sartori M, Hubbard MD. Global diversity of mayflies (Ephemeroptera, Insecta) in freshwater. Hydrobiologia. 2008;595:339–350. doi: 10.1007/s10750-007-9028-y. [DOI] [Google Scholar]
- 34.Hitchings TR. Leptophlebiidae (Ephemeroptera) of the alpine region of the Southern Alps, New Zealand. Aquat Insects. 2009;31(Sup 1):595–601. doi: 10.1080/01650420802666868. [DOI] [Google Scholar]
- 35.Ross HH. Evolution and classification of the mountain caddisflies. Urbana: Univ. of Illinois Press; 1956. [Google Scholar]
- 36.Illies J. Versuch einer allgemeinen biozönotischen Gliederung der Fließgewässer. Intern Revue Hydrobiol Hydrogr. 1961;46(2):205–213. doi: 10.1002/iroh.19610460205. [DOI] [Google Scholar]
- 37.Wiggins GB, Wichard W. Phylogeny of pupation in Trichoptera, with proposals on the origin and higher classification of the Order. J N Am Benthol Soc. 1989;8(3):260–276. doi: 10.2307/1467330. [DOI] [Google Scholar]
- 38.Statzner B, Douady CJ, Konecny L, Doledec S. Unravelling phylogenetic relationships among regionally co-existing species: Hydropsyche species (Trichoptera: Hydropsychidae) in the Loire River. Zootaxa. 2010;2556:51–68. [Google Scholar]
- 39.Statzner B, Doledec S. Phylogenetic, spatial, and species-trait patterns across environmental gradients: the case of Hydropsyche (Trichoptera) along the Loire River. Int Rev Hydrobiol. 2011;96(2):121–140. doi: 10.1002/iroh.201111325. [DOI] [Google Scholar]
- 40.Toussaint EF, Hall R, Monaghan MT, Sagata K, Ibalim S, Shaverdo HV, Vogler AP, Pons J, Balke M. The towering orogeny of New Guinea as a trigger for arthropod megadiversity. Nat Commun. 2014;5:4001. doi: 10.1038/ncomms5001. [DOI] [PubMed] [Google Scholar]
- 41.Vuataz L, Sartori M, Gattolliat J-L, Monaghan MT. Endemism and diversification in freshwater insects of Madagascar revealed by coalescent and phylogenetic analysis of museum and field collections. Mol Phylogenet Evol. 2013;66(3):979–991. doi: 10.1016/j.ympev.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 42.Soldán T. Status of the systematic knowledge and priorities in Ephemeroptera studies: the Oriental region. In: Dominguez E, editor. Trends in Research in Ephemeroptera and Plecoptera. New York: Kluwer Academic/Plenum Publishers; 2001. pp. 53–65. [Google Scholar]
- 43.Soldán T, Landa V. A key to the Central European species of the genus Rhithrogena (Ephemeroptera: Heptageniidae) Klapalekiana. 1999;38:25–37. [Google Scholar]
- 44.Webb JM, McCafferty WP. Heptageniidae of the world. Part II: Key to the Genera. Can J Arthropod Identif. 2008;7:1–55. [Google Scholar]
- 45.Sartori M. What is Ecdyonurus sumatranus Ulmer, 1939? A contribution to the knowledge of the genus Rhithrogena in the Oriental Region (Ephemeroptera, Heptageniidae) Zootaxa. 2014;3802(2):193–208. doi: 10.11646/zootaxa.3802.2.3. [DOI] [PubMed] [Google Scholar]
- 46.Barber-James H, Sartori M, Gattolliat J, Webb J. World checklist of freshwater Ephemeroptera species. 2013. World Wide Web electronic publication. Available online at http://fada.biodiversity.be/group/show/35. Accessed 30 Aug 2016.
- 47.Bauernfeind E, Soldán T. The Mayflies of Europe. Ollerup, Denmark: Apollo Books; 2012. [Google Scholar]
- 48.Lubini V, Knispel S, Sartori M, Vicentini H, Wagner A. Listes rouges Ephémères, Plécoptères, Trichoptères. Espèces menacées en Suisse, état 2010. Office fédéral de l’environnement, Berne, et Centre Suisse de Cartographie de la Faune. Neuchâtel: L’environnement pratique n° 1212; 2012. [Google Scholar]
- 49.Brittain JE, Sartori M. Ephemeroptera. In: Resh VH, Cardé R, editors. Encyclopedia of insects. New York: Academic; 2003. pp. 373–380. [Google Scholar]
- 50.Belfiore C, Picariello O, Scillitani G, Cretella M. Genetic divergence between two insular species of the genus Rhithrogena (Ephemeroptera, Heptageniidae) Fragm Entomol. 1992;23(2):235–242. [Google Scholar]
- 51.Vuataz L, Sartori M, Wagner A, Monaghan MT. Toward a DNA taxonomy of Alpine Rhithrogena (Ephemeroptera: Heptageniidae) using a mixed Yule-coalescent analysis of mitochondrial and nuclear DNA. PLoS One. 2011;6(5):e19728. doi: 10.1371/journal.pone.0019728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wiens JJ, Chippindale PT, Hillis DM. When are phylogenetic analyses misled by convergence? A case study in Texas cave salamanders. Syst Biol. 2003;52(4):501–514. doi: 10.1080/10635150390218222. [DOI] [PubMed] [Google Scholar]
- 53.Renoult JP, Geniez P, Bacquet P, Benoit L, Crochet PA. Morphology and nuclear markers reveal extensive mitochondrial introgressions in the Iberian Wall Lizard species complex. Mol Ecol. 2009;18(20):4298–4315. doi: 10.1111/j.1365-294X.2009.04351.x. [DOI] [PubMed] [Google Scholar]
- 54.Tao WJ, Zou M, Wang XZ, Gan XN, Mayden RL, He SP. Phylogenomic analysis resolves the formerly intractable adaptive diversification of the endemic clade of East Asian Cyprinidae (Cypriniformes) PLoS One. 2010;5(10):e13508. doi: 10.1371/journal.pone.0013508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaliontzopoulou A, Carretero MA, Llorente GA. Morphology of the Podarcis wall lizards (Squamata: Lacertidae) from the Iberian Peninsula and North Africa: patterns of variation in a putative cryptic species complex. Zool J Linn Soc. 2012;164(1):173–193. doi: 10.1111/j.1096-3642.2011.00760.x. [DOI] [Google Scholar]
- 56.Jacob U. Rhithrogena braaschi n.sp., eine neue Heptageniide aus Bulgarien (Insecta, Ephemeroptera) Entomol Nachr. 1974;11(12):167–173. [Google Scholar]
- 57.Sowa R. Contribution à la connaissance des espèces européennes de Rhithrogena Eaton (Ephemeroptera, Heptageniidae) avec le rapport particulier aux espèces des Alpes et des Carpates. In: Landa V, Soldán T, Tonner M, editors. Proceeding of the fourth international conference on Ephemeroptera. Bechyne: CSAV; 1984. pp. 37–52. [Google Scholar]
- 58.Zurwerra A, Metzler M, Tomka I. Biochemical systematics and evolution of the European Heptageniidae (Ephemeroptera) Arch Hydrobiol. 1987;109(4):481–510. [Google Scholar]
- 59.Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33(2):511–518. doi: 10.1093/nar/gki198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004;20(3):426–427. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- 61.Posada D. Collapse: describing haplotypes from sequence alignments. Vigo: Computation Evolutionary Biology Lab, University of Vigo; 2004. [Google Scholar]
- 62.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nylander JAA. MrAIC.pl. Program distributed by the author. Evolutionary Biology Centre, Uppsala University; 2004.
- 64.Brandley MC, Schmitz A, Reeder TW. Partitioned Bayesian analyses, partition choice, and the phylogenetic relationships of scincid lizards. Syst Biol. 2005;54(3):373–390. doi: 10.1080/10635150590946808. [DOI] [PubMed] [Google Scholar]
- 65.Shapiro B, Rambaut A, Drummond AJ. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol Biol Evol. 2006;23(1):7–9. doi: 10.1093/molbev/msj021. [DOI] [PubMed] [Google Scholar]
- 66.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heled J, Drummond AJ. Bayesian inference of species trees from multilocus data. Mol Biol Evol. 2010;27(3):570–580. doi: 10.1093/molbev/msp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10(4):e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68(4):978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73(5):1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harrigan RJ, Mazza ME, Sorenson MD. Computation vs. cloning: evaluation of two methods for haplotype determination. Mol Ecol Resour. 2008;8(6):1239–1248. doi: 10.1111/j.1755-0998.2008.02241.x. [DOI] [PubMed] [Google Scholar]
- 72.Garrick RC, Sunnucks P, Dyer RJ. Nuclear gene phylogeography using PHASE: dealing with unresolved genotypes, lost alleles, and systematic bias in parameter estimation. BMC Evol Biol. 2010;10(1):1. doi: 10.1186/1471-2148-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Flot JF. SeqPHASE: a web tool for interconverting PHASE input/output files and FASTA sequence alignments. Mol Ecol Resour. 2010;10(1):162–166. doi: 10.1111/j.1755-0998.2009.02732.x. [DOI] [PubMed] [Google Scholar]
- 74.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 75.Zwickl DJ. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Austin: the University of Texas; 2006. [Google Scholar]
- 76.Bazinet AL, Zwickl DJ, Cummings MP. A gateway for phylogenetic analysis powered by grid computing featuring GARLI 2.0. Syst Biol. 2014;63(5):812–818. doi: 10.1093/sysbio/syu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marshall DC, Simon C, Buckley TR. Accurate branch length estimation in partitioned Bayesian analyses requires accommodation of among-partition rate variation and attention to branch length priors. Syst Biol. 2006;55(6):993–1003. doi: 10.1080/10635150601087641. [DOI] [PubMed] [Google Scholar]
- 78.Rambaut A, Suchard MA, Xie D, Drummond AJ. Tracer v1.6. 2014. Available from http://beast.bio.ed.ac.uk/Tracer.
- 79.Sukumaran J, Holder MT. DendroPy: a Python library for phylogenetic computing. Bioinformatics. 2010;26(12):1569–1571. doi: 10.1093/bioinformatics/btq228. [DOI] [PubMed] [Google Scholar]
- 80.Brower AVZ. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial-DNA evolution. Proc Natl Acad Sci U S A. 1994;91(14):6491–6495. doi: 10.1073/pnas.91.14.6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Papadopoulou A, Anastasiou I, Vogler AP. Revisiting the insect mitochondrial molecular clock: the mid-Aegean trench calibration. Mol Biol Evol. 2010;27(7):1659–1672. doi: 10.1093/molbev/msq051. [DOI] [PubMed] [Google Scholar]
- 82.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4(5):699–710:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pybus OG, Harvey PH. Testing macro-evolutionary models using incomplete molecular phylogenies. P Roy Soc B-Biol Sci. 2000;267(1459):2267–2272. doi: 10.1098/rspb.2000.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rabosky DL. Likelihood methods for detecting temporal shifts in diversification rates. Evolution. 2006;60(6):1152–1164. doi: 10.1111/j.0014-3820.2006.tb01194.x. [DOI] [PubMed] [Google Scholar]
- 86.Nee S, Holmes EC, May RM, Harvey PH. Extinction rates can be estimated from molecular phylogenies. Philos T Roy Soc B. 1994;344(1307):77–82. doi: 10.1098/rstb.1994.0054. [DOI] [PubMed] [Google Scholar]
- 87.Barraclough TG, Nee S. Phylogenetics and speciation. Trends Ecol Evol. 2001;16(7):391–399. doi: 10.1016/S0169-5347(01)02161-9. [DOI] [PubMed] [Google Scholar]
- 88.Brock CD, Harmon LJ, Alfaro ME. Testing for temporal variation in diversification rates when sampling is incomplete and nonrandom. Syst Biol. 2011;60(4):410–419. doi: 10.1093/sysbio/syr007. [DOI] [PubMed] [Google Scholar]
- 89.Stadler T. Simulating trees with a fixed number of extant species. Syst Biol. 2011;60(5):676–684. doi: 10.1093/sysbio/syr029. [DOI] [PubMed] [Google Scholar]
- 90.Cusimano N, Stadler T, Renner SS. A new method for handling missing species in diversification analysis applicable to randomly or nonrandomly sampled phylogenies. Syst Biol. 2012;61(5):785–792. doi: 10.1093/sysbio/sys031. [DOI] [PubMed] [Google Scholar]
- 91.R Core Team . R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 92.Schluter D, Price T, Mooers AØ, Ludwig D. Likelihood of ancestor states in adaptive radiation. Evolution 1997;51(6):1699–1711. [DOI] [PubMed]
- 93.Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20(2):289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 94.Sartori M, Oswald R. Rhithrogena grischuna nov. sp., a new mayfly species from eastern Switzerland related to Rh. hercynia Landa, 1969 (Ephemeroptera; Heptageniidae) Ann Limnol. 1988;24(3):261–268. doi: 10.1051/limn/1988022. [DOI] [Google Scholar]
- 95.Belfiore C. Heptageniidae from Corsica and Sardinia (Italy): Rhithrogena nuragica, new species, Rhithrogena eatoni Esben-Petersen 1912, and Rhithrogena insularis Esben-Petersen 1913 (Ephemeroptera) Ann Limnol. 1987;23(2):87–94. doi: 10.1051/limn/1987013. [DOI] [Google Scholar]
- 96.Zrelli S, Sartori M, Bejaoui M, Boumaiza M. Rhithrogena sartorii, a new mayfly species (Ephemeroptera: Heptageniidae) from North Africa. Zootaxa. 2011;3139:63–68. [Google Scholar]
- 97.Sowa R. Notes sur quelques espèces paléarctiques de Rhithrogena Eaton (Ephemeroptera, Heptageniidae) Bull Acad Pol Sci Biol. 1973;21(1):21–26. [Google Scholar]
- 98.Alba-Tercedor J, Sowa R. Two interesting Rhithrogena Eaton from Spain: R. thomasi sp.n. and R. monserrati sp.n. (Ephemeroptera: Heptageniidae) Aquat Insects. 1986;8(3):185–189. doi: 10.1080/01650428609361250. [DOI] [Google Scholar]
- 99.Demoulin G. Deuxième contribution à la connaissance des Ephéméroptères de l'ambre oligocène de la baltique. Dtsc Entomol Z. 1968;15(I-III):233–276. [Google Scholar]
- 100.Ritzkowski S. K-Ar-Altersbestimmung der bernsteinführenden Sedimente des Samlandes (Paläogen, Bezirk Kaliningrad) Metalla (Sonderheft) 1997;66:19–23. [Google Scholar]
- 101.Kluge NJ. A recent may fly species (Ephemeroptera, Heptageniidae) in Baltic amber. Paleontol J. 1986;2:106–107. [Google Scholar]
- 102.Monaghan MT, Gattolliat JL, Sartori M, Elouard JM, James H, Derleth P, Glaizot O, de Moor F, Vogler AP. Trans-oceanic and endemic origins of the small minnow mayflies (Ephemeroptera, Baetidae) of Madagascar. P Roy Soc B-Biol Sci. 2005;272(1574):1829–1836. doi: 10.1098/rspb.2005.3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rutschmann S, Gattolliat JL, Hughes SJ, Báez M, Sartori M, Monaghan MT. Evolution and island endemism of morphologically cryptic Baetis and Cloeon species (Ephemeroptera, Baetidae) on the Canary Islands and Madeira. Freshwater Biol. 2014;59(12):2516–2527. doi: 10.1111/fwb.12450. [DOI] [Google Scholar]
- 104.Britten RJ. Rates of DNA-sequence evolution differ between taxonomic groups. Science. 1986;231(4744):1393–1398. doi: 10.1126/science.3082006. [DOI] [PubMed] [Google Scholar]
- 105.Andersen NM, Cheng L, Damgaard J, Sperling FAH. Mitochondrial DNA sequence variation and phylogeography of oceanic insects (Hemiptera: Gerridae: Halobates spp.) Mar Biol. 2000;136(3):421–430. doi: 10.1007/s002270050701. [DOI] [Google Scholar]
- 106.Shapiro LH, Strazanac JS, Roderick GK. Molecular phylogeny of Banza (Orthoptera: Tettigoniidae), the endemic katydids of the Hawaiian Archipelago. Mol Phylogenet Evol. 2006;41(1):53–63. doi: 10.1016/j.ympev.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 107.Brumfield RT. Inferring the origins of lowland Neotropical birds. Auk. 2012;129(3):367–376. doi: 10.1525/auk.2012.129.3.367. [DOI] [Google Scholar]
- 108.Miller KG, Wright JD, Fairbanks RG. Unlocking the ice house - Oligocene-Miocene oxygen isotopes, eustasy, and margin erosion. J Geophys Res. 1991;96:6829–6848. doi: 10.1029/90JB02015. [DOI] [Google Scholar]
- 109.Zachos J, Pagani M, Sloan L, Thomas E, Billups K. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science. 2001;292(5517):686–693. doi: 10.1126/science.1059412. [DOI] [PubMed] [Google Scholar]
- 110.Hugueney M, Berthet D, Bodergat AM, Escuillie F, Mourer-Chauvire C, Wattinne A. The Oligocene-Miocene boundary in Limagne: faunal changes in the mammals, birds and ostracods from the different levels of Billy-Crechy (Allier, France) Geobios-Lyon. 2003;36(6):719–731. doi: 10.1016/j.geobios.2003.01.002. [DOI] [Google Scholar]
- 111.Kamikuri SI, Nishi H, Moore TC, Nigrini CA, Motoyama I. Radiolarian faunal turnover across the Oligocene/Miocene boundary in the equatorial Pacific Ocean. Mar Micropaleontol. 2005;57(3-4):74–96. doi: 10.1016/j.marmicro.2005.07.004. [DOI] [Google Scholar]
- 112.Suto I. The explosive diversification of the diatom genus Chaetoceros across the Eocene/Oligocene and Oligocene/Miocene boundaries in the Norwegian Sea. Mar Micropaleontol. 2006;58(4):259–269. doi: 10.1016/j.marmicro.2005.11.004. [DOI] [Google Scholar]
- 113.Wade BS, Pearson PN. Planktonic foraminiferal turnover, diversity fluctuations and geochemical signals across the Eocene/Oligocene boundary in Tanzania. Mar Micropaleontol. 2008;68(3-4):244–255. doi: 10.1016/j.marmicro.2008.04.002. [DOI] [Google Scholar]
- 114.Seiffert ER. Evolution and extinction of Afro-Arabian primates near the Eocene-Oligocene boundary. Folia Primatol. 2007;78(5-6):314–327. doi: 10.1159/000105147. [DOI] [PubMed] [Google Scholar]
- 115.Ivany LC, Patterson WP, Lohmann KC. Cooler winters as a possible cause of mass extinctions at the Eocene/Oligocene boundary. Nature. 2000;407(6806):887–890. doi: 10.1038/35038044. [DOI] [PubMed] [Google Scholar]
- 116.Jansen G, Savolainen R, Vepsalainen K. Phylogeny, divergence-time estimation, biogeography and social parasite-host relationships of the Holarctic ant genus Myrmica (Hymenoptera: Formicidae) Mol Phylogenet Evol. 2010;56(1):294–304. doi: 10.1016/j.ympev.2010.01.029. [DOI] [PubMed] [Google Scholar]
- 117.Hsü KJ, Montadert L, Bernoulli D, Cita MB, Erickson A, Garrison RE, Kidd RB, Mèlierés F, Müller C, Wright R. History of the Mediterranean salinity crisis. Nature. 1977;267(5610):399–403. doi: 10.1038/267399a0. [DOI] [Google Scholar]
- 118.Gliozzi E, Ceci ME, Grossi F, Ligios S. Paratethyan ostracod immigrants in Italy during the Late Miocene. Geobios-Lyon. 2007;40(3):325–337. doi: 10.1016/j.geobios.2006.10.004. [DOI] [Google Scholar]
- 119.Sola E, Sluys R, Gritzalis K, Riutort M. Fluvial basin history in the northeastern Mediterranean region underlies dispersal and speciation patterns in the genus Dugesia (Platyhelminthes, Tricladida, Dugesiidae) Mol Phylogenet Evol. 2013;66(3):877–888. doi: 10.1016/j.ympev.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 120.Gattolliat J-L, Cavallo E, Vuataz L, Sartori M. DNA barcoding of Corsican mayflies (Ephemeroptera) with implications on biogeography, systematics and biodiversity. Arthropod Syst Phylogeny. 2015;73(1):3–18. [Google Scholar]
- 121.Hewitt GM. Quaternary phylogeography: the roots of hybrid zones. Genetica. 2011;139(5):617–638. doi: 10.1007/s10709-011-9547-3. [DOI] [PubMed] [Google Scholar]
- 122.Sartori M, Landolt P. Atlas de distribution des Ephémères de Suisse (Insecta, Ephemeroptera) Neuchâtel: SEG-CSCF; 1999. [Google Scholar]
- 123.Brulin M. Atlas de distribution des Éphémères de France. 2ème partie: famille des Heptageniidae (Ephemeroptera) Ephemera. 2010;11(2):71–133. [Google Scholar]
- 124.Sowa R, Degrange C. Taxonomie et répartition des Rhithrogena Eaton du groupe alpestris (Ephemeroptera: Heptageniidae) des Alpes et des Carpates. Pol Pis Entomol. 1987;57:475–494. [Google Scholar]
- 125.Lubini V, Sartori M. Current status, distribution, life cycle and ecology of Rhithrogena germanica Eaton, 1885 in Switzerland: preliminary results (Ephemeroptera, Heptageniidae) Aquat Sci. 1994;56(4):388–397. doi: 10.1007/BF00877184. [DOI] [Google Scholar]
- 126.Belfiore C. Notes on Italian Heptageniidae (Ephemroptera). Rhithrogena fiorii, Grandi, 1953 and R. adrianae sp.n. Aquat Insects. 1983;5(2):69–76. doi: 10.1080/01650428309361129. [DOI] [Google Scholar]
- 127.Alba-Tercedor J, Sowa R. New representatives of the Rhithrogena diaphana-group from continental Europe, with a redescription of R. diaphana Navás, 1917 (Ephemeroptera: Heptageniidae) Aquat Insects. 1987;9(2):65–83. doi: 10.1080/01650428709361274. [DOI] [Google Scholar]
- 128.Vuataz L, Rutschmann S, Monaghan MT, Sartori M. Data from: Molecular phylogeny and timing of diversification in Alpine Rhithrogena (Ephemeroptera: Heptageniidae). Dryad Digit Respository. 2016. doi:10.5061/dryad.q0j90. [DOI] [PMC free article] [PubMed]
- 129.Revell LJ. phytools: an R package for phylogenetic comparative biology (and other things) Methods Ecol Evol. 2012;3(2):217–223. doi: 10.1111/j.2041-210X.2011.00169.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Extracted DNA, individuals and photographs are deposited at the Museum of Zoology, Lausanne, Switzerland (MZL). All sequences are available from Genbank/ENA database (see Additional file 1: Table S2, for accession numbers). The matrices (fasta files) corresponding to the five gene fragments, the *BEAST input (xml) and tree files, the MrBayes input and tree files (nexus) including the five gene trees, the Garli tree files, the BEAST input (xml) and tree files, and the R script of the CorSiM analyses, are available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.q0j90 [128].