

Abstract
Although there are a number of published trials of human leukocyte antigen (HLA) – identical sibling hematopoietic cell transplantation (HCT) for sickle cell disease (SCD) that span 2 decades, when and for whom this therapy should be pursued is a subject of debate. Assessments of the risks of transplant-related complications that include infertility and debilitating graft-versus-host disease and long-term quality of life after successful HCT are difficult to perform without prospective trials in transplant and non-transplant cohorts. However, it is possible to assess the risk of mortality and to compare published rates of survival in individuals with sickle cell disease treated and not treated by HCT. In this brief review, projections about mortality risk based upon recent published reports are reviewed and summarized. The published data show overall survival and event-survival rates of 95% and 92%, respectively, in children treated by HLA-identical sibling HCT. The overall survival in the Center for International Blood and Marrow Transplant Research (CIBMTR) (N=412) and European Blood and Marrow Transplant (EBMT) (N=487) registries was 91% and 95%, respectively. These results provide broad support for the therapeutic value of HLA-identical sibling HCT for children with sickle cell disease and serve as the basis for a strong recommendation in favor of the option of HCT when a suitable donor is available. The experience of HLA-identical sibling HCT in adults with SCD is limited but appears similar to results in children, and these preliminary observations warrant further investigation.
Introduction
Hematopoietic cell transplantation (HCT) for sickle cell disease (SCD) is curative in the majority of individuals who receive this treatment, but it is a treatment option that very few families and patients pursue1–4. The principal reason why so few transplants are performed is that most affected people lack a suitable donor. Even so, it is estimated that 18% of affected individuals will have an HLA-identical (HLA-ID) sibling donor5. Yet, far less than 1% of the SCD population in the US has received a transplant. Many barriers to transplant exist, and these are detailed in other reports6, 7,8. A key barrier is a prevailing assumption that HCT is risky and carries a mortality rate that exceeds mortality experienced with a supportive care approach. In addition, there are risks of infertility and of graft-versus-host disease (GVHD) that can cause a chronic debilitating disorder. In this brief review, the basis for assumptions about mortality risk is examined in an update of the contemporary experience of HLA-ID sibling HCT for SCD.
An important benefit of successful HCT is the elimination of sickle erythropoiesis, thereby significantly reducing or in most instances, ending the risk of sickle-related complications1, 9, 10. Thus, most agree that quality of life after successful HCT should be very much improved compared to that in individuals who continue to live with SCD. These comparisons about protection from sickle-related damage are difficult because prospective comparisons between groups of subjects treated by HCT and by supportive care have never been conducted. Thus, most analyses rely upon comparisons to historical controls, which weakens their impact. In addition, some recipients have experienced events soon after HCT such as pain, intra-cranial hemorrhage, and infection; thus, protection is not universal in those who survive with engraftment of donor cells, although these events eventually resolve. For these reasons, the benefit of HCT with regard to symptom abatement and organ function will not be the focus of this review. However, the importance of conducting studies that systematically monitor prospective outcomes in comparison cohorts that might establish unequivocal indications for HCT cannot be overstated.
HCT in Children with SCD
In developing eligibility criteria for HCT in early studies, investigators selected clinical features of SCD that carry a high burden of ongoing supportive care with a risk of cumulative organ injury and an association with early mortality11. In childhood, supportive care delivered at comprehensive sickle cell centers is currently associated with excellent survival to adulthood, with a risk of mortality before age 18 that ranges from 1–2% by age 20 in the East London Cohort to 6.1% in the Dallas Newborn Cohort12, 13. Thus, transplantation studies in childhood focused initially on minimizing the risks of early transplant-related death and of sickle-related clinical complications after successful transplantation. Currently, HCT in children with SCD is typically restricted to those with a clinical stroke or who have experienced recurrent vaso-occlusive complications such as pain and/or acute chest syndrome despite receiving optimal supportive care. The eligibility criteria used in the largest pediatric clinical trials completed 10 to 15 years ago are presented in Table 1. In the current era, as the survival rates in transplant and non-transplant cohorts converge, restricting HCT solely in children who have had a significant complication such as stroke is no longer appropriate, as suggested below. A liberalized approach to indications for HCT would also increase its utilization.
Table 1.
Indications for hematopoietic cell transplantation in children with sickle cell disease
| Patients with sickle cell disease (HbSS or HbSβ°-thalassemia) less than 16 years of age |
| One or more of the following complications: |
| Stroke or central nervous system event lasting longer than 24 hours |
| Impaired neuropsychologic function with abnormal cerebral magnetic resonance imaging and angiography |
| Recurrent acute chest syndrome |
| Stage I or II sickle lung disease (defined in43) |
| Recurrent vaso-occlusive painful episodes or recurrent priapism |
| Sickle nephropathy (glomerular filtration rate 30–50% of predicted normal) |
Transplant Results in Children: HLA-ID Sibling HCT
A compilation of the most recent single center patient series in children with SCD treated by HLA-ID sibling HCT is presented in Table 2. In a series of 40 patients treated in Rome, Italy, the overall survival and event-free survival were both 91% after an HLA-ID sibling bone marrow (BM) transplantation with a conventional preparative regimen of busulfan (BU)/cyclophosphamide (CY)/horse anti-thymocyte globulin (ATG) with or without fludarabine (Flu)14. In a series from Belgium, 37 of 38 children treated since 1995, who also received hydroxyurea (HU) well before HCT, survive free of SCD with an 8-year estimate of event-free survival (EFS) of 97.1%15. In a series of children treated in New York who received a combination of BU, Flu and alemtuzumab (Alem) before HLA-ID sibling HCT, all 18 children survived free of SCD after HCT16. Another single center series from Atlanta observed 24 of 25 patients who were treated by HLA-ID sibling HCT between 1993 and 2007, after preparation with BU/CY/horse ATG17 survived free of SCD. A recent multicenter investigation of 43 children with SCD who received Alem/Flu/melphalan (Mel) before HLA-ID sibling bone marrow transplantation reported survival and EFS probabilities of 93% and 90.7%, respectively18. Finally, the experience from Pavia was also recently reported in which all 30 recipients survive after HLA-ID HCT after preparation with BU/thiotepa(TT)/Flu or Treosulfan(Treo)/TT/Flu and ATG. Together these combined series include 218 recipients, of whom 208 (95%) survive after transplantation, and 200 (92%) survive free of SCD. These updated published results strongly suggest that survival after HCT from an HLA-ID sibling in children with SCD is not inferior to survival among those treated by standard supportive care. At last follow-up, only 6 survivors (3%) were receiving immunosuppressive therapy to treat chronic GVHD.
Table 2.
Clinical outcome of HLA-ID sibling HCT for children with SCD
| Center | Preparative regimen | GvHD prophyl | n | Age range (yrs) | Published outcomes | Latest follow up | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Follow up (yrs) | Death (mos) | Complications | IST | GvHD | % donor chimerism | |||||
| Rome (Lucarelli, 2014)14 | BU14 mg/kg, CY 200 mg/kg/rATG 10 mg/kg, ±Flu 150 mg/m2 | CsA, MTX, pred | 40 | 2–17 | 1 – 10 | 3 (2.5, 6, 15) | 3 deaths from GVHD | 1 of 40 | 17.5% aGVHD, 5% cGVHD | 25 – 100% |
| Brussels (Dedeken, 2014)15 | BU 13–18 mg/kg, CY 200 mg/kg, ±rATG (10–20 mg/kg), ±HU | CsA, MTX Or MMF (UCB) | 50 | 1.7 – 15.3 | 0.4 – 21.3 | 2 (6, 78) | 4, sepsis, 1 IMI, seizures 21%, 6, PRES | 1 of 50 | 20.5% aGVHD, 20% cGVHD | 15 – 100% |
| New York (Bhatia, 2014)16 | BU 12.8 – 16 mg/kg, Flu 180 mg/m2, Alem 54 mg/m2 | Tacrolimus, MMF | 18 | 2.3 – 20.2 | 0.4 – 7.5 | none | ICH in 1, PRES in 1, CMV react in 4 | none | 17% aGVHD, 11% cGVHD | Mean 88% at 1 year |
| Mississippi (Majumdar, 2010)44 | BU 14 mg/kg, CY 200 mg/kg, ATG 90 mg/kg | CsA, Pred | 10 | 2.8 – 16.3 | 2.9 – 9.9 | 1 | 1 death from sepsis, 1 AIHA | 1 of 10 | 40% aGVHD 10% ceGVHD | 15 – 100% |
| Atlanta (McPherson, 2011)17 | BU 14 mg/kg, CY 200 mg/kg, ATG 90 mg/kg | CsA, MTX | 27 | 3.3 – 17.4 | 0.1 – 10 | 1 (3) | 8 VOD, 16% seizures, 2 ICH | none | 12% aGVHD, 1 death from cGVHD | 62 – 100% |
| Pavia (Strocchio, 2015)45 | BU 16 mg/kg, TT 10 mg/kg, Flu 160 mg/m2 or Treo 14 gm/m2, TT 10 mg/kg, Flu 160 mg/m2, ATG | CsA, MTX or MMF | 30 | 1.7 – 18.8 | 0.5 – 14 | none | Stomatitis (43%), GI toxicity (17%); no VOD | none | 7% Gr I–II aGVHD, 7% cGVHD in BU group, none in treo group | 50% (BU) and 36% (Treo) at 1 year |
| USA (King, 2015)18 | Alem 48 mg, Flu140–150 mg/m2, Mel 140 mg/m2 | CsA or tacrolimus | 43 | 3 – 20.3 | 0.75 – 11.83 | 3 (11, 18, 21) | 3 deaths from GVHD | 19% by 1yr; 9% by 2 yrs | 23% aGVHD, 13.4% cGVHD | 29 – 100% |
Abbreviations: Alem, alemtuzumab; ATG, anti-thymocyte globulin [r(rabbit)]; BU, busulfan; CMV, cytomegalovirus; CY, cyclophosphamide; Flu, fludarabine; Treo, treosulfan; TT, thiotepa; TBI, total body irradiation; aGVHD, acute GVHD; cGVHD, chronic GVHD; ceGVHD, chronic extensive GVHD; CsA, cyclosporine; MMF, mycophenolate mofetil; MTX, methotrexate; Pred, prednisone; VOD, veno-occlusive disease; PRES, posterior reversible encephalopathy syndrome
Similar results have been reported from transplant registry data where there are approximately 1200 SCD transplant cases from related and alternate donor sources19. Among the HLA-ID sibling donor cohorts, the overall survival in the Center for International Blood and Marrow Transplant Research (CIBMTR) (N=412) and European Blood and Marrow Transplant (EBMT) (N=487) registries was 91% and 95%, respectively (Figure 1). In a separate analysis comparing outcomes in 160 children with SCD who received HLA-ID sibling BM and umbilical cord blood (UCB) transplantation between 1994 and 2005 in the US and Europe, there was no statistically significant difference in overall survival and EFS between these 2 donor sources20. Moreover, the combined 6-year disease-free survival was 92%. These retrospective registry data also were reviewed critically by an international expert panel on behalf of the EBMT Inborn Error and EBMT Paediatric Working Parties21 which made the following recommendations about HLA-ID HCT for children:
Young patients with symptomatic SCD who have an HLA-ID sibling donor should be transplanted as early as possible, preferably at pre-school age.
Unmanipulated BM or UCB (whenever available) from HLA-ID sibling donors are the recommended stem cell source.
Figure 1.
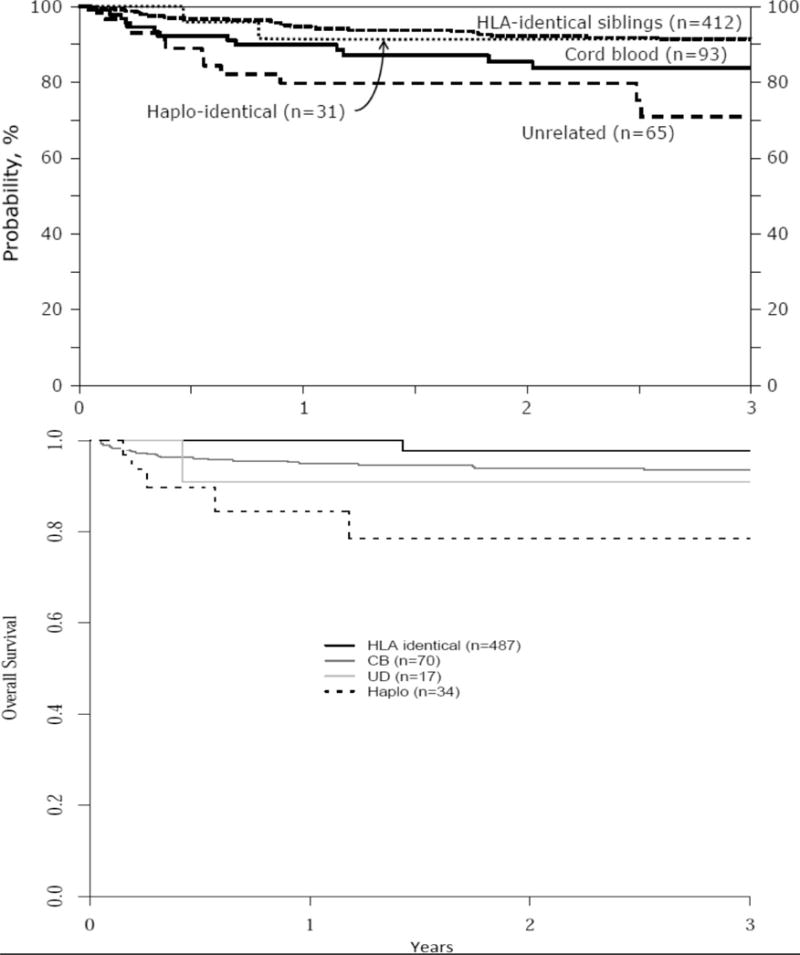
Depictions of overall survival after HCT for sickle cell disease according to donor source, as analyzed from the CIBMTR (upper panel) and European (EBMT) (lower panel) registries. Hematopoietic cell source is indicated in both panels. Reproduced with permission19.
However, an alternate view recently expressed by sickle cell disease experts is that “additional research is still needed that addresses the potential risks of this therapy (e.g., failure of engraftment and chronic graft-versus-host disease, GVHD) before HCT can become a widely used therapy”22. The objective of reducing the risk of HCT in children with SCD has been largely accomplished when an HLA-ID sibling donor is used, which challenges this opinion. The 2-year transplant-related mortality risk after HLA-ID sibling donor HCT appears very similar to the risk in children with SCD who receive standard supportive care. This notion is further strengthened by a recent report that described a cohort of 469 children and adults with SCD in Belgium in which the 15-year survival after transplantation compared to supportive care was not significantly different (mortality rate of 0.38 and 0.36/100 patient years, respectively, in supportive care and transplant groups), although survival was superior in a group treated by HU23. Novel methods in preserving fertility after HCT should hasten a revision of the current indications for HLA-ID sibling HCT, extending this therapeutic option to all children who have a sickle cell anemia genotype. Oocyte cryopreservation has been reported in a patient with sickle cell disease24. Cryopreservation of ovarian tissue is available as a research procedure, and this tissue might be utilized to restore endocrine and reproductive function after HCT25.
Eligibility Criteria – Adults with SCD
There is limited but growing transplant experience in adults with SCD. The eligibility criteria used in 2 active trials are shown in Table 3. There is a potential for a wider acceptability of HCT in adults with severe SCD for several reasons. First, unlike the experience in childhood during which survival to adulthood improved significantly after the institution of comprehensive care, the mortality rate among adults has changed very little in the past several decades26, 27. Patients with symptomatic SCD who participated in the Multicenter Study of Hydroxyurea (MSH) represent a cohort that mirrors adult patients referred for HCT.28 The annual mortality rate in the long-term (17.5 years) follow-up cohort from the MSH study was 4.4 per 100 person-years. The mortality rate was quite similar across groups who received HU for less than 5 years, 5 to <10 years, and 10 to <15 years. After roughly 13 years of continuous HU treatment, however, the mortality rate declined to 2.5% per year and 2.25% per year in the groups of patients treated for the longest periods of time. Another recent report of a cohort that included 534 adults with SCD showed 25% mortality at the end of the 10-year study. The mortality was even higher among those having >4 pain crisis per year or those with a higher organ severity score27. Thus, if the 2-year mortality probability after HCT in adults is <20%, it is very likely that transplantation will confer a long-term survival advantage in adult recipients.
Table 3.
Indications of HCT in adult recipients with SCD
| Age, 15 – 40 years with SCD AND one or more of the following |
| Clinically significant neurologic event (stroke) or any neurological deficit lasting >24 hours |
| History of 2 or more episodes of acute chest syndrome (ACS) in the 2-year period preceding HCT despite the institution of supportive care measures (i.e. asthma therapy and/or HU) |
| History of 3 or more severe pain crises per year in the 2-year period preceding enrollment despite the institution of supportive care measures (i.e. a pain management pain and/or treatment with HU) |
| Administration of regular RBC transfusion therapy, defined as receiving 8 or more transfusions per year for≥1 year to prevent vaso-occlusive clinical complications (i.e. pain, stroke and acute chest syndrome) |
| An echocardiographic finding of tricuspid valve regurgitant jet (TRJ) ≥ 2.7 m/sec |
Doppler trans-thoracic echocardiography has been validated in several cohorts as a screening tool to identify high-risk patients for early mortality. In 3 large screening studies, approximately 30% of patients had a tricuspid valve regurgitant jet velocity (TRV) >2.5 m/s and 10% had a TRV >3 m/s. In all the epidemiological studies performed to date, the risk ratio of early death in adults with a TRV > 2.5 m/s ranged from 9.24 to 15. 9 fold29–31. More recently, a larger cohort of 483 patients was screened in the Walk-PHASST (Walk-Treatment of Pulmonary Hypertension and Sickle Cell Disease with Sildenafil Therapy) screening study32, 33. In this cohort, 67 participants (9.1%) had TRV was > 3 m/sec which was associated with a 2-year mortality of 11.9%. Using a two-variable positive-predictive value assessment of 2-year mortality risk in this same cohort, it was possible to identify very high-risk patients. Subjects who had a combination of TRV >3 m/sec with WBC> 13,500 or chronic transfusion therapy had a 2-year mortality rate that exceeded 20%34. Finally, 43 of 240 subjects from a cohort of adults with SCD died between 2000–2005 with a median survival of 40 years35. The authors concluded that HU protected from acute sickle-related events but not from cardiopulmonary complications that were the leading cause of death. Thus, in selected groups of adults with SCD, even if the 2-year probability of transplant-related mortality is approximately 20%, survival in the short-term compared to those who lack a donor might be acceptable.
Transplant Results in Adults: HLA-ID Sibling HCT
The possibility of successful HCT in young adults with SCD was suggested by a report of 15 patients from a French group and by parallel efforts in thalassemia major in which myeloablative regimens with reduced toxicity have been developed to lower the risk of transplant-related mortality36, 37. While reduced intensity and non-myeloablative conditioning regimens have been successful in treating hematological malignancies where there are co-morbidities, these have been less successful in hemoglobinopathies38, 39. However, results of recent trials suggest progress that might be extended to clinical practice. Between 2004 and 2013, 30 patients with severe disease who were 16 to 65 years of age were treated with Alem, total-body irradiation (TBI, 300 cGy), and sirolimus followed by HLA-ID sibling filgrastim-mobilized peripheral blood stem cell transplantation in a single center trial9, 40. With a median follow-up of 3.6 (range, 1.8 – 6) years, 87% of recipients had long-term engraftment without acute or chronic GVHD and overall survival was 97%. However, 11 of 26 surviving patients who had mixed donor-host chimerism were still receiving immunosuppressive therapy at last follow-up due to concerns about late graft failure. More recently, a multicenter pilot trial of HCT for SCD reported results in 22 adults with severe sickle cell disease treated by HLA-ID related (N=17) or unrelated (N=5) donor HCT after a myeloablative combination of BU, Flu, and rabbit ATG. Twenty-one of 22 patients survive after HCT, all with engraftment of donor cells41. Together, these initial series have generated very good results and if confirmed in a larger, multicenter investigation of HLA-ID sibling HCT in adults, could support the notion that survival after transplantation is not inferior to survival in those not treated by HCT.
Summary
Just as refinement in supportive care for sickle cell disease has improved the likelihood of survival to adulthood, results after HLA-ID sibling HCT have also improved significantly in the past 15 years. In addition, the goal of cure is achieved in 90% or more of transplant recipients. Because there appears to be very little if any survival disadvantage after HCT compared to those who receive supportive care, a broadened view about transplant eligibility is warranted42. The role of HCT in adults with symptomatic SCD is less well defined because of the small number of published reports; however, projections based upon the risk of early mortality in adults with severe SCD also warrants broader endorsement of HCT as a therapeutic option, particularly when investigated in NIH-funded prospective clinical trials.
Table 4.
Recent results of HLA-matched HCT in adults with SCD
| Center | Preparative regimen | GvHD prophyl | n | Age range (yrs) | Published outcomes | Latest follow up | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Follow up (yrs) | Death (mos) | Complications | IST | GvHD | % donor chimerism | |||||
| NHLBI (Hsieh, 2009, 2014) | Alem 1mg/kg,TBI 300 cGy | Sirolimus | 30 | 17–65 | 1.8–6 | 1 (ICH) after GR | GR (n=4) | 11 of 26 | none | Median T-Cell 48% |
| France (Kuentz, 2011) | BU 14 mg/kg, CY 200 mg/kg, rATG 10 mg/kg | CsA, MTX | 15 | 16 – 27.5 | 1 – 16.1 | 1 (ICH) | SDH (n=1), seizures | – | 8 acute GVHD, 2 chronic GVHD | 75 – 100% |
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bernaudin F, Socie G, Kuentz M, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110:2749–2756. doi: 10.1182/blood-2007-03-079665. [DOI] [PubMed] [Google Scholar]
- 2.Vermylen C, Cornu G, Ferster A, et al. Haematopoietic stem cell transplantation for sickle cell anaemia: the first 50 patients transplanted in Belgium. Bone Marrow Transplant. 1998;22:1–6. doi: 10.1038/sj.bmt.1701291. [DOI] [PubMed] [Google Scholar]
- 3.van Besien K, Koshy M, Anderson-Shaw L, et al. Allogeneic stem cell transplantation for sickle cell disease. A study of patients’ decisions. Bone Marrow Transplant. 2001;28:545–549. doi: 10.1038/sj.bmt.1703208. [DOI] [PubMed] [Google Scholar]
- 4.Walters MC, Patience M, Leisenring W, et al. Barriers to bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. 1996;2:100–104. [PubMed] [Google Scholar]
- 5.Mentzer WC, Heller S, Pearle PR, Hackney E, Vichinsky E. Availability of related donors for bone marrow transplantation in sickle cell anemia. The American journal of pediatric hematology/oncology. 1994;16:27–29. [PubMed] [Google Scholar]
- 6.Hankins J, Hinds P, Day S, et al. Therapy preference and decision-making among patients with severe sickle cell anemia and their families&. Pediatr Blood Cancer. 2007;48:705–710. doi: 10.1002/pbc.20903. [DOI] [PubMed] [Google Scholar]
- 7.Roth M, Krystal J, Manwani D, Driscoll C, Ricafort R. Stem cell transplant for children with sickle cell anemia: parent and patient interest. Biol Blood Marrow Transplant. 2012;18:1709–1715. doi: 10.1016/j.bbmt.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 8.Omondi NA, Ferguson SE, Majhail NS, et al. Barriers to hematopoietic cell transplantation clinical trial participation of african american and black youth with sickle cell disease and their parents. Journal of pediatric hematology/oncology. 2013;35:289–298. doi: 10.1097/MPH.0b013e31828d5e6a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. Jama. 2014;312:48–56. doi: 10.1001/jama.2014.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walters MC, Hardy K, Edwards S, et al. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2010;16:263–272. doi: 10.1016/j.bbmt.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. The New England journal of medicine. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 12.Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115:3447–3452. doi: 10.1182/blood-2009-07-233700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Telfer P, Coen P, Chakravorty S, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica. 2007;92:905–912. doi: 10.3324/haematol.10937. [DOI] [PubMed] [Google Scholar]
- 14.Lucarelli G, Isgro A, Sodani P, et al. Hematopoietic SCT for the Black African and non-Black African variants of sickle cell anemia. Bone Marrow Transplant. 2014;49:1376–1381. doi: 10.1038/bmt.2014.167. [DOI] [PubMed] [Google Scholar]
- 15.Dedeken L, Le PQ, Azzi N, et al. Haematopoietic stem cell transplantation for severe sickle cell disease in childhood: a single centre experience of 50 patients. Br J Haematol. 2014;165:402–408. doi: 10.1111/bjh.12737. [DOI] [PubMed] [Google Scholar]
- 16.Bhatia M, Jin Z, Baker C, et al. Reduced toxicity, myeloablative conditioning with BU, fludarabine, alemtuzumab and SCT from sibling donors in children with sickle cell disease. Bone Marrow Transplant. 2014;49:913–920. doi: 10.1038/bmt.2014.84. [DOI] [PubMed] [Google Scholar]
- 17.McPherson ME, Hutcherson D, Olson E, Haight AE, Horan J, Chiang KY. Safety and efficacy of targeted busulfan therapy in children undergoing myeloablative matched sibling donor BMT for sickle cell disease. Bone Marrow Transplant. 2011;46:27–33. doi: 10.1038/bmt.2010.60. [DOI] [PubMed] [Google Scholar]
- 18.King AA, Kamani N, Bunin N, et al. Successful Matched Sibling Donor Marrow Transplantation Following Reduced Intensity Conditioning in Children with Hemoglobinopathies. American journal of hematology. 2015 doi: 10.1002/ajh.24183. [DOI] [PubMed] [Google Scholar]
- 19.Gluckman E. Allogeneic transplantation strategies including haploidentical transplantation in sickle cell disease. Hematology/the Education Program of the American Society of Hematology. American Society of Hematology. Education Program. 2013;2013:370–376. doi: 10.1182/asheducation-2013.1.370. [DOI] [PubMed] [Google Scholar]
- 20.Locatelli F, Kabbara N, Ruggeri A, et al. Outcome of patients with hemoglobinopathies given either cord blood or bone marrow transplantation from an HLA-identical sibling. Blood. 2013;122:1072–1078. doi: 10.1182/blood-2013-03-489112. [DOI] [PubMed] [Google Scholar]
- 21.Angelucci E, Matthes-Martin S, Baronciani D, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99:811–820. doi: 10.3324/haematol.2013.099747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. Jama. 2014;312:1033–1048. doi: 10.1001/jama.2014.10517. [DOI] [PubMed] [Google Scholar]
- 23.Le PQ, Gulbis B, Dedeken L, et al. Survival among children and adults with sickle cell disease in Belgium: Benefit from hydroxyurea treatment. Pediatr Blood Cancer. 2015 doi: 10.1002/pbc.25608. [DOI] [PubMed] [Google Scholar]
- 24.Dovey S, Krishnamurti L, Sanfilippo J, et al. Oocyte cryopreservation in a patient with sickle cell disease prior to hematopoietic stem cell transplantation: first report. Journal of assisted reproduction and genetics. 2012;29:265–269. doi: 10.1007/s10815-011-9698-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demeestere I, Simon P, Dedeken L, et al. Live birth after autograft of ovarian tissue cryopreserved during childhood. Human reproduction. 2015;30:2107–2109. doi: 10.1093/humrep/dev128. [DOI] [PubMed] [Google Scholar]
- 26.Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine. 2005;84:363–376. doi: 10.1097/01.md.0000189089.45003.52. [DOI] [PubMed] [Google Scholar]
- 27.Elmariah H, Garrett ME, De Castro LM, et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. American journal of hematology. 2014;89:530–535. doi: 10.1002/ajh.23683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. American journal of hematology. 2010;85:403–408. doi: 10.1002/ajh.21699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ataga KI, Moore CG, Jones S, et al. Pulmonary hypertension in patients with sickle cell disease: a longitudinal study. British journal of haematology. 2006;134:109–115. doi: 10.1111/j.1365-2141.2006.06110.x. [DOI] [PubMed] [Google Scholar]
- 30.De Castro LM, Jonassaint JC, Graham FL, Ashley-Koch A, Telen MJ. Pulmonary hypertension associated with sickle cell disease: clinical and laboratory endpoints and disease outcomes. American journal of hematology. 2008;83:19–25. doi: 10.1002/ajh.21058. [DOI] [PubMed] [Google Scholar]
- 31.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. The New England journal of medicine. 2004;350:886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 32.Machado RF, Barst RJ, Yovetich NA, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood. 2011;118:855–864. doi: 10.1182/blood-2010-09-306167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sachdev V, Kato GJ, Gibbs JS, et al. Echocardiographic markers of elevated pulmonary pressure and left ventricular diastolic dysfunction are associated with exercise intolerance in adults and adolescents with homozygous sickle cell anemia in the United States and United Kingdom. Circulation. 2011;124:1452–1460. doi: 10.1161/CIRCULATIONAHA.111.032920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rotz SJ, O’Riordan MA, Kim C, de Lima M, Gladwin MT, Little JA. Traffic Light: prognosis-based eligibility for clinical trials of hematopoietic SCT in adults with sickle cell anemia. Bone Marrow Transplant. 2015;50:918–923. doi: 10.1038/bmt.2015.11. [DOI] [PubMed] [Google Scholar]
- 35.Fitzhugh CD, Lauder N, Jonassaint JC, et al. Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. American journal of hematology. 2010;85:36–40. doi: 10.1002/ajh.21569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bernardo ME, Piras E, Vacca A, et al. Allogeneic hematopoietic stem cell transplantation in thalassemia major: results of a reduced-toxicity conditioning regimen based on the use of treosulfan. Blood. 2012;120:473–476. doi: 10.1182/blood-2012-04-423822. [DOI] [PubMed] [Google Scholar]
- 37.Kuentz M, Robin M, Dhedin N, et al. Is there still a place for myeloablative regimen to transplant young adults with sickle cell disease? Blood. 2011;118:4491–4492. doi: 10.1182/blood-2011-07-367490. author reply 4492–4493. [DOI] [PubMed] [Google Scholar]
- 38.Horan JT, Liesveld JL, Fenton P, Blumberg N, Walters MC. Hematopoietic stem cell transplantation for multiply transfused patients with sickle cell disease and thalassemia after low-dose total body irradiation, fludarabine, and rabbit anti-thymocyte globulin. Bone Marrow Transplant. 2005;35:171–177. doi: 10.1038/sj.bmt.1704745. [DOI] [PubMed] [Google Scholar]
- 39.Iannone R, Casella JF, Fuchs EJ, et al. Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and beta-thalassemia. Biol Blood Marrow Transplant. 2003;9:519–528. doi: 10.1016/s1083-8791(03)00192-7. [DOI] [PubMed] [Google Scholar]
- 40.Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. The New England journal of medicine. 2009;361:2309–2317. doi: 10.1056/NEJMoa0904971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krishnamurti L, et al. Pilot Investigation of Bone Marrow Transplantation in Adults with Severe Sickle Cell Disease (STRIDE) Blood. 2015 [Google Scholar]
- 42.Majhail NS, Farnia SH, Carpenter PA, et al. Indications for Autologous and Allogeneic Hematopoietic Cell Transplantation: Guidelines from the American Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2015 doi: 10.1016/j.bbmt.2015.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Powars D, Weidman JA, Odom-Maryon T, Niland JC, Johnson C. Sickle cell chronic lung disease: prior morbidity and the risk of pulmonary failure. Medicine. 1988;67:66–76. [PubMed] [Google Scholar]
- 44.Majumdar S, Robertson Z, Robinson A, Starnes S, Iyer R, Megason G. Outcome of hematopoietic cell transplantation in children with sickle cell disease, a single center’s experience. Bone Marrow Transplant. 2010;45:895–900. doi: 10.1038/bmt.2009.244. [DOI] [PubMed] [Google Scholar]
- 45.Strocchio L, Zecca M, Comoli P, et al. Treosulfan-based conditioning regimen for allogeneic haematopoietic stem cell transplantation in children with sickle cell disease. British journal of haematology. 2015;169:726–736. doi: 10.1111/bjh.13352. [DOI] [PubMed] [Google Scholar]