

Synopsis
The number of available symptomatic treatments has markedly enhanced the care of patients with Amyotrophic Lateral Sclerosis (ALS). Once thought to be “untreatable”, patients with ALS today clearly benefit from multidisciplinary care. The impact of such care on the disease course, including rate of progression and mortality, has surpassed the treatment effects commonly sought in clinical drug trials. Unfortunately, there are few randomized controlled trials of medications or interventions addressing symptom management which has resulted in the need for physicians to base their selection of specific therapies upon personal experience and anecdotal reports (1 Forshew). In this review, we will provide the level of evidence, when available, for each intervention that is currently considered “standard of care” by consensus opinion.
Keywords: Sialorrhea, pseudobulbar affect, non-invasive ventilation, secretion management, laryngospasm, edema, urinary urgency, constipation, depression, anxiety and end of life issues
Sialorrhea
Sialorrhea is a socially embarrassing symptom related to pharyngeal muscle weakness which can lead to aspiration pneumonia, the most common cause of death in ALS other than respiratory failure. The prevalence of the sialorrhea among ALS patients is estimated at 50% (2 Sufit). Patients frequently have to wipe their mouth with a tissue, or in extreme cases, may need to insert a paper towel or washcloth into their mouths to absorb the saliva.
The American Academy of Neurology practice parameter for the care of ALS patients published in 1999 recommended both pharmacologic interventions and non-pharmacologic approaches such as suctioning (3 Miller). Treatment with anticholinergic medication (Table 1) (4 Jackson) is considered “first line” pharmacologic therapy, however, the benefits of this class of medication can be insufficient to completely address the symptom. In addition, common side effects associated with anticholinergic therapy include constipation, fatigue, impotence, urinary retention, blurred vision, tachycardia, orthostatic hypotension and dizziness. These side effects occur most commonly in elderly patients. In addition, anticholinergic medications are relatively contraindicated in patients with a history of glaucoma, benign prostatic hypertrophy, or cardiac conduction disorders (especially bifascicular block, left bundle-branch block and a prolonged QT interval).
Table 1.
Medications Commonly Used For Sialorrhea
| Medication | Dose |
|---|---|
| Amitriptyline | 25–75 mg qhs |
| Nortriptyline | 20–100 mg qhs |
| Atropine | 0.4 mg q 4–6 h |
| 1–2 ophthalmic drops SL q 4–6 h | |
| Glycopyrrolate | 1–2 mg TID |
| Scopolamine patch | Apply behind ear q 3 days |
| Hyoscyamine sulfate | 0.125 mg – 0.25 mg q 4–6 h (available as oral tabs, elixir or sub-lingual tabs) |
| Botulinum Toxin Type A | |
| Botulinum Toxin Type B |
Selection of a particular medication often depends upon the severity and frequency of the drooling. Sialorrhea associated with mealtimes or a particular time of day may be treated with “prn” administration of atropine, hyoscyamine or glycopyrrolate. Transdermal scopolamine, botulinum toxin or antidepressant medications provide a more continuous effect. Patients who have difficulty swallowing medications may prefer an agent that can be given sublingually, transdermally, or is available in a liquid form that can be administered directly through a PEG tube.
Data from the national ALS Patient CARE database indicate that over 70% of ALS patients treated with atropine, glycopyrrolate, or amitriptyline reported these modalities were helpful (2 (2 Sufit, 4 Bradley). By inference, approximately 30% of patients were not helped by these therapies. There remains, to date, no randomized trial comparing the efficacy of these different agents in the ALS population. In general, all of these medications may cause or aggravate existing problems with constipation, and therefore it is recommended that a stool softener be initiated at the same time the anti-cholinergic agent is prescribed.
Botulinum toxin is the newest mode of sialorrhea therapy and has, thus far, shown to have great promise in patients resistant to conventional medical therapy (6 Portis, 7 Geiss, 8 Rowe, 9 Bhatia, 10 Jackson). In a double-blind, controlled trial of botulinum toxin type B (BTxB) in 20 patients with ALS with refractory sialorrhea (10 Jackson), patients were randomized to 2,500 U of BTxB or placebo into bilateral parotid and submandibular glands. Treated patients reported a global improvement of 82% at 2 and 4 weeks compared to 38% in placebo (p < 0.05). At 12 weeks, 50% of patients receiving BTxB were improved compared to 14% receiving placebo. There were no significant adverse events.
Radiation therapy for medically refractory sialorrhea reduced salivary production, but side effects included erythema, sore throat, and nausea (11 Harriman). A “satisfactory response” was observed and saliva secretion rate diminished with a single dose of 7–7.5 Gy bilaterally (12 Andersen).
Based on the AAN Practice Guidelines published in 2009 (13 Miller), in patients with ALS who have medically refractory sialorrhea, BTxB should be considered (Level B) and low-dose radiation therapy to the salivary glands may be considered (Level C).
Sticky secretions can also be problematic for a patient with ALS. These secretions may originate not only from the salivary glands but mucous from the nose and mouth may also contribute to them. (12 A Marin 1986) Sticky secretions can be a symptom of dehydration and encouraging patient to increase fluid intake may be helpful. Using a room humidifier for those not using noninvasive ventilation and bleeding humidified air into the machine for those who do may reduce their tenacity. An open label trial of 16 patients with thick secretions that had failed anticholinergic therapy were treated with either propanolol 10 mg twice a day or metoprolol 25 mg twice a day found that 75% of patients reported decreased secretions with sustained short-term efficacy in most. (12 B Newall 1996) Drinking papaya or pineapple juice, and reducing caffeine, milk or alcohol have been anecdotally suggested (Radunovic 12 C).
Pseudobulbar Affect
Pseudobulbar affect (PBA) affects 20%–50% of patients with ALS, especially in patients with bulbar onset. (14 McCullagh) The characteristics of PBA include uncontrolled laughter or crying, often with minimal or no provocation. Episodes are often sudden, involuntary outbursts of emotion inappropriate to the context of the situation. Patients experiencing uncontrolled crying are more common than those with uncontrolled laughter, however, the symptoms can result in significant disability, limiting social interactions and impairing quality of life (QoL).
Although it is not a mood disorder, selective serotonin reuptake inhibitors, tricyclic antidepressants and some serotonin-norepinephrine reuptake inhibitors have been used for treatment of PBA (Table 2) (4 Jackson).
Table 2.
Medications For Treatment of Pseudobulbar Affect
| Drug | Dose | Common Side Effects |
|---|---|---|
SSRI antidepressants:
|
10–50 mg qd 10–30 mg qd 50mg qd - BID 50–100 qd - BID 20–60 mg qd |
Sexual dysfunction, akathisia, sleep disturbance, anxiety |
Tricyclic antidepressants:
|
25–75mg qhs |
Dry mouth, fatigue, dizziness, urinary retention, constipation |
| Mirtazapine | 15mg qhs | Abnormal dreams, mild cognitive change, constipation, |
| Venlafaxine | 37.5–75mg BID -TID | Anorexia, constipation, weight loss, impotence, anxiety, dizziness |
| Dextromethorphan/Quinidine | 20mg/10mg 1 tab BID | Nausea, dizziness, somnolence, loose stools |
A novel combination of dextromethorphan (20mg) and quinidine sulfate (10mg) (Nuedexta™) has been shown to be effective in a large phase three multicenter randomized trial (15 Brooks). Nuedexta patients reported significantly less emotional lability, improved QoL and improved quality of relationship scores. Side effects included dizziness, nausea, and somnolence. These side effects can be minimized by initiating the dose at 1 tablet at bedtime for 7 days followed by twice a day dosing. The AAN Practice Guidelines (13 Miller) recommend that if side effects are acceptable, dextromethorphan/quinidine should be considered for symptoms of pseudobulbar affect in patients with ALS (Level B).
Sleep disruption
Sleep disruption in patients with ALS is frequently multifactorial in etiology and may be due to respiratory muscle weakness, difficulty re-positioning in bed, anxiety, depression and pain. Nocturnal hypoventilation results in frequent arousals and decreased total sleep time resulting in daytime fatigue and poor concentration. Overall, impaired sleep can markedly affect QoL and likely impact prognosis.
Available solutions to address impaired sleep are as varied as the diverse issues causing it. Simple physical adaptations such as an electric hospital bed can be ideal to enhance mobility and positioning. An alternating pressure air mattress or gel overlay mattress can lessen the discomfort from limited mobility. Noninvasive positive pressure ventilation can significantly affect respiratory hygiene and improve sleep quality. Antidepressant medications are also effective at reducing anxiety, depression and promoting sleep. In that regard, mirtazapine (15mg qhs) can be especially helpful. At higher doses (≥ 30 mg) mirtazapine may have a confounding effect due to enhancement of its noradrenergic effect. The anticholinergic action of the tricyclic antidepressant group can also be especially helpful, particularly in patients with sialorrhea.
Anxiolytic medications such as benzodiazepines, used specifically to induce sleep, can be helpful when used selectively. Zolpidem tartrate (10mg qhs) is often effective and preferred due to the low risk of respiratory depression. Pharmacological tolerance and withdrawal symptoms can become evident, however, with chronic use. Addressing the underlying cause of such anxiety (depression, fear, pain etc) is a preferred method of treatment.
Alternative pharmacologic agents such as melatonin, passionflower, lavender, and hops have been effective for individual patients, however, their benefits are quite variable and untested.
Respiratory Insufficiency
Patients with ALS commonly develop respiratory failure as a result of decreased diaphragmatic and intercostal muscle strength and impaired glottis function resulting in an ineffective cough. (16 Arnulf). Dyspnea is an uncommon presenting symptom of chronic respiratory failure. Rather, patients usually present insidiously with symptoms related to nocturnal oxygen desaturations such as frequent nighttime arousals, morning headache, excessive daytime sleepiness, orthopnea and fatigue.
Patients with ALS should have an assessment of pulmonary function every 3–6 months. FVC is the most commonly used respiratory measurement in ALS and is a significant predictor of survival (17 Czaplinski). In most cases, however, the earliest detectable abnormality is a reduction in maximum inspiratory pressure (MIP), sniff nasal pressure (SNP), sniff trandiaphragmatic pressure (sniff Pdi) or nocturnal oximetry (desaturations <90% for 1 cumulative minute) (18 Jackson). Supine FVC, although more difficult to perform, may be a better predictor of diaphragm weakness than erect FVC (19 Lechtzin, 20 Varrato).
When FVC falls below 50% of normal or the MIP is < 60cm, non-invasive ventilation should be initiated. Discussions about respiratory interventions should always take place in concert with a pulmonologist or respiratory therapist expert in managing neuromuscular patients, and the opinions and wishes of patients and family should be the prime consideration. If the topics of a living will and durable power of attorney for health care have not yet been addressed, they should be discussed. (21 Vaszar, 22 Hull)
Non-invasive ventilation (NIV) should be considered to treat respiratory insufficiency in ALS, both to lengthen survival and to slow the rate of FVC decline (Level B) (23 Miller). In a randomized controlled study, patients using NIV experienced a median survival benefit of 205 days (24 Bourke). NIV was initiated based on orthopnea with an MIP <−60 cm or symptomatic hypercapnia. FVC declined more slowly after introducing NIV (pre −2.2%/month compared to post −1.1%/month) and the decline was slower in those who used NIV >4 hours/day.
NIV has demonstrated a positive impact on QOL in several studies (Level C). (23 Miller, 18 Jackson, 25 Bourke, 26 Butz) There were improvements noted in energy, vitality, (25 Bourke, 27 Lyall) shortness of breath, daytime somnolence, depression, concentration, sleep quality, and physical fatigue for 10 months or more. (26 Butz)
NIV may be considered at the earliest sign of nocturnal hypoventilation or respiratory insufficiency in order to improve compliance (Level C) (23 Miller, 28 Pinto, 29 Aboussouan). Orthopnea has been shown to be a strong predictor of benefit and also better compliance with NIV (24 Bourke). NIV use correlated with symptoms of orthopnea and dyspnea as well as with the use of PEG, speech devices, and riluzole (30 Jackson). Young age and preserved upper limb function have also predicted better compliance.
Noncompliance with NIV was seen in 75% of patients with ALS and frontotemporal dysfunction vs 38% in patients with classic ALS (relative risk 2.0) (31 Olney). Low compliance has also been reported in bulbar patients (24 Bourke, 32 Gruis) but cognitive/executive function was not described. Compliance can be improved by prescribing a variety of different interfaces including nasal mask, nasal pillows, full-face masks or mouth pieces in order to maximize patient comfort and minimize pressure sores. NIV should be started at low pressures to maximize a patient’s tolerance to the device. Generally the inspiratory pressure (IPAP) is set at 6–8 cm H20 and the expiratory pressure (EPAP) at 3–5 cm H20. The IPAP can then be titrated in 1–2 cm increments based on patient tolerance and clinical status. The device should be placed on a spontaneous timed mode with a backup rate set at the patient’s nocturnal respiratory rate (generally 8–10). Frequent follow-up by a trained respiratory therapist is crucial to deal with any problems affecting patient tolerance. A heated humidifier should be prescribed if the patient develops symptoms of nasal dryness or congestion.
Expiratory respiratory muscle weakness can lead to ineffective cough, retained upper airway secretions, and pulmonary infections. PCEFs greater than 160 L/min are needed to clear secretions, (33 Bach) and when the PCEF falls below 270 L/min, (34 Tzeng) mechanical insufflation/exsufflation (MIE) may be considered to clear secretions, particularly during an acute chest infection (Level C). (23 Miller) These devices allow delivery of pressurized air (30–50 cm H2O) followed by an immediate forced exsufflation of negative pressure (30–50 cm H2O) for several seconds via a mouthpiece or naso-oral mask. The use of MIE safely allows patients with respiratory tract infections and profuse mucous production to clear their secretions and permit the ongoing use of NIV. MIE pressures should be set to the maximum comfortable level tolerated by each patient in order to eliminate airway secretions. In general, inspiratory pressures are set at 20–40 cm H20 and expiratory pressures are set at 5–20 cm H20. Another device useful for helping patients clear secretions is the “chest vest” that rapidly inflates and deflates resulting in a vibratory like manner up to 25 chest wall compressions/second. This helps dislodge mucous from the bronchial walls for removal via cough or a cough assist device. There is currently insufficient data to support or refute HFCWO for clearing airway secretions in patients with ALS (Level U). (Miller 23)
Medications with mucolytics like guaifenesin or N-acetylcysteine, a β-receptor antagonist (such as metoprolol or propanolol), nebulized saline, or an anticholinergic bronchodilator such as ipratropium are widely used; however, no controlled studies exist in ALS.
The Centers for Disease Control recommends that essentially all patients with significant neuromuscular illness should receive one or more doses of the pneumococcal vaccine and annual vaccines for the pertinent influenza virus. Adequate hydration must also be maintained to assure easy mobilization of secretions.
Acute respiratory failure in ALS patients is usually precipitated by an acute infection or pulmonary embolism and is seldom due to muscle weakness alone. For dyspneic or obtunded patients, emergency evaluation for endotracheal intubation may be necessary. If hypoxia alone is present and the patient is alert, oxygen at low-flow rates (< 1 L/min) may correct hypoxia although such therapy must be administered with caution, since patients with neuromuscular disease are often hypercapneic, and oxygen often depresses ventilatory drive and exacerbates hypoventilation. (35 Mangera) NIV is usually tried initially if control of secretions and cough assistance is successful. (36 Garpestad) Whether intubation or NIV is utilized, patients sometimes can be weaned following the precipitating respiratory crisis; others may require ventilation assistance indefinitely. Pulmonary consultation is always indicated in these patients.
NIV is contraindicated in patients with copious secretions, cognitive impairment, or lack of caregiver support. (37 Perrin, 38 Park) In these instances, ventilation may be more easily managed with tracheostomy and positive-pressure ventilation. (figure 1) Initiation of chronic respiratory support poses major practical, financial, medical, and ethical responsibilities for patients and families and it is often helpful for patients to discuss the procedure with others receiving such support. The input of experienced pulmonary consultants and palliative medicine services can be invaluable in these instances.
Figure 1. Respiratory management algorithm.
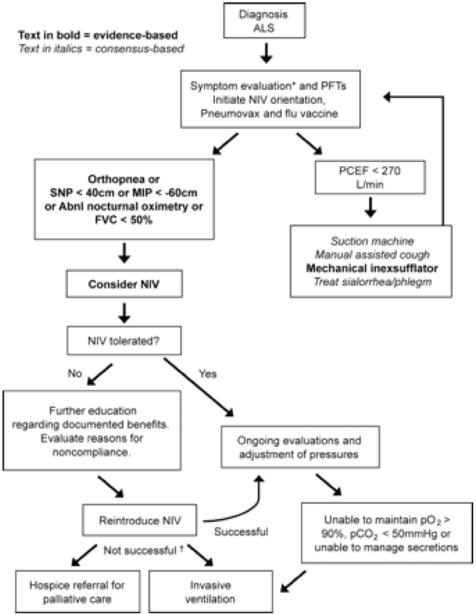
PFT = pulmonary function tests; PCEF = peak cough expiratory flow; NIV = noninvasive ventilation; SNP = sniff nasal pressure; MIP = maximal inspiratory pressure; FVC = forced vital capacity (supine or erect); Abnl.nocturnal oximetry = pO2 <4% from baseline. *Symptoms suggestive of nocturnal hypoventilation: frequent arousals, morning headaches, excessive daytime sleepiness, vivid dreams, †If NIV is not tolerated or accepted in the setting of advancing respiratory compromise, consider invasive ventilation or referral to hospice.
Taken from 23 Miller
Fatigue
Fatigue is reported in 44–83% of patient with ALS and likely is multifactorial with sleep disruption, nocturnal complaints such as nocturia and cramps, nutritional status, weakness, vital capacity, functional status, depression, and medications including riluzole all potentially playing a role. (39 McElhiney 2009, 40 LoCoco 2012, 41 Ramirez 2008, 42 Bensimon 1994)) In a given patient, it may be challenging to tease out the biggest contributors, but treating those that are readily identified to see if fatigue improves is a practical approach. Overall and physical fatigue but not mental fatigue was found more often in ALS patients compared to controls. (43 Lou J-S 2003) Modafinil in doses ranging from 100 to 300 mg daily in a placebo controlled trial for 4 weeks resulted in a statistically significant improvement in the Clinical Global Impression score (the study’s primary endpoint) in 19/25 on active drug and in 1/7 on placebo (p=0.003) as well as improvement on the Visual Analogue Scale (VAS) for energy (p=0.039) and the VAS for stamina (p=0.009) but not in the Fatigue Severity Scale (p=0.066. (44 Rabkin 2009).
Nutrition
Weight loss in ALS patients may occur because of dysphagia, muscle atrophy, poor appetite related to depression, and hypermetabolic state. (45 Mascaritioli, 46 Desport, 47 Dupuis) In addition, caloric needs in an ALS patient may be underestimated using traditional equations in part because of increased work of using weak muscles, as well as energy expenditure associated with cramps, spasticity, fasciculations, and pseudobulbar manifestations. A potentially more accurate way to measure an ALS patient’s total daily caloric needs uses the Harris-Benedict equation but also incorporates scores of 6 items from the ALS functional rating scale relating to speech, handwriting, dressing and hygiene, turning in bed, walking and dyspnea. (48 Kasarskis)
Weight loss of greater than 10%, (49 Limousin) rapid changes in BMI from time of symptom onset to diagnosis, (50 Shimuzu) as well as greater BMI changes in the first 2 years of being diagnosed with ALS (51 Jawaid) are all associated with a worse prognosis. Patients with a BMI classification of mild obesity at time of study entry had a better prognosis (52 Paganoni), and in a prospective nutritional and cancer study of over a half a million subjects followed for 14–28 years, higher body fat was associated with a lower risk of dying from ALS. (Gallo 2013)
Despite this data, there is mixed evidence regarding survival benefits of providing enteral nutrition through gastrostomy tubes in patients with ALS. In the absence of randomized controlled trials, physicians are left with case controlled studies and prospective and retrospective cohort studies. (Katzberg 54). Survival benefits for those undergoing a gastrostomy include studies authored by Chiò (55) and Czaplinski (56) while examples of those finding no benefit have been reported by Forbes (57) and Mitchell (58). Possible reasons for failure to always demonstrate benefit may be patient’s reluctance to do the procedure until they have lost a substantial amount of weight and so are already malnourished when the procedure is performed. Another challenge with enteral nutrition is that noncompliance with the nutritionist’s recommendations is not uncommon. (61)
Enteral nutrition has resulted in stabilization and or improvement in the BMI following gastrostomy placement. (59 Desport 2000, 60 Mazzini 1995). It also allows patients to stay hydrated and provides a safe manner to administer medications. Radiographically inserted gastrostomy tubes may confer both a survival benefit (62 Chio 2004) as well as be associated with fewer insertional failures and post procedure episodes of aspiration (63 Allen 2013) compared to endoscopically placed tubes.
Only small relatively short duration studies have investigated different types of diets in patients with ALS to determine if there is a preferred diet that could influence disease progression, with variable findings as described in Table 3.
Table 3.
Diets studied in ALS
| Author | Diet | N= | Duration | Results |
|---|---|---|---|---|
| Stanich 64 | High protein | 20 | 6 months | No change in muscle mass or disease progression |
| Silva 65 | High protein | 16 | 4 months | Stabilization of ALS-FRS-R |
| Dorst 66 | High fat, high calorie High carbohydrate, high calorie |
22 16 |
3 months | Wt stabilized ALS-FRS progressed High dropout rate with High carbohydrate/high calorie |
| Willis 67 | High fat, high calorie High carbohydrate, high calorie Control |
8 9 7 |
3 months | High carbohydrate, high calorie group had fewer adverse events, dropouts, and deaths |
Early swallowing problems are addressed with changes made while eating, including smaller bites, slowing down, and avoiding talking while eating. Consistency changes may be suggested, including blending solid foods or adding thickener to liquids. Speech therapists may provide instructions in chin tuck or head turning to improve the ease and safety of a swallow. As the disease progresses, these are frequently not sufficient, and a feeding tube is considered. Reasons to recommend a gastrostomy tube include malnutrition/weight loss, long meal times, evidence for aspiration, and dehydration. (Miller 1999 3) Although the AAN practice guidelines supports placement when the vital capacity is above 50% because of concerns of increased risk as this value falls, (Miller 23) recent data provides evidence that a gastrostomy tube can be safely placed with vital capacities below this value, particularly when noninvasive ventilation can be used during the procedure. (Czell 68)
Pain
Pain is reported in 57 – 72% of patients with ALS and may involve the extremities, neck, back or trunk. (Pizziementi (69) Chio 2012 (70)) Pain can occur at any time during the course of the disease, and does not necessarily correlate with depression but is associated with more impaired functional status and worse quality of life. (Pizziementi (69) Ajroud (71) Pagnini (72)) While nonsteroidal anti-inflammatory drugs are frequently used, a number of therapies are prescribed including non-opioid analgesics, opioids, muscle relaxants, quinine sulfate, gabapentin, steroids, botulinum toxin, and physical therapy. (Chio 2012 (70)) The more common causes of pain are felt to be related to limited range of motion in joints, general immobility, spasticity, and or cramps. Descriptions of pain include burning, aching, cramping, and shock-like. (Newrick 73)
A Cochrane review on this subject in 2013 found no controlled or quasi-controlled studies of treating pain in patients with ALS. (74). Therefore, one cannot endorse specific ways to treat pain in ALS, though sorting out the type of pain may help the physician develop a rational approach to developing a treatment plan. In addition, if pain is related to immobility, trying to improve the comfort of what the patient sits or sleeps on, such as changing the cushion on their wheelchair or suggesting an alternating pressure mattress may be practical interventions to consider.
Spasticity
Spasticity, in addition to being a source of pain in ALS patients, can also limit mobility and function. Studies specifically investigating treatment of spasticity in ALS patients are limited. Common medications used to treat spasticity in ALS patients include baclofen, tizanidine, benzodiazepines, and dantrolene. None of these have been specifically studied in ALS, but rather in other disorders such as multiple sclerosis and cerebral palsy associated with spasticity. (Miller 23 Radunokovic 12C) One concern in using these medications in ALS patients is that they can potentially worsen weakness, which could adversely impact function. Eight patients with pain attributed to spasticity who had failed oral medication management received an intrathecal baclofen pump. There was a significant reduction in the mean pain score, and 6/8 subjects had a reduction in their pain score including 3 who became pain free. Mean pre-operative Ashworth spasticity score was 2.93 and a year following pump placement the mean score was 1.72. (75 McClelland) In an unblinded trial, 14 patients were randomized to a moderate exercise program and 11 to their usual activities. At 3 months but not at 6 months patients performing moderate exercise showed less deterioration on the Ashworth spasticity scale; at 9 and 12 months patient dropouts made it impossible to compare the groups. (76 Drory 2001) An open label trial of levetiracetam in 20 patients found it improved cramps as well as the phasic but not tonic spasticity. (Bedlack 77) Marked spasticity of masseter muscles preventing jaw opening has been effectively treated in open label studies with botulinum toxin. (78, 79) Hydrotherapy, cryotherapy, heat, and ultrasound have also been used despite lack of controlled trials. (Anderson 2012 80)
LARYNGOSPASM
Laryngospasm is the sudden sensation that air can’t be moved in and out, usually lasting seconds accompanied by inspiratory stridor, or audible respirations due to a rapid and forceful contraction of laryngeal adductors. It is extremely frightening since the patient can’t take a breath or call for help. In a study on laryngospasm, 2 percent of a control group of 122 patients in the early stages of ALS reported laryngospasm (81 Sperfeld), though up to 19% of patients have reported it in later stages (1 Forshew). Common causes of laryngospasm in ALS are liquid or saliva in contact with the larynx, acid reflux, smoke, strong smells, emotion, alcohol, cold bursts of air, and even spicy foods. Non-pharmacologic measures help in many patients. A rapid change to the upright position of the upper body, fixation of the arms to stabilize the body, breathing through the nose, swallowing repetitively and breathing with slow exhalation through lips have been reported to shorten episodes. If frequent enough and non-pharmacologic measures are ineffective, benzodiazepines bid or tid can be used. (4 Jackson, 85 Kuhnlein) In the case of GERD, treat with anti-reflux therapy and prokinetic drugs like metoclopramide before meals and at bedtime.
JAW QUIVERING/CHEEK BITING
ALS patients may report jaw quivering or clenching due to spasticity, precipitated by pain, anxiety or cold. Jaw clenching can be severe enough to interfere with oral hygiene. Treatment includes benzodiazepines as listed in Table 4. Botulinum toxin injected at 2 sites within the masseters on each side has been reported anecdotally to be effective. (4) Dental guards can alleviate cheek biting at night.
Table 4.
Medications Used for Jaw Clenching
| Medication | Dose |
|---|---|
| Clonazepam | 0.5 –1 mg tid |
| Diazepam | 2.5 –5 mg tid |
| Lorazepam | 0.5 –1 mg tid |
| Alprazolam | 0.25–0.5 mg tid |
EDEMA
Dependent edema of the hands and feet occurs in weak limbs because of immobility and reduced muscle pump activity. If severe, it can be associated with painful burning sensations and fragile, easily damaged skin. Limb elevation is the primary intervention. A reclining wheelchair with leg elevation above the heart and hospital bed are probably the most comfortable and efficient, especially since the patient can reposition frequently. Moving the legs as much as possible, passive range of motion and stretching exercises performed by a caregiver several times a day, though labor intensive, are also helpful. Some patients can benefit from compression stockings, but they require proper fitting and are very difficult to put on and take off. Diuretics are avoided unless edema is severe, because fluid overload is not the issue and patients with ALS already have trouble with maintaining good hydration and diuretics worsen muscle cramps. (82 Borasio) If edema is not improved by elevating the limbs or asymmetrical, consider evaluating the patient for deep venous thrombosis since if chronic, patients may not complain of calf pain or have erythema.
CONSTIPATION AND URINARY URGENCY
Autonomic symptoms have been reported in up to 29% of patients with ALS. (89 Piccione) Common symptoms involve urinary and gastrointestinal dysfunction. (88 Rocha) Changes in the intermediolateral columns and the Onuf nucleus provide an anatomical explanation. Causes of constipation are multifactorial and include reduced mobility, reduced fluid and food intake and medications such as anticholinergics, tricyclic antidepressants, narcotics, muscle relaxants, and sedatives. Long delays in getting to the bathroom contribute to the problem. Abdominal muscles weaken and make it difficult to expel stool, even if soft. Treatment can start with increased dietary fiber and water, as well as prune juice. Fiber laxatives (methylcellulose, bran, psyllium) are bulk forming agents and require 8 ounces of fluid (preferably water) taken immediately with each dose and maintaining good hydration to prevent intestinal blockage. In patients with dysphagia, failure to drink enough water to swallow the fiber laxative might allow swelling in the esophagus. They can also cause abdominal distension. Stool softeners are taken on a daily basis and reduce the amount of straining needed for a bowel movement. Stimulant or irritant laxatives like senna, cascara, and bisacodyl tablets and suppositories should be used at the lowest effective dose and not in slowly progressive disease as they can cause an atonic colon if used for years. They can cause abdominal cramps after each dose and if overused, fluid and electrolyte disturbances. Lactulose and polyethylene glycol are effective osmotic agents. They can cause abdominal distension, diarrhea, flatulence and nausea. More intensive treatments such as enemas, magnesium citrate and manual disimpaction are last resorts. Another option is to adjust the tube feeding formula to one with higher fiber content. Patients who are bed-ridden or receiving narcotics should be given mild laxatives prophylactically.
Urinary frequency and urgency are common in ALS patients. Some patients feel the need to urinate every 1–2 hours. For weak patients transferring to and from a toilet every few hours can be difficult and time consuming. Some patients won’t leave their home because they cannot get to a bathroom fast enough and do not want to wear a diaper. Non-pharmacologic interventions include avoiding caffeine, alcohol, timed voidings, and the use of a urinal and condom catheter for men. Though physicians are reluctant to use foley catheters or refer a patient to the urologist for a suprapubic catheter, many patients are willing to accept the risk of infection and nursing care as they are no longer confined to home. Anticholinergic medications are used after urinary tract infections or an enlarged prostate is ruled out. Medications may improve urinary urgency and frequency in some patients, but not all. It is uncommon for symptoms to resolve completely. Oxybutynin is commonly tried first at 5 mg bid or tid. It is inexpensive and can be crushed and used with a PEG tube. Extended release oxybutynin at doses of 5, 10 and 15 mg are used once a day but can’t be crushed for PEG tube use. Oxytrol skin patch delivers a continuous amount of medication. Tolterodine tartrate (Detrol), Darifenacin (Enablex), Solifenacin (Vesicare), Tropsium (Sanctura) and Fesoterodine (Toviaz) are other options. Although beneficial, anticholinergics may produce dry mouth, constipation, blurred vision and drowsiness as well as confusion in elderly patients with dementia.
DEPRESSION AND ANXIETY
The reported prevalence of depression in ALS patients varies widely and depends on which assessment measure is used. (91 Atassi 2011, 92 Rabkin 2014) The ALS Depression Inventory (ADI-12) is a self-reported questionnaire developed specifically to screen for depression in ALS and does not refer to motor related symptoms in ALS. Using the ADI-12, Atassi et al in 2011 reported that the prevalence of mild and severe depression was 29% and 6% respectively compared to 10% of the general population with major depression. The lower rate of severe depression was attributed to patients receiving anti-depressants to treat sialorrhea, pseudobulbar affect, and insomnia, (91 Atassi 2011) The ALS Cosmos study reported that 88% of 329 patients had no depressive disorder based on structured telephone interviews. Minor depression was found in 7% and 5% had a current major depressive disorder by DSM-IV criteria. Demographic, financial and employment factors were unrelated to depression, as were duration of ALS symptoms and respiratory status. Also of the 19% of the sample who expressed a wish to die, only 37% were clinically depressed. (92 Rabkin 2014). Several classes of medications are used for the treatment of depression in ALS and are used depending on their side effect profile. There have been no controlled clinical studies of these medications in ALS patients. Tricyclic antidepressants like amitriptyline can be prescribed if anticholinergic effects are desired simultaneously for treating sialorrhea, pseudobulbar affect or insomnia.
Prevalence rates for anxiety in patients with ALS range from 0–30%. (93 Kurt 2007) The early disease phase, especially during the diagnostic period, has been reported to be associated with a higher level of stress and anxiety. (96 Vignola 2008) Anxiety is usually treated with anxiolytics like benzodiazepines, but again there have been no systematic studies of these drugs in patients with ALS. Treatment of depression and anxiety can involve both cognitive behavioral therapy and pharmacological intervention. There are no clinical trials that allow a recommendation of pharmacotherapy over psychotherapy. A positive patient-physician relationship and easy access in communicating with their physician reduces levels of anxiety in patients. Patients may benefit from a range of approaches, including relaxation strategies such as meditation and biofeedback. Cognitive behavioral therapy equips patients with the skills to overcome maladaptive thought patterns and encourages emotional readjustment.
END OF LIFE
End of life conversations can be a source of great anxiety and discomfort for the patient, their families and health care professionals, and as a result such discussions may be avoided. The issue of advance directives should be raised soon after the diagnosis. Palliative care can start any time after the diagnosis of ALS and permits the use of high tech assistive communication devices, power wheelchairs, hospitalization, non-invasive ventilation and PEG tubes. The referring physician is responsible initially for managing all medical treatment until the patient is admitted to hospice for continued palliative care. Dyspnea, despite liberal use of NIV, can be treated with morphine starting at 2.5–5 mg every 4 hours and increased in dose and frequency as needed. Anxiety and restlessness can be treated with lorazepam 0.5–2 mg every 4 hours as needed. (88 Rocha) Opiates and anxiolytics doses should be increased if they are not providing satisfactory control.
Palliative care is most often done in the hospital setting, though more community-based outpatient services have become available in large metropolitan areas. Hospice care provides interdisciplinary case management, medications, durable medical equipment and supplies. The hospice medical director manages medical treatment. Some hospices offer hospice residential care. It is appropriate for patients with FVC less than 30% of predicted, and an estimated 6 months of life. In a study of the last month of life in ALS patients, the most common symptoms were difficulty communicating, dyspnea, choking episodes, insomnia, and pain. Many of these symptoms were often inadequately controlled. (98 Ganzini) A significant number of caregivers reported depressed mood (40%), anxiety (30%) and confusion (10%) in patients. (98 Ganzini) Hospice services are essential in the management of end of life in patients with ALS and optimize the likelihood of a peaceful and dignified death. Despite its advantages, it is generally underused or initiated too late in the disease course. (99 Blackhall)
Failure to address advance directives can result in a difficult situation in which a patient who did not want tracheostomy mechanical ventilation (TMV) is intubated. Less than 10% of ALS patients in the United States use invasive ventilation (100 Rabkin 2013). Factors influencing patients against selecting invasive ventilation include high cost for home care on a ventilator, increased caregiver burden, and continued progression of the disease. Patients with TMV may end up in an extended care facility if round the clock care is not available in their home. Many caregivers report TMV at home a major burden which impacts their own health. The locked-in state occurs in 18% of ALS patients surviving more than 5 years on TMV. (101 Hayashi) Withdrawal of invasive ventilation is usually carried out with the use of high-dose opiates and benzodiazepines. A Danish palliative care study on withdrawal of invasive mechanical ventilation at home in patients with advanced ALS reported median time from discontinuation to apnea was 15 minutes (range 0–18 hours). (102 Dreyer) Doses of morphine were in the range of 60–400 mg (median 100 mg) and doses of diazepam in the range of 20–120 mg (median of 100 mg) before the ventilator was disconnected.
Summary
Patients with ALS should be provided opportunities for multidisciplinary care since the spectrum of available treatments can markedly impact both quality of life and survival. ALS should no longer be considered “untreatable” based on the broad range of interventions that can be prescribed for symptom management.
Table 5.
Medications Used For Urinary Urgency and Frequency
(lower doses with co-morbidities)
| Amitriptyline | 12.5–75 mg qhs | |
|---|---|---|
| Oxybutynin (Ditropan) Oxytrol |
2.5–5 mg bid 3.9 mg patch qd gel |
Dry mouth can be significant Skin irritation, minimal dry mouth Gel without skin irritation |
| Tolterodine (Detrol LA) | 1–2 mg bid | |
| Darifenacin hydrobromide (Enablex ER) | 7.5–15 mg qd | Less impact on cognitive function |
| Solifenacin (Vesicare) | 5–10 mg qd | |
| Tropsium (Sanctura) | 20 mg bid | Less interaction with CYP-450 drug |
| Tropsium (Sanctura XR) | 60 mg qd | |
| Fesoterodine (Toviaz) | 4–8 mg qd | Superior to tolterodine |
| Mirabegron (Myrbetriq) | 25–50 mg qd | Can’t use in renal, hepatic or uncont |
Table 6.
Medications Used for Depression and Anxiety (Also see side effects under Table 2)
| DEPRESSION | Dose |
|---|---|
| SSRI antidepressants | |
| Sertraline (Zoloft) | 50–200 mg/d |
| Fluoxetine (Prozac) | 10–60 mg/d |
| Paroxetine (Paxil) | 10–50 mg/d |
| Paroxetine CR | 12.5–62.5 mg/d |
| Citalopram (Celexa) | 20–40 mg/d |
| Escitalopram (Lexapro) | 10–20 mg/d |
| Tricyclic antidepressants | 12.5–150 mg qhs |
| Amitriptyline (Elavil) | 50-200 mg qhs |
| Nortriptyline (Pamelor) | 50–150 mg qhs |
| Serotonin norepinephrine reuptake inhibitor | |
| Venlafaxine (Effexor) | 37.5–225 mg qd |
| Duloxetine (Cymbalta) | |
| Norepinephrine dopamine reuptake inhibitor | |
| Bupropion hydrochloride (Welbutrin) | 300–450 mg qd |
| Norepinephrine serotonin modulator | |
| Mirtazapine (Remeron) | 15–45 mg qhs |
| ANXIETY | |
| Benzodiazepines | |
| Alprazolam (Xanax) | 0.25–1 mg tid |
| Lorazepam (Ativan) | 0.5–2 mg tid |
| Diazepam (Valium) | 2–10 mg tid |
| Clonazepam (Klonopin) | 0.5–2 mg qhs |
| 5HT1A agonists | |
| Buspirone | 10 mg tid |
Key Points.
Sialorrhea should initially be treated with anticholinergic medications. For patients who remain “medically refractory”, treatment with botulinum toxin type B (Level B; recommendation by the American Academy of Neurology Practice Paremeters Committee) or low-dose radiation therapy to the salivary glands (Level C) should be considered.
Pseudobulbar affect can be successfully treated with selective serotonin reuptake inhibitors, tricyclic antidepressants, and serotonin-norepinephrine reuptake inhibitors. A combination of dextromethorphan/quinidine should also be considered (Level B).
Non-invasive ventilation (NIV) should be considered to treat respiratory insufficiency, both to lengthen survival and to slow the rate of FVC decline (Level B). NIV has demonstrated a positive impact on quality of life (Level C). NIV may be considered at the earliest sign of nocturnal hypoventilation in order to improve compliance (Level C).
Mechanical insufflation/exsufflation may be considered to clear secretions, particularly during an acute chest infection (Level C).
Modafinl may be considered to treat fatigue if managing other contributing features fails to be effective. (Level C)
While enteral nutrition may help with weight stabilization, studies that have investigated possible survival benefits have provided mixed results. There is no conclusive data regarding specific diets and possible benefits in ALS. Multiple studies have found that weight loss is associated with a worse prognosis.
While pain is a common complaint in ALS, no controlled studies for managing it have been reported. Identifying the type and cause of the pain may be helpful in its management.
No controlled trials have been done investigating treatment of spasticity in ALS; medications used based upon their benefits in other disorders associated with spasticity or in open label trials in ALS include baclofen (both orally and intrathecally), tizanidine, benzodiazepines, botulinum toxin, dantrolene, and levetiracetam. Exercise intervention has also been attempted, but given the disease progression, it has been challenging to study because of high dropout rates.
No controlled trials have reported on the effect of treating depression and anxiety in ALS; anti-depressants are selected based upon their side effect profile. Anxiolytics and cognitive behavioral therapy are helpful in the treatment of anxiety.
End of life discussions are difficult for the patient, family and physician, but must not be avoided. Advance directives should be addressed soon after the diagnosis. Hospice services are essential in the management of end of life issues in patients with ALS and optimize the likelihood of a peaceful and dignified death. Despite its advantages, it is generally underused or initiated too late in the disease course.
Acknowledgments
This publication was also supported by an Institutional Clinical and Translational Science Award, NIH/NCATS Grant Numbers UL1TR000001. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
A.M. has received research grants from Cytokinetics.
C.J. is a consultant and has received research support from OneWorld Meds regarding use of Myobloc for sialorrhea. She also serves on Speaker’s bureau for Nuedexta for PBA.
M.D. is on the speaker’s bureau or is a consultant for Baxter, Biomarin, Catalyst, CSL-Behring, Depomed, Genzyme, Merck, NuFactor and Pfizer. He has also received grants from Catalyst, CSL-Behring, FDA/OPD, GSK, MDA, NIH and TMA.
R.B. has served as a consultant and received consulting fees from Baxter, CSL Behring, Genzyme, Grifols, Novartis and NuFactor. He has received research grants from Biomarin, Cytokinetics, Eli Lilly, FDA/OPD, GSK, MDA, MGFA, Neals, NIH, NINDS, Novartis, PTC, Sanofi/Genzyme, and Teva.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Stacy Rudnicki, Email: rudnickistacy@uams.edu, University of Arkansas for Medical Sciences, 501 Jackson Stephens Drive, Room 769, Little Rock, Arkansas 72205-7199.
April L. McVey, Email: amcvey@kumc.edu, University of Kansas Medical Center, Dept. of Neurology, 3901 Rainbow Blvd, Mailstop 2012, Kansas City, KS 66160.
Carlayne E. Jackson, Email: jacksonce@uthscsa.edu, University of Texas Health Science Center, 8300 Floyd Curl Drive, Mail Code 7883, San Antonio, TX 78229-3900.
Mazen M. Dimachkie, Email: mdmachkie@kumc.edu, University of Kansas Medical Center, Dept. of Neurology, 3901 Rainbow Blvd, Mailstop 2012, Kansas City, KS 66160.
Richard J. Barohn, Email: rbarohn@kumc.edu, University of Kansas Medical Center, Dept. of Neurology, 3901 Rainbow Blvd, Mailstop 2012, Kansas City, KS 66160.
References
- 1.Forshew DA, Bromberg MB. A survey of clinicians’ practice in the symptomatic treatment of ALS. Amyotrophic Lateral Sclerosis & Other Motor Neuron Disorders. 2003;4:258–63. doi: 10.1080/14660820310017344. [DOI] [PubMed] [Google Scholar]
- 2.Sufit R, Miller R, Mitsumoto H, et al. Prevalence and treatment outcomes of sialorrhea in Amyotrophic Lateral Sclerosis patients as assessed by the ALS Patient Care Database. Annals of Neurology. 1999;46:506. [Google Scholar]
- 3.Miller RG, Rosenberg JA, Gelinas DF, et al. Practice parameter: the care of the patient with amyotrophic lateral sclerosis (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology: ALS Practice Parameters Task Force. Neurology. 1999;52:1311–1323. doi: 10.1212/wnl.52.7.1311. [DOI] [PubMed] [Google Scholar]
- 4.Jackson CE, Rosenfeld J. Amyotrophic Lateral Sclerosis. New York, NY: Taylor &Francis Group; 2006. Symptomatic Pharmacotherapy: Bulbar and Constitutional Symptoms; pp. 649–664. [Google Scholar]
- 5.Bradley WG, Anderson F, Bromberg M, et al. Current management of ALS: comparison of the ALS CARE Database and the AAN Practice Parameter. The American Academy of Neurology. Neurology. 2001;57:500–4. doi: 10.1212/wnl.57.3.500. [DOI] [PubMed] [Google Scholar]
- 6.Portis M, Gamba M, Bertaccji G, Vaj P. Treatment of sialorrhea with ultrasound guided botulinium toxin type A injection in patietns with neurological disorders. J Neurol Neurosurg Psychiatry. 2001:538–540. doi: 10.1136/jnnp.70.4.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giess R, Naumann M, Werner E, et al. Injections of botulinum toxin A into the salivary glands improve sialorrhoea in amyotrophic lateral sclerosis. [see comment] Journal of Neurology, Neurosurgery & Psychiatry. 2000;69:121–3. doi: 10.1136/jnnp.69.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rowe D, Erjavec S. An open-label pilot study of intra-parotid botulinum toxin A injections in the treatment of sialorrhea in motor neuron disease. Amyotrophic Lateral Sclerosis & Other Motor Neuron Disorders. 2003;4:53–54. [Google Scholar]
- 9.Bhatia KP, Munchau A, Brown P. Botulinum toxin is a useful treatment in excessive drooling in saliva. Journal of Neurology, Neurosurgery & Psychiatry. 1999:67. doi: 10.1136/jnnp.67.5.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson CE, Gronseth G, Rosenfeld J, et al. Randomized double-blind study of botulinum toxin type B in ALS patients. Muscle & Nerve. 2009;39:137–143. doi: 10.1002/mus.21213. [DOI] [PubMed] [Google Scholar]
- 11.Harriman M, Morrison M, Hay J, et al. Use of radiotherapy for control of sialorrhea in patients with amyotrophic lateral sclerosis. J Otolaryngology. 2001;30:242–249. doi: 10.2310/7070.2001.19779. [DOI] [PubMed] [Google Scholar]
- 12.Andersen PM, Gronberg H, Franze L, Funegard U. External radiation of the parotid glands significantly reduces drooling in patients with motor neuron disease with bulbar paresis. J Neurol Sci. 2001;191:111–114. doi: 10.1016/s0022-510x(01)00631-1. [DOI] [PubMed] [Google Scholar]
- 12A.Marin MG. Pharmacology of airway secretion. Pharmaocl Rev. 1986;38:273–289. [PubMed] [Google Scholar]
- 12B.Newall AR, Orser R, Hunt M. The control of oral secretions in bulbar ALS/MND. J Neurol Sci. 1996;139:43–44. doi: 10.1016/0022-510x(96)00104-9. [DOI] [PubMed] [Google Scholar]
- 12C.Radunovic A, Mitsumotor H, Leigh PN. Clinical care of patients with amyotrophic lateral sclerosis. The Lancet Neurology. 2007;6(10):913–925. doi: 10.1016/S1474-4422(07)70244-2. [DOI] [PubMed] [Google Scholar]
- 13.Miller RG, Jackson CE, Kasarskis EJ, et al. Practice Parameter update: The care of the patient with amyotrophic lateral sclerosis: Multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review) Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1227–1233. doi: 10.1212/WNL.0b013e3181bc01a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCullagh S, Moore M, Gawel M, Feinstein A. Pathological laughing and crying in amyotrophic lateral sclerosis: an association with prefrontal cognitive dysfunction. J Neurol Sci. 1999;169:43–48. doi: 10.1016/s0022-510x(99)00214-2. [DOI] [PubMed] [Google Scholar]
- 15.Brooks BR, Thisted RA, Appel SH, et al. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial. Neurology. 2004;63:1364–70. doi: 10.1212/01.wnl.0000142042.50528.2f. [DOI] [PubMed] [Google Scholar]
- 16.Arnulf I, Similowski T, Salachas F, et al. Sleep disorders and diaphragmatic function in patients with amyotrophic lateral sclerosis. American Journal of Respiratory & Critical Care Medicine. 2000;161:849–56. doi: 10.1164/ajrccm.161.3.9805008. [DOI] [PubMed] [Google Scholar]
- 17.Czaplinski A, Yen AA, Appel SH. Forced vital capacity (FVC) as an indicator of survival and disease progression in an ALS clinic population. J Neurol Neurosurg Psychiatry. 2006;77:390–392. doi: 10.1136/jnnp.2005.072660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson CE, Rosenfeld J, Moore DH, et al. A preliminary evaluation of a prospective study of pulmonary function studies and symptoms of hypoventilation in ALS/MND patients. J Neurol Sci. 2001;191:75–78. doi: 10.1016/s0022-510x(01)00617-7. [DOI] [PubMed] [Google Scholar]
- 19.Lechtzin N, Wiener CM, Shade DM, Clawson L, Diette GB. Spirometry in the supine position improves the detection of diaphragmatic weakness in patients with amyotrophic lateral sclerosis. Chest. 2002;121:436–442. doi: 10.1378/chest.121.2.436. [DOI] [PubMed] [Google Scholar]
- 20.Varrato J, Siderowf A, Damiano P, Gregory S, Feinberg D, McCluskey L. Postural change of forced vital capacity predicts some respiratory symptoms in ALS. Neurology. 2001;57:357–359. doi: 10.1212/wnl.57.2.357. [DOI] [PubMed] [Google Scholar]
- 21.Vaszar LT, Weinacker AB, Henig NR, Raffin TA. Ethical issues in the long-term management of progressive degenerative neuromuscular diseases. Semin Respir Crit Care Med. 2002;23:307–314. doi: 10.1055/s-2002-33039. [DOI] [PubMed] [Google Scholar]
- 22.Hull J, Aniapravan R, Chan E, Chatwin M, Forton J, Gallagher J, Gibson N, Gordon J, Hughes I, McCulloch R, Russell RR, Simonds A. British Thoracic Society guideline for respiratory management of children with neuromuscular weakness. Thorax. 2012;67(Suppl 1):i1–40. doi: 10.1136/thoraxjnl-2012-201964. [DOI] [PubMed] [Google Scholar]
- 23.Miller RG, Jackson CE, Kasarskis EJ, et al. Practice Parameter update: The care of the patient with amyotrophic lateral sclerosis: Drug, nutritional, and respiratory therapies (an evidence-based review) Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1218–1226. doi: 10.1212/WNL.0b013e3181bc0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol. 2006;5:140–147. doi: 10.1016/S1474-4422(05)70326-4. [DOI] [PubMed] [Google Scholar]
- 25.Bourke SC, Bullock RE, Williams TL, Shaw PJ, Gibson GJ. Noninvasive ventilation in ALS: indications and effect on quality of life. Neurology. 2003;61:171–177. doi: 10.1212/01.wnl.0000076182.13137.38. [DOI] [PubMed] [Google Scholar]
- 26.Butz M, Wollinsky KH, Wiedemuth-Catrinescu U, et al. Longitudinal effects of noninvasive positive-pressure ventilation in patients with amyotrophic lateral sclerosis. Am J Phys Med Rehabil. 2003;82:597–604. doi: 10.1097/01.PHM.0000078239.83545.D0. [DOI] [PubMed] [Google Scholar]
- 27.Lyall RA, Donaldson N, Fleming T, et al. A prospective study of quality of life in ALS patients treated with noninvasive ventilation. Neurology. 2001;57:153–156. doi: 10.1212/wnl.57.1.153. [DOI] [PubMed] [Google Scholar]
- 28.Pinto A, de Carvalho M, Evangelista T, Lopes A, Sales-Luis L. Nocturnal pulse oximetry: a new approach to establish the appropriate time for non-invasive ventilation in ALS patients. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:31–35. doi: 10.1080/14660820310006706. [DOI] [PubMed] [Google Scholar]
- 29.Aboussouan LS, Khan SU, Meeker DP, Stelmach K, Mitsumoto H. Effect of noninvasive positive-pressure ventilation on survival in amyotrophic lateral sclerosis. Ann Intern Med. 1997;127:450–453. doi: 10.7326/0003-4819-127-6-199709150-00006. [DOI] [PubMed] [Google Scholar]
- 30.Jackson CE, Lovitt S, Gowda N, Anderson F, Miller RG. Factors correlated with NPPV use in ALS. Amyotroph Lateral Scler. 2006;7:80–85. doi: 10.1080/14660820500504587. [DOI] [PubMed] [Google Scholar]
- 31.Olney RK, Murphy J, Forshew D, et al. The effects of executive and behavioral dysfunction on the course of ALS. Neurology. 2005;65:1774–1777. doi: 10.1212/01.wnl.0000188759.87240.8b. [DOI] [PubMed] [Google Scholar]
- 32.Gruis KL, Brown DL, Schoennemann A, Zebarah VA, Feldman EL. Predictors of noninvasive ventilation tolerance in patients with amyotrophic lateral sclerosis. Muscle Nerve. 2005;32:808–811. doi: 10.1002/mus.20415. [DOI] [PubMed] [Google Scholar]
- 33.Bach JR. Amyotrophic lateral sclerosis: predictors for prolongation of life by noninvasive respiratory aids. Arch Phys Med Rehabil. 1995;76:828–832. doi: 10.1016/s0003-9993(95)80547-8. [DOI] [PubMed] [Google Scholar]
- 34.Tzeng AC, Bach JR. Prevention of pulmonary morbidity for patients with neuromuscular disease. Chest. 2000;118:1390–1396. doi: 10.1378/chest.118.5.1390. [DOI] [PubMed] [Google Scholar]
- 35.Mangera Z, Panesar G, Makker H. Practical approach to management of respiratory complications in neurological disorders. Intl J Gen Med. 2012;5:255–263. doi: 10.2147/IJGM.S26333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garpestad E, Hill N. Noninvasive ventilation for patients with neuromuscular disease and acute respiratory failure. Chest. 2008;133:315. doi: 10.1378/chest.07-2180. Author Reply. [DOI] [PubMed] [Google Scholar]
- 37.Perrin C, Unterborn JN, Ambrosio CD, Hill NS. Pulmonary complications of chronic neuromuscular diseases and their management. Muscle Nerve. 2004;29:5–27. doi: 10.1002/mus.10487. [DOI] [PubMed] [Google Scholar]
- 38.Park JH, Kang SW, Lee SC, Choi WA, Kim DH. How respiratory muscle strength correlates with cough capacity in patients with respiratory muscle weakness. Yonsei Med J. 2010;51:392–397. doi: 10.3349/ymj.2010.51.3.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McElhiney MC, Rabkin JG, Gordon PH, Goetz R, Mitsumoto H. Prevalence of fatigue and depression in ALS patients and change over time. J Neurol Neurosurg Psychiatry. 2009;80:1146–1149. doi: 10.1136/jnnp.2008.163246. [DOI] [PubMed] [Google Scholar]
- 40.Lo Coco D, La Bella V. Fatigue, sleep, and nocturnal complaints in patients with amyotrophic lateral sclerosis. Eur J Neurol. 2012;19:760–63. doi: 10.1111/j.1468-1331.2011.03637.x. [DOI] [PubMed] [Google Scholar]
- 41.Ramirez C, Piemonte ME, Callegaro D, DaSilva HC. Fatigue in amyotrophic lateral sclerosis: frequency and associated factors. Amyotroph Lateral Scler. 2008;9:75–80. doi: 10.1080/17482960701642502. [DOI] [PubMed] [Google Scholar]
- 42.Bensimon G, Lacomblez L, Meininger VA. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- 43.Lou J-S, Reeves A, Benice T, Seton G. Fatigue and depression are associated with poor quality of life in ALS. Neurology. 2003;60:122–123. doi: 10.1212/01.wnl.0000042781.22278.0a. [DOI] [PubMed] [Google Scholar]
- 44.Rabkin JG, Gordon PH, McElhiney M, Rabkin R, Chew S, Mitsumoto H. Modafinil treatment of fatigue in patients with ALS: a placebo controlled study. Muscle Nerve. 2009;39:297–303. doi: 10.1002/mus.21245. [DOI] [PubMed] [Google Scholar]
- 45.Mascaritioli M, Kushta I, Molfino A, Inghilleri M, Sabatelli M, Fanelli F. Nutritional and metabolic support in patients with amyotrophic lateral sclerois. Nutrition. 2012;28:959–966. doi: 10.1016/j.nut.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 46.Desport JC, Preux PM, Magy L, et al. Factors associated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am J Clin Nutri. 2001;74:328–334. doi: 10.1093/ajcn/74.3.328. [DOI] [PubMed] [Google Scholar]
- 47.Dupuis L, Pradat P-F, Ludolph AC, Loeffler J-P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurology. 2011;10:75–82. doi: 10.1016/S1474-4422(10)70224-6. [DOI] [PubMed] [Google Scholar]
- 48.Kasarskis EF, Mendiondo MS, Matthews DE, et al. Estimating deaily energy expenditure in individuals with amyotrophic lateral sclerosis. Am J Clin Nutri. 2014;99:792–803. doi: 10.3945/ajcn.113.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Limousin N, Blasco H, Corcia P, et al. Malnutrition at the time of diagnosis is associated with a shorter disease duration in ALS. J Neurol Sci. 2010;297:36–39. doi: 10.1016/j.jns.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 50.Shimuzu T, Nagaoka U, Nakayama Y, et al. Reduction rate of body mass index predicts prognosis for survival in amyotrophic lateral sclerosis: a multicenter study in Japan. Amyotoph Lat Scler. 2012;13:363–366. doi: 10.3109/17482968.2012.678366. [DOI] [PubMed] [Google Scholar]
- 51.Jawaid A, Murphy SB, Wilson AM, et al. A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotroph Lat Scler. 2010;11:542–548. doi: 10.3109/17482968.2010.482592. [DOI] [PubMed] [Google Scholar]
- 52.Paganoni S, Deng J, Jaffa M, Cudkowicz ME, Willis A-M. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve. 2011;44:20–24. doi: 10.1002/mus.22114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gallo V, Wark P, Jenab M, et al. Prediagnostic body fat and risk of death from amyotrophic lateral sclerosis. Neurology. 2013;80:829–838. doi: 10.1212/WNL.0b013e3182840689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katzberg HD, Benatar M. Enteral tube feeding for amyotrophic lateral sclerosis/motor. neuron disease. The Cochrane Collaboration. 2011;1:1–13. doi: 10.1002/14651858.CD004030.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chiò A, Bottacchi E, Buffa C, Mutani R, Mora G, PARALS Positive effects of tertiary centres for amyotrophic lateral sclerosis on outcomes and use of hospital facilities. J Neurol Neurosurg Psychiatry. 2006;77:948–950. doi: 10.1136/jnnp.2005.083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Czaplinsi A, Yen AA, Simpson ER, Appel SH. Slower disease progression and prolonged survival in contemporary patients with amyotrophic lateral sclerosis: is the natural history of amyotrophic lateral sclerosis changing? Arch Neurol. 2006;63:1139–1143. doi: 10.1001/archneur.63.8.1139. [DOI] [PubMed] [Google Scholar]
- 57.Forbes RB, Colvisse S, Swingler RJ, Scottish Motor Neurone Disease Research Group Frequency, timing and outcome of gastrostomy tubes for amyotrophic lateral sclerosis/motor neurone disease - a record linkage study from the Scottish Motor Neuron Disease Registry. J Neurol. 2004;251(7):813–817. doi: 10.1007/s00415-004-0429-9. [DOI] [PubMed] [Google Scholar]
- 58.Mitchell JD, O’Brien MR, Joshi M. Audit of outcomes in motor neuron disease (MND) patients treated with riluzole. Amyotrophic Lateral Sclerosis. 2006;7(2):67–71. doi: 10.1080/14660820500396984. [DOI] [PubMed] [Google Scholar]
- 59.Desport JC, Preux PM, Truong CT, Courat L, Vallat JM, Couratier P. Nutritional assessment and survival in ALS patients. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. 2000;1(2):91–6. doi: 10.1080/14660820050515386. [DOI] [PubMed] [Google Scholar]
- 60.Mazzini L, Corra T, Zaccala M, Mora G, Del Piano M, Galante M. Percutaneous endoscopic gastrostomy and enteral nutrition in myotrophic lateral sclerosis. Journal of Neurolog y. 1995;242(10):695–698. doi: 10.1007/BF00866922. [DOI] [PubMed] [Google Scholar]
- 61.Zhang M, Hubbard J, Rudnicki SA, Johansen CS, Dalton K, Heiman-Patterson T, Forshew DA, Willis AM. ee-SPEN Journal. Survey of current enteral nutritio practices in treatment of amyotrophic lateral sclerosis. Available online 1 December 2012. http://www.sciencedirect.com/science/article/pii/S2212826312000619. [DOI] [PMC free article] [PubMed]
- 62.Chiò A, Galetti R, Finocchiaro C, et al. Percutaneous radiological gastrostomy: a safe and effective methodof nutritional tube placement in advanced ALS. J Neurol Neurosurg Psychiatry. 2004;75:645–646. doi: 10.1136/jnnp.2003.020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allen JA, Chen R, Ajroud-Driss S, Sufit RL, Heller S, Siddique T, Wolfe L. Gastrostomy tube placement by endoscopty vs radiologic methods in patients with ALS: a retrospective study of complications and outcomes. Amyotrophic Lateral sclerosis & Frontotemporal Degeneration. 2013 May;14(4):308–14. doi: 10.3109/21678421.2012.751613. [DOI] [PubMed] [Google Scholar]
- 64.Stanich P, Chiapetta A, Oliviera A, Gabbai A. Nutritional supplementation in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler and Other Motor Neuron Disord. 2002;3:119. [Google Scholar]
- 65.Silva LB, Mourao LF, Silva AA, Lima NM, Almeida SR, Franca MC, Jr, et al. Effect of nutritional supplementation with milk whey proteins in amyotrophic lateral sclerosis patients. Arq Neuropsiquiatr. 2010;68(2):263–8. doi: 10.1590/s0004-282x2010000200021. [DOI] [PubMed] [Google Scholar]
- 66.Dorst J, Cypionka J, Ludolph AC. High-caloric food supplements in the treatment of amyotrophic lateral sclerosis: A prospective interventional study. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(7–8):533–6. doi: 10.3109/21678421.2013.823999. [DOI] [PubMed] [Google Scholar]
- 67.Wills AM, Hubbard J, Macklin EA, Glass J, Tandan R, Simpson EP, et al. Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet. 2014 doi: 10.1016/S0140-6736(14)60222-1. http://dx.doi.org/10.1016/S0140-6736(14)60222- [DOI] [PMC free article] [PubMed]
- 68.Czell D, Bauer M, Binek J, Schoch OT, Weber M. Outcomes of percutaneous endoscopic gastrostomy tube isertion in respiratory impaired amyotrophic lateral sclerosis patients under noninvasive ventilation. Resp Care. 2013;58(5):838–844. doi: 10.4187/respcare.02024. [DOI] [PubMed] [Google Scholar]
- 69.Pizziementia A, Aragona M, Onesti E, Inghilleri M. Depression, pain, and quality of life in patients with amyotrophic lateral sclerosis: a cross sectional study. Functional neurology. 2013;28(2):115–119. doi: 10.11138/FNeur/2013.28.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chio A, Canosa A, Gallo S, Moglia C, Ilardi A, Cammarosano S, Papurello D, Calvo A. Pain in amyotrophic lateral sclerosis: a population-based controlled study. European Journal of Neurology. 2012;19(4):551–5. doi: 10.1111/j.1468-1331.2011.03540.x. [DOI] [PubMed] [Google Scholar]
- 71.Ajroud-Driss S, Casey P, Heller S, Allen J, Siddique T, Sufit R. Prevalance and characteristics of pain in early and late stages of ALS. Amyotrophic Lateral sclerosis & Frontotemporal Degeneration. 2013;14(5–6):369–72. doi: 10.3109/21678421.2012.751614. [DOI] [PubMed] [Google Scholar]
- 72.Pagnini F, Lunetta C, Banfi P, Rossi G, Fossati F, Marconi A, Castelnuovo G, Corbo M, Molinari E. Pain in amyotrophic lateral sclerosis: a psychological perspective. Neurological Sciences. 2012;33(5):1193–1196. doi: 10.1007/s10072-011-0888-6. [DOI] [PubMed] [Google Scholar]
- 73.Newrick PG, Langton-Hewer R. Pain in motor neuron disease. J Neurol Neurosurgery, Psych. 1985;48:838–840. doi: 10.1136/jnnp.48.8.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brettschneider J, Kurent J, Ludolph A. Drug therapy for pain in amyotrophic lateral sclerosis or motor neuron disease (Review) The Cochrane Library. 2013;(6):1–19. doi: 10.1002/14651858.CD005226.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McClelland S, Bethoux FA, Boulis NM, et al. Intrathecal baclofen for spasticity-related pain in amyotrophic lateral sclerosis: efficacy and factors associated with pain relief. Muscle Nerve. 2008;37(3):396–398. doi: 10.1002/mus.20900. [DOI] [PubMed] [Google Scholar]
- 76.Drory VE, Goltsman E, Reznik JG, Mosek A, Korxzyn AD. The value of muscle exercise in patients with amyotrophic lateral sclerosis. J Neurol Sci. 2001;191:133–137. doi: 10.1016/s0022-510x(01)00610-4. [DOI] [PubMed] [Google Scholar]
- 77.Bedlack RS, Pastula DM, Hawes J, Heydt D. Open-lable pilot trial of leetiracetam for cramps and spasticity in patients with motor neuron disease. Amytrophic lateral Sclerosis. 2009;10(4):210–215. doi: 10.1080/17482960802430773. [DOI] [PubMed] [Google Scholar]
- 78.Winterholler MG, Heckman JG, Hecht M, Erbguth FJ. Recurrent trismus and stridor in an ALS patient: successfuk treatment with botulinum toxin. Neurology. 2002;58:502–503. doi: 10.1212/wnl.58.3.502. [DOI] [PubMed] [Google Scholar]
- 79.Restivo DA, Lanza S, Marchese-Raguna R, Plameri A. Improvement of masseter spasticity by botulinum toxin facilitates PEG placement in amyotrophic lateral sclerosis. doi: 10.1053/gast.2002.36877. [DOI] [PubMed] [Google Scholar]
- 80.Anderson PM, Abrahams S, Borasio G, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS) - revised report of an EFNS task force. European J Neurol. 2012;19:360–375. doi: 10.1111/j.1468-1331.2011.03501.x. [DOI] [PubMed] [Google Scholar]
- 81.Sperfeld AD, Hanemann OC, Ludolph AC, Kassubek J. Laryngospasm: An underdiagnosed symptom of X-linked spinobulbar muscular atrophy. Neurology. 2005 Feb 22;64(4):753–754. doi: 10.1212/01.WNL.0000151978.74467.E7. [DOI] [PubMed] [Google Scholar]
- 82.Borasio GD, Oliver D. The Control of Other Symptoms. In: Oliver D, Borasio GD, Walsh D, editors. Amyotrophic Lateral Sclerosis. Oxford: Oxford University Press; 2000. pp. 72–79. [Google Scholar]
- 84.Jackson CE, Rosenfeld J. Amyotrophic Lateral Sclerosis. New York, NY: Talylor & Francis Group; 2006. Symptomatic Pharmacotherapy: Bulbar and Constitutional Symptoms; pp. 649–664. [Google Scholar]
- 85.Kuhnlein P, Gdynia HJ, Sperfeld AD, Lindner-Pfleghar B, Ludolph AC, Prodiegel M, Riecker A. Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nature Clinical Practice Neurology. 2008 Jul;4(7) doi: 10.1038/ncpneuro0853. [DOI] [PubMed] [Google Scholar]
- 86.Jenkins TM, Hollinger H, McDermott CJ. The evidence for symptomatic treatments in amyotrophic lateral sclerosis. Curr Opin Neurol. 2014;27:524–531. doi: 10.1097/WCO.0000000000000135. [DOI] [PubMed] [Google Scholar]
- 87.Simmons Z. Management Strategies for Patients With Amyotrophic Lateral Sclerosis From Diagnosis Through Death. The Neurologist. 2005 Sep;11(5):257–270. doi: 10.1097/01.nrl.0000178758.30374.34. [DOI] [PubMed] [Google Scholar]
- 88.Rocha JA, Reis C, Simoes F, Fonseca J, Mendes-Ribeiro J. Diagnostic Investigation and Multidisciplinary Management in Motor Neuron Disease. J Neurol. 2005;252:1435–1447. doi: 10.1007/s00415-005-0007-9. [DOI] [PubMed] [Google Scholar]
- 89.Piccione EA, Sletten DM, Staff NP, Low PA. Autonomic System and ALS. Muscle and Nerve. doi: 10.1002/mus.24457. Accepted Article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nubling GS, Bauer M, Bauer Rm, Hensler M, Lorenzi S, Hapfelmeier A, Irwin DE, Borasio GD, Winkler AS. Increased Prevalence of Bladder and Intestinal Dysfunction in Amyotrophic Lateral Sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2014 Jun;15(3–4):174–9. doi: 10.3109/21678421.2013.868001. [DOI] [PubMed] [Google Scholar]
- 91.Atassi N, Cook A, Pineda CM, Yerramilli-Rao P, Pulley D, Cudkowicz M. Depression in Amyotrophic Lateral Sclerosis. Amyotroph Lateral Scler. 2011 Mar;12(2):109–112. doi: 10.3109/17482968.2010.536839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rabkin JG, Goetz R, Factor-Litvak P, Hupf J, McElhiney M, Singelton J, Mitsumoto H, The ALS Cosmos Study Group Depression and Wish to Die in a Multicenter Cohort of ALS patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014 Dec 8;:1–9. doi: 10.3109/21678421.2014.980428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kurt A, Nijboer F, Maatuz T, Kubler A. Depression and Anxiety in Individuals with Amyotrophic Lateral Sclerosis: epidemiology and management. CNS Drugs. 2007;21(4):279–91. doi: 10.2165/00023210-200721040-00003. [DOI] [PubMed] [Google Scholar]
- 94.Chen D, Xiaoyan G, Sheng Z, Wei Q, Song W, Cao B, Huang F, Yang F, Shang H. Depression and Anxiety in Amyotrophic Lateral Sclerosis: Correlations Between the Distress of Patients and Caregivers. Muscle Nerve. 2015;51:353–357. doi: 10.1002/mus.24325. [DOI] [PubMed] [Google Scholar]
- 95.Taylor L, Wicks P, Leigh PN, Goldstein LH. Prevalence of Depression in Amyotrophic Lateral Sclerosis and other motor disorders. Eur J Neurol. 2010;17:1047–1053. doi: 10.1111/j.1468-1331.2010.02960.x. [DOI] [PubMed] [Google Scholar]
- 96.Vignola A, Guzzo A, Calvo A, Moglia C, Pessia A, Cavallo E, Cammarosano S, Giacone S, Ghiglione P, Chio A. Anxiety Undermines Quality of Life in ALS Patients and Caregivers. European Journal of Neurology. 2008 Nov;15(11):1231–1236. doi: 10.1111/j.1468-1331.2008.02303.x. [DOI] [PubMed] [Google Scholar]
- 97.Tagami M, Kimura F, Nakajima H, Ishida S, Fujiwara S, Doi Y, Hosokawa T, Yamane K, Unoda K, Hirose T, Tani H, Ota S, Sugina M, Shinoda K, Hanfusa T. Tracheostomy and Invasive Ventilation in Japanese ALS Patients: Decision Making and Survival Analysis: 1990–2010. J Neurol Sci. 2014 Sep 15;344(1–2):158–164. doi: 10.1016/j.jns.2014.06.047. [DOI] [PubMed] [Google Scholar]
- 98.Ganzini L, Johnston WS, Silveira MJ. The Final Month of Life in Patients with ALS. Neurology. 2002;59:428–431. doi: 10.1212/wnl.59.3.428. [DOI] [PubMed] [Google Scholar]
- 99.Blackhall LJ. Amyotrophic Lateral Sclerosis and Palliative Care: Where We Are, and the Road Ahead. Muscle Nerve. 2012;45:311–318. doi: 10.1002/mus.22305. [DOI] [PubMed] [Google Scholar]
- 100.Rabkin J, Ogino M, Goetz R, McElhiney M, Marziliano A, Imai T, Atsuta N, Mortia M, Tateishi T, Mastumura T, Mistumoto H. Tracheostomy with Invasive Ventilation for ALS Patients: Neurologist’s roles in the US and Japan. Amyotroph Lateral Scler Frontotemporal Degener. 2013 Mar;14(2):116–123. doi: 10.3109/17482968.2012.726226. [DOI] [PubMed] [Google Scholar]
- 101.Hayashi H, Oppenheier EA. ALS Patients on TPPV: Totally Locked-In State, Neurologic Findings and Ethical Implications. Neurology. 2003;61:135–137. doi: 10.1212/01.wnl.0000069925.02052.1f. [DOI] [PubMed] [Google Scholar]
- 102.Dreyer PS, Felding M, Klitnaes CS, Lorenzen CK. Withdrawal of Invasive Home Mechanical Ventilation in Patients with Advanced Amyotrophic Lateral Sclerosis: Ten Years of Danish Experience. Journal of Palliative Medicine. 2012 Feb 17;15(2):205–209. doi: 10.1089/jpm.2011.0133. [DOI] [PubMed] [Google Scholar]