

Abstract
Alopecia areata (AA) is an autoimmune disease of the hair follicle (HF) that results in hair loss of varying severity. Recently, we showed that IFN-γ-producing NKG2D+CD8+ T cells actively infiltrate the HF, and are responsible for its destruction in C3H/HeJ AA mice. Our transcriptional profiling of human and mouse alopecic skin showed that the IFN pathway is the dominant signaling pathway involved in AA. We showed that IFN inducible chemokines (CXCL9/10/11) are markedly upregulated in the skin of AA lesions, and further, that the IFN inducible chemokine receptor, CXCR3, is upregulated on alopecic effector T cells. To demonstrate whether CXCL9/10/11 chemokines were required for development of AA, we treated mice with blocking antibodies to CXCR3, which prevented the development of AA in the graft model, inhibiting the accumulation of NKG2D+CD8+ T cells in the skin and cutaneous lymph nodes. These data demonstrate proof of concept that interfering with the Tc1 response in AA via blockade of IFN inducible chemokines can prevent the onset of AA. CXCR3 blockade could be approached clinically in human AA with either biologic or small molecule inhibition, the latter being particularly intriguing as a topical therapeutic.
Introduction
Alopecia areata (AA) is one of the most prevalent autoimmune diseases with a lifetime incidence of 1.7% (1). AA is characterized by an extensive localized inflammatory T cell infiltrate around the hair follicle (HF) and focal, extensive, or complete hair loss in both males and females. The C3H/HeJ mouse model of AA, as well as the humanized rodent model with human alopecic skin explanted onto NOD/SCID mice, supported a T cell-dependent, autoimmune mechanism in which the breakdown of immune privilege is followed by an attack on anagen HF (2–4). We and others have observed that both human AA patients, as well as the C3H/HeJ mouse model of AA, exhibit a striking IFN-γ-specific Th1 cytokine signature in the skin (4–6). IFN-γ is prominently expressed in AA lesions and may contribute to the collapse of HF immune privilege by upregulating MHC class I expression in the HF, which has been implicated in the pathogenesis of AA (7, 8). In C3H/HeJ mice, administration of IFN-γ has been shown to induce follicular expression of MHC class I and II, leading to the loss of HF immune privilege and induction of autoimmune hair loss (9). Likewise, it has been shown that blockade of the function of IFN-γ inhibits the development of alopecia in C3H/HeJ mice (5, 10).
Recently, we determined that NK-type CD8+NKG2D+ T cells are the dominant immune effectors infiltrating the HF in both humans and C3H/HeJ mice with AA (5, 11). The signals that recruit autoreactive T cell migration into skin and HF leading to AA are unknown. Leukocyte infiltration into inflammatory sites is critical for the initiation and progression of a variety of inflammatory disorders and is controlled via the activation and signaling of specific cell surface chemokine receptors (12–14). Chemokines are a superfamily of chemotactic cytokines that play important roles in the generation and maintenance of immune and inflammatory responses. They are also involved in a wide range of disease processes, including infection, autoimmune, inflammatory, and malignant diseases (12–14).
The CXCR3 receptor and its cognate ligands, CXCL9, CXCL10 and CXCL11 have been implicated in directing a Th1 inflammatory response (15–18). Recent studies support the notion that the CXCR3 receptor is an attractive therapeutic target for treating autoimmune diseases, such as rheumatoid arthritis (RA), vitiligo and psoriasis (19–22). In humans, the efficiency of a blocking CXCL10 antibody (MDX-1100) was reported in a phase 2 clinical trial for RA, and underscored the therapeutic potential blocking the CXCR3-CXCL10 axis in autoimmunity (23).
We and others found that many of the upregulated genes in alopecic skin of both species were IFN-response genes, including the IFN-inducible chemokines CXCL9-11 (5, 24). Chemokines, CXCL9 and CXCL10 are elevated in the serum of AA patients (25). The marked upregulation of CXCR3 ligand expression, together with the increased number of CXCR3+ lymphocytes on infiltrating T cells, suggests further interrogation of the CXCR3 pathway in AA (25–27). To define the role of CXCR3 in AA pathogenesis, we investigated chemokine expression in lesional skin of patients with AA, as well as in C3H/HeJ mice and studied the consequences of CXCR3 blockade in mice with AA.
We demonstrate that CXCR3 ligands are highly expressed in lesional skin of human AA patients and in C3H/HeJ mice with AA. Further, we found that blockade of the CXCR3-ligand interaction prevents the development of AA by markedly reducing the accumulation of CD8+NKG2D+ T cell in lesional skin and inhibiting expansion of CD8+NKG2D+ T cell in skin draining lymph nodes (SDLNs). This study invites further investigation of CXCR3 blockade as a new therapeutic target for AA.
Materials and Methods
Mice
C3H/HeJ mice (Jackson Laboratories) were maintained under specific pathogen-free conditions at the animal facility at the Columbia University Medical Center (CUMC). Transfer of AA was performed using grafted alopecic C3H/HeJ skin, as described previously (23). In brief, mice spontaneously affected with AA were euthanized and full thickness skin grafts of approximately 2 cm in diameter were grafted onto 7–10 week old normal-haired female C3H/HeJ mice. Hair loss typically began at around 4–6 weeks after grafting. All experiments were performed in compliance with institutional guidelines as approved by the Institutional Animal Care and Use Committee of CUMC.
Antibodies and Reagents
Multiparameter flow cytometric analysis of murine immune cell phenotype was performed by staining with the following fluorochrome-conjugated Abs mAbs: CD3 (17A2), CD4 (GK1.5), CD8 (53–6.7), CD11b (M1/70), CD11c (N418), CD19 (6D5), CD25 (PC61.5), CD44 (IM7), CD45 (30-F11), CD62L (MEL-14), B220 (RA3-6B2), NKG2D (CX5), I-A/I-E (M5/114.15.2), IFN-γ (XMG1.2), pan NK cells (DX5, eBioscience), CXCR3 (CXCR3-173, eBioscience), Foxp3 (FJK-16s, eBioscience) and Ki-67 (SoIA15, eBioscience). Unless stated otherwise, Abs were purchased from Biolegend.
For immunohistochemistry and immunofluorescence staining of human tissue sections, we used Abs against CXCL9 (AF392, R&D Systems), CXCL10 (AF-266, R&D Systems), CXCL11 (FL-94, SCBT), CD3 (EPR4517, Abcam), CD4 (SP35, Spring Bioscience), CD8 (SP16, Spring Bioscience) and CXCR3 (1C6, BD Biosciences). For immunohistochemistry and immunofluorescence staining of mouse tissue sections, we used Abs against CXCL9 (AF-492, R&D Systems), CXCL10 (AF-466, R&D Systems), CXCL11 (FL-94, SCBT), CXCR3 (C-20, SCBT), CD4 (RM4-5, Biolegend), CD8 (53-6.7, Biolegend), H-2Kk, biotin labeled (36-7-5, Biolegend), I-A/I-E (M5/114.15.2, Biolegend).
Purified recombinant human or mouse TNF-α and IFN-γ (Peprotech) were used for in vitro HF organ culture or in vivo injection.
Preparation of single-cell suspensions
To prepare skin single-cell suspensions, mice skin was cut into small pieces and digested in a solution of RPMI-1640 medium (Life Technologies) with 2 mg/ml collagenase type 1 (Worthington) supplemented with 5% FBS for 75 min at 32 °C. The digested skin was then minced, passed over 70 μm cell strainer (BD Biosciences) and washed before staining. Whole spleens or LNs were dissociated and filtered with a 40 μm cell strainer. Spleen or blood cells were depleted of erythrocytes by ammonium chloride lysis and washed before staining.
Cell stimulation and flow cytometry
The single cell suspensions from the indicated tissues were first pre-incubated with anti-CD16/32 (93, Biolegend) in FACS buffer (PBS containing 2% FBS and 0.1% NaN3) for 20 min at 4° C to block the nonspecific binding of Abs to FcγR. For surface markers staining, the cells were incubated with various combinations of fluorochrome-conjugated Abs in FACS buffer for 30 min at 4° C, after which the cells were washed with FACS buffer three times. For detection of intracellular CXCL9 (AF-492, R&D Systems), CXCL10 (AF-466, R&D Systems), FOXP3 or Ki-67, the cells were fixed after surface markers staining, and then permeabilized using the FOXP3 fixation/permeabilization kit (eBioscience) and stained intracellularly with the indicated Abs for 30 min at 4° C, followed by fluorochrome-conjugated donkey anti-goat Ab (Life Technologies) for detection of CXCL9 and CXCL10. For intracellular detection of IFN-γ, 2 × 106 LN cells or skin single cell suspensions were cultured with 1× Cell Stimulation Cocktail (eBioscience) in RPMI-1640 medium with 10% FBS. After 1 h, Brefeldin A (BD Biosciences) was added, followed by additional 4 h incubation at 37° C. The cells were then fixed, and permeabilized using the BD Cytofix/Cytoperm (BD Biosciences) and stained intracellularly with anti-IFN-γ for 30 min at 4° C. Flow cytometric analysis was performed on BD LSRII flow cytometer (BD Biosciences) and analyzed with FlowJo software (Treestar). Viable cell populations were gated based on forward and side scatters and by DAPI (Biolegend) staining.
Immunohistochemistry and immunofluorescence staining
Immunofluorescence staining of tissue sections was performed as previously described (5). The immunofluorescence staining of mouse MHC class I was performed using biotin labeled mouse anti-H-2Kk, followed by fluorochrome-conjugated streptavidin (Life Technologies). The endogenous biotin was blocked using a streptavidin/biotin blocking kit (Vector Laboratories). Immunohistochemical stainin g of formalin-fixed, paraffin-embedded tissue sections were performed using the microwave antigen retrieval method as previously described (5). Briefly, human CD3 or CD8 antigen was retrieved with 10 mM citrate buffer (pH 6.0), and 1 mM EDTA (pH 8.0) for human CD4 or CXCR3 antigen. For both frozen tissue and formalin-fixed, paraffin-embedded tissue section staining procedures, the individual primary Ab was used at optimal concentrations for detection (final concentration ~10ug/ml), followed by ImmPRESS HRP secondary Abs (Vector Laboratories). The detection of Ab complexes was followed by incubation with ImmPACT NovaRED Peroxidase (HRP) Substrate (Vector Laboratories).
CXCR3 ligand detection assay
For mouse CXCR3 ligand detection, capture ELISA was performed on serum using an ELISA kit (CXCL9 (R&D Systems), CXCL10 (Peprotech) and CXCL11 (Abcam)) according to the manufacture’s instruction.
Human HF organ culture
Human scalp skin specimens were obtained with informed consent during routine hair restoration surgery. Anagen HFs were micro-dissected, and cultured within 6 h after surgery (28). Isolated normal human scalp HFs were cultured in William’s E medium (Life Technologies) supplemented with 10 μg/ml insulin (Sigma),10 ng /ml hydrocortisone (Sigma), 50 U/ml penicillin, and 50 μg/ml streptomycin using 24-well culture plates (28). Three HFs per well were cultured in 2 ml William’s E medium with PBS or IFN-γ (50 ng/ml) or IFN-γ (50 ng/ml) plus TNF-α (25 ng/ml) for 48 h at 37°C. The specimens used for cryosections or RNA extractions were snap-frozen in liquid nitrogen, and stored at −80°C until use. Before staining, specimens were embedded and processed for longitudinal cryosections (8 μm). Cryosections were fixed with acetone at −20°C for 10 min for immunohistochemistry, and stored at −20°C until use.
Hair plucking for induction of synchronous hair cycle and intradermal cytokine injection
Synchronous hair cycles were induced by plucking the dorsal hair during the normally synchronized second post-natal telogen phase (29). At day 14, mice received a single intradermal injection of 200 ng IFN-γ or 200 ng IFN-γ with 50 ng TNF-α in areas of the skin that were depilated. PBS was used as a control. After 48 h, biopsies were taken and each biopsy was cut into two: half was snap-frozen in Tissue-Tek O.C.T. compound (Sakura Finetek) for cryosectioning and the other half was snap-frozen in liquid nitrogen for RNA extraction.
In vivo measurement of lymphocyte migration and proliferation with CFSE
For positive selection of T cell populations, CD8+ T cells from SDLNs from C3H/HeJ AA mice were first enriched by untouched isolation of CD8+ T cells kit (Miltenyi Biotec) and the CD8+NKG2D+ or CD8+NKG2D− T cells were further purified on an Influx cell sorter (BD Biosciences). The purity was more than 95%. Lymphocyte migration was measured using CFSE-labeled T cells as tracers. Briefly, CD8+NKG2D+ or CD8+NKG2D− T cells were isolated as above, stained with 5 μM CFSE (eBioscience), and 4 × 106 T cells were injected i.v. per mouse. In cell transfer experiments, 1mg of anti-CXCR3 (CXCR3-173) mAb or control hamster IgG was given i.v. just prior to intravenous administration of labeled T cells. For lymphocyte migration, the skin was removed for snap frozen in Tissue-Tek O.C.T. compound after 48 h. For T cells proliferation and trafficking to other tissues, mononuclear cells from SDLN and spleen were isolated and the CFSE dilution of CFSE-labeled T cells was measured on a BD LSR II flow cytometer (BD Biosciences) at day 7 after adoptive transfer of T cells.
qRT-PCR
Total cellular RNA was extracted using RNeasy Kit (Qiagen) from skin or HF homogenates. Subsequently, reverse transcription and quantitative RT-PCR was performed using real-time PCR detection system and an SYBR green amplification kit (Life Technologies) to determine the expression of CXCL9, CXCL10 and CXCL11 mRNA in the skin and HF. The quantity was standardized using GAPDH primers. Primers (forward and reverse listed 5′-3′) used for mouse qRT-PCR were as follows: CXCL9 (5′-GCACGATCCACTACAAATCCC-3′ and 5′-GGTTTGATCTCCGTTCTTCAGT-3′), CXCL10 (5′-CCAAGTGCTGCCGTCATTTC-3′ and 5′-TCCCTATGGCCCTCATTCTCA-3′), CXCL11 (5′-TGTAATTTACCCGAGTAACGGC- 3′ and 5′-CACCTTTGTCGTTTATGAGCCTT-3′) and GAPDH (5′-GAAGGTCGGTGTGAACGGA-3′ and 5′-GAAGGTCGGTGTGAACGGA-3′). Primers (forward and reverse listed 5′-3′) used for human qRT-PCR were as follows: CXCL9 (5′-GTAGTGAGAAAGGGTCGCTGT-3′ and 5′-AGGGCTTGGGGCAAATTGTT-3′), CXCL10 (5′-GTGGCATTCAAGGAGTACCTC-3′ and 5′-TGATGGCCTTCGATTCTGGATT-3′), CXCL11 (5′-ATGAGTGTGAAGGGCATGGC-3′ and 5′-TCACTGCTTTTACCCCAGGG-3′) and GAPDH (5′-TCACCAGGGCTGCTTTTAACTC-3′ and 5′-GGGTGGAATCATATTGGAACA-3′).
Prevention studies using CXCR3 neutralizing Ab
For prevention studies, mice were given treatment beginning the day of grafting (5–10 mice per group). Anti-CXCR3 (CXCR3-173, hamster) mAb (30) or control hamster IgG was administrated by i.p. injection (200 μg in 100μl PBS; eBioscience and BioXcell, respectively) two times weekly for 12 wk. Full-thickness skin biopsies were excised from the dorsal surface of each mouse at interim time points, and skin samples were either snap frozen in liquid nitrogen for RNA extraction or snap frozen in Tissue-Tek O.C.T. compound for immunostaining. Hair status was examined twice weekly and hair growth index calculated as previously described (5).
Statistical analysis
All data are present as the mean ± SD. Groups of data were compared using a two-tailed Student t test. Values of p < 0.05 were considered significant.
Results
IFN-inducible chemokines are dramatically upregulated in human and mouse with AA
AA is a T cell-mediated autoimmune disease with evidence implicating inflammatory cytokines in the pathogenesis of the disease. To identify key cytokine pathways in AA, we used comparative genomics to characterize the transcriptional landscape of alopecic lesional skin both from humans and mice with AA. Gene expression profiling revealed that the most upregulated genes in alopecic skin were IFN-response genes, including the IFN-inducible chemokines CXCL9, CXCL10 and CXCL11 (5). Consistent with previous studies (24, 26, 31), qPCR results showed that CXCL9, CXCL10 and CXCL11 mRNAs were highly expressed in the alopecic skin of C3H/HeJ AA mice compared to controls (Fig. 1A). IFN-inducible chemokines CXCL9, CXCL10 and CXCL11 each bind to a common primary receptor, CXCR3. These chemokines are up-regulated in a proinflammatory cytokine milieu, and their major function is to selectively recruit immune cells that express CXCR3 at sites of inflammation.
FIGURE 1.
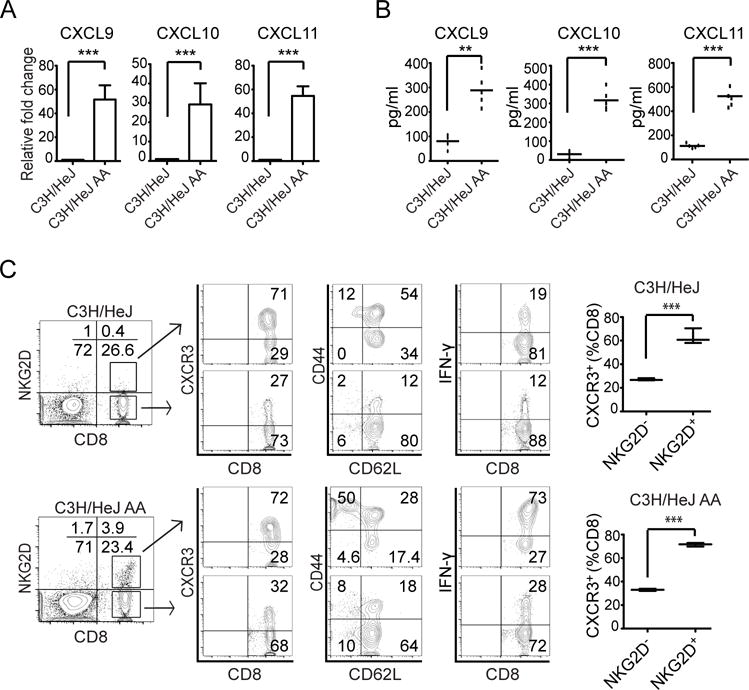
Expression of CXCR3 ligands and phenotype of CD8+NKG2D+ T cells in C3H/HeJ mice with AA. (A) Relative expression of CXCL9, CXCL10, and CXCL11 genes in lesional skin from C3H/HeJ mice (8 weeks after AA skin grafting) and in skin from non-alopecic C3H/HeJ mice was determined by quantitative RT-PCR. Quantitative RT-PCR results were normalized to GAPDH and represent mean ± SEM (n = 4–5 per group). (B) Serum samples from C3H/HeJ AA and aged-matched non-alopecic C3H/HeJ mice were analyzed to determine levels of CXCL9, CXCL10, and CXCL11 (n = 5 per group). The CXCR3 ligands CXCL9, CXCL10, and CXCL11 were significantly elevated in C3H/HeJ mice with AA. (C) NKG2D+CD8+ T cells express CXCR3 receptor within SDLNs. The left panels illustrate the percentage of CD8+NKG2D+ T cells within the SDLNs from alopecic C3H/HeJ mice and non-alopecic C3H/HeJ mice. The middle panels depict flow cytometric analysis of CD44, CD62L, and CXCR3 expression by CD8+NKG2D+ T cells and CD8+NKG2D− T cells within the SDLN from alopecic C3H/HeJ mice and non-alopecic C3H/HeJ mice. In alopecic mice CD8+NKG2D+ T cells in SDLNs have effector memory CD44hiCD62Llow phenotype and are CXCR3high. The right panels illustrate cytokine production by CD8+NKG2D− T cells or CD8+NKG2D+ T cells. SDLN cells were stimulated by PMA and ionomycin for 5 hours. Percentage of intracellular IFN-γ-producing T cells among CD8+NKG2D− T cells or CD8+NKG2D+ T cells were quantitated by flow cytometric analysis. Data are expressed as mean ± SEM, and results are combined from three independent experiments. *indicates p <0.05, ** indicates p<0.01.
Increased levels of circulating CXCL9 and 10 have been detected in inflammatory processes and autoimmunity (18, 19, 25). To determine whether these chemokines could be detected in C3H/HeJ AA mice, blood samples were collected from C3H/HeJ AA mice and age-matched control, and serum levels of CXCL9, CXCL10 and CXCL11 were measured by ELISA. Mice with AA had significantly elevated serum levels of CXCL9, CXCL10 and CXCL11 compared to controls (Fig. 1B).
CXCR3 positive CD8 T cells infiltrate lesional skin in human and mouse with AA
We have previously shown that C3H/HeJ mice with AA exhibit a striking cutaneous lymphadenopathy with increased total cellularity. High frequencies of CD8+NKG2D+ T cells were detected in the SDLNs and alopecic skin from C3H/HeJ AA mice (5). To further characterize the CD8+NKG2D+ T cells in C3H/HeJ AA mice, we determined the expression of molecules associated with T cell migration and trafficking, such as CD44, CD62L and CXCR3, on T cells isolated from SDLNs. Our flow cytometry data showed that most CD8+NKG2D+ T cells from SDLNs in C3H/HeJ AA mice were CD44high, CD62Llow and CXCR3high, and expressed high levels intra-cytoplasmic IFN-γ after stimulation (Fig. 1C). Interestingly, a small population of CD8+ T cells coexpressing NKG2D (0.3 ± 0.1%) was also detected in the SDLNs from non-alopecic C3H/HeJ mice (Fig. 1C). Although CD8+NKG2D+ T cells from non-alopecic C3H/HeJ mice and AA mice all demonstrated higher proportions of cells expressing CXCR3, the CD8+NKG2D+ T cells from AA mice were dominated by an effector memory phenotype (CD44highCD62Llow) and possessed a greater ability to produce IFN-γ than the CD8+NKG2D+ T cells from non-alopecic C3H/HeJ mice (Fig. 1C). The expression of CXCR3 ligands in AA alopecic skin and expression of CXCR3 on SDLNs CD8+NKG2D+ T cells pointed to a role of CXCR3 ligands in the recruitment of CD8+NKG2D+ effector memory T cells to inflamed AA skin. These results suggest that elevated CXCR3 ligand expression in C3H/HeJ AA mice may be involved in the recruitment of CXCR3 expressing CD8+NKG2D+ T cells to inflamed skin and HF.
CXCR3 expression on activated T cells is important for the amplification of IFN-γ-dependent recruitment into peripheral tissues during infection and autoimmune responses (15–18). To determine whether CXCR3 is expressed on infiltrating T cells in the alopecic skin of patients with AA, skin biopsies from patients were examined by immunostaining. Consistent with previous reports (24, 26), we found that CXCR3+ T cells markedly infiltrated in and around HFs from AA patients (Fig. 2A). Likewise, CXCR3+CD8+ T cells that infiltrated in and around HFs were also detected in the alopecic skin of C3H/HeJ mice with AA (Fig. 2B).
FIGURE 2.
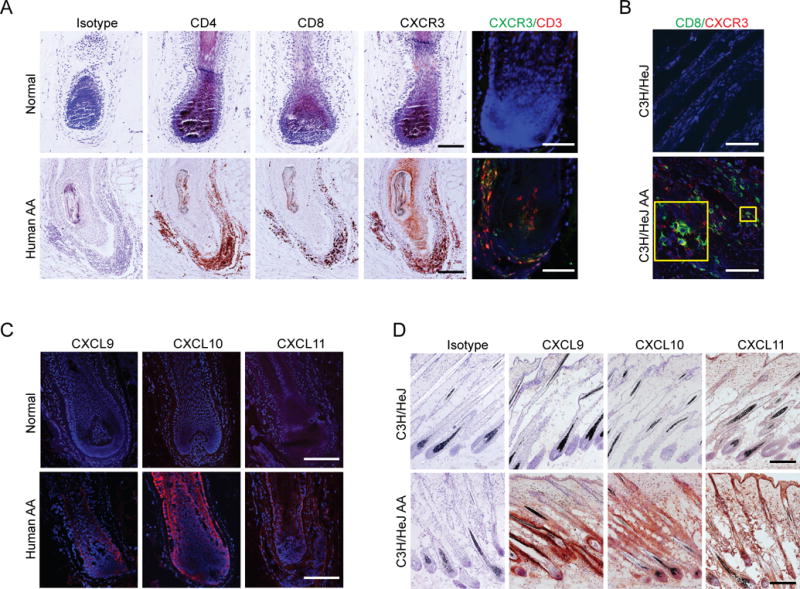
Expression of CXCR3 ligands and their common receptor in AA lesional skin. (A) Detection of CD4, CD8, and CXCR3 expression on HF infiltrating cells in human AA scalp and normal control by immunohistochemistry. Double immunofluorescent staining of CXCR3 (green) and CD3 (red) is also shown. (B) Immunofluorescent staining of CXCL9 (red), CXCL10 (red) and CXCL1 (red) expression in human AA scalp and normal control. (C) Double immunofluorescent staining of CD8 (green) and CXCR3 (red) on HF infiltrating cells in skin sections from C3H/HeJ AA and non-alopecia C3H/HeJ mice. (D) Detection of CXCL9, CXCL10, and CXCL11 expression in skin sections from C3H/HeJ AA and non-alopecia C3H/HeJ mice by immunohistochemistry where CXCL9, CXCL10, and CXCL11 expression is indicated by the presence of red precipitants. Data are representative of four separate samples in each staining. Scale bar = 200μM.
Production of CXCR3 ligands by AA hair follicles
CXCR3 ligands are IFN-inducible chemokines that are highly expressed in inflamed tissues in a wide range of disease process (16–22). The infiltration of CXCR3+ T cells in and around HF raises the possibility that its ligands might be secreted by HFs and/or surrounding inflammatory cells for further recruitment of CXCR3+ T cells into the inflamed skin. Although we showed the upregulation of CXCR3 ligands in human and mouse alopecic skin (Fig. 1A), the cellular source of the ligands is unknown. Our immunohistochemical results showed that in human alopecic skin, a strong CXCL10, and weak CXCL9 and CXCL11 immunostaining was detected in the HFs compared to normal skin. We found that CXCL10 and CXCL11 were prominently expressed in the proximal outer root sheath (ORS) and inner root sheath (IRS), whereas CXCL9 was observed predominantly in the inner root sheath (IRS) (Fig. 2C). In C3H/HeJ AA mice alopecic skin, striking expression of CXCL9, CXCL10 and CXCL11 immunostaining was mostly detected in the ORS of inflamed HFs (Fig. 2D).
IFN-γ can induce CXCL9 and CXCL10, and activated CD8+ T cells are an important source of IFN-γ (27). TNF-α has been shown to play a role in the pathogenesis of AA, and TNF-α potentiates IFN-γ-induced CXCL9, CXCL10 production in keratinocytes (32). The epithelium of human scalp HFs expresses both IFN-γ Rα and IFN-γ Rβ (28). Next, we asked whether IFN-γ might upregulate the expression CXCL9, CXCL10 and CXCL11 in the HF. To address this, we treated micro-dissected normal human HFs with IFN-γ or IFN-γ plus TNF-α in vitro using a HF organ culture assay (28). Consistent with the upreguation of mRNA level, immunostaining results indicated that the expression of CXCL9, CXCL10 and CXCL11 protein was markedly upregulated in human HFs after IFN-γ treatment and TNF-α potentiates IFN-γ-induced CXCL9, CXCL10 and CXCL11 production (Fig. 3A). We also detected a dramatic upregulation of CXCL9, CXCL10 and CXCL11 both at protein and mRNA level in non-alopecic C3H/HeJ mice after IFN-γ treatment and TNF-α potentiates IFN-γ-induced CXCL9, CXCL10 and CXCL11 production, compared to lower expression in control, PBS-treated mice (Fig. 3B). Therefore, TNF-α may synergize with IFN-γ, secreted by activated T cells (33), to induce chemokine expression in the HF that can then further enhance the recruitment of T cell into the HF during pathogenesis of AA.
FIGURE 3.
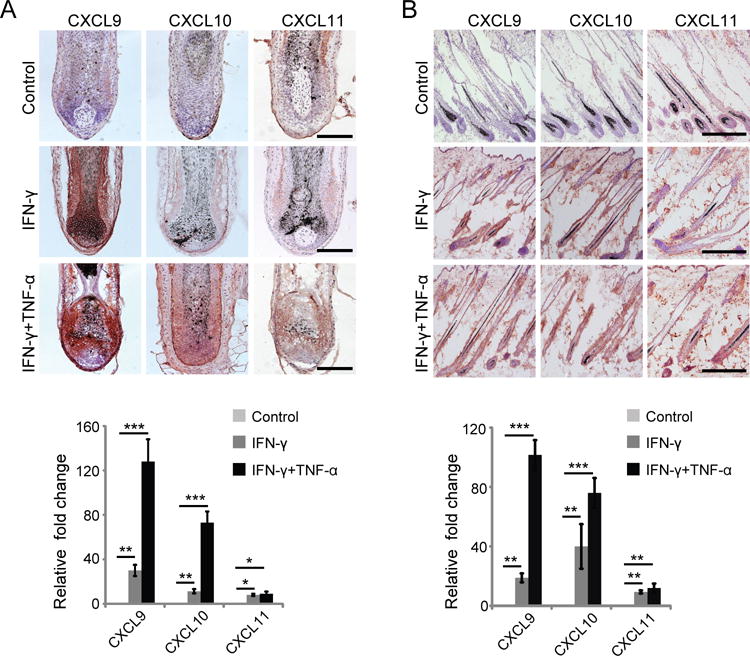
IFN-γ upregulates CXCR3 ligand expression in HF. (A) Normal human anagen HFs were microdissected and cultured in vitro for 48 hours in the presence of 50 ng/ml rhIFN-γ and 25 ng/ml rhTNF-α or 50 ng/ml rhIFN-γ alone (B). Upregulation of CXCL9, CXCL10, and CXCL11 in HF was determined by immunohistochemistry. Relative expression of CXCL9, CXCL10, and CXCL11 genes in normal human HF was determined by quantitative RT-PCR. Quantitative RT-PCR results were normalized to GAPDH and represent mean ± SEM relative expression from PBS and IFN-γ and TNF-α treatment. Data are representative of four independent experiments. Scale bar = 200μM. * indicates p <0.05, ** indicates p<0.01.
CXCR3 plays a key role in skin NKG2D+CD8+ T cell trafficking
Having established that CD8+NKG2D+ T cells from SDLNs express CXCR3, and that they can induce AA upon transfer into C3H/HeJ mice, we next investigated the contribution of CXCR3 to the recruitment of CD8+NKG2D+ T cells into inflamed skin. To determine whether CXCR3 plays a role in CD8+NKG2D+ T cell migration into AA alopecic skin, we isolated CD8+NKG2D+ T and CD8+NKG2D− T cells, and after CFSE-labeling, injected them into recipient C3H/HeJ mice within 2–3 weeks after grafting. At this stage, inflammation was limited to AA-affected HFs within the grafted skin and around HFs in host skin immediately adjacent to the graft and provided a model to test migration into active AA lesions. We quantitated accumulation of the labeled cells in inflammatory sites using immunofluorescence microscopy. We observed much greater migration of CD8+NKG2D+ T cells than CD8+NKG2D− T cells into grafted AA alopecic skin (Fig. 4A). To test whether their migration was CXCR3-dependent, we administered a neutralizing anti-CXCR3 mAb. We found that CXCR3 blockade significantly inhibited T cell migration into the active AA alopecic skin (Fig. 4A). By flow cytometry, we further demonstrated that CD8+NKG2D+ T cells migrated into the active AA alopecic skin in a CXCR3-dependent manner (Fig. 4B). Collectively, these data support the hypothesis that CXCR3 is required for migration of NKG2D+CD8+ T cells into inflamed AA alopecic skin in response to induction of CXCR3 ligands.
FIGURE 4.
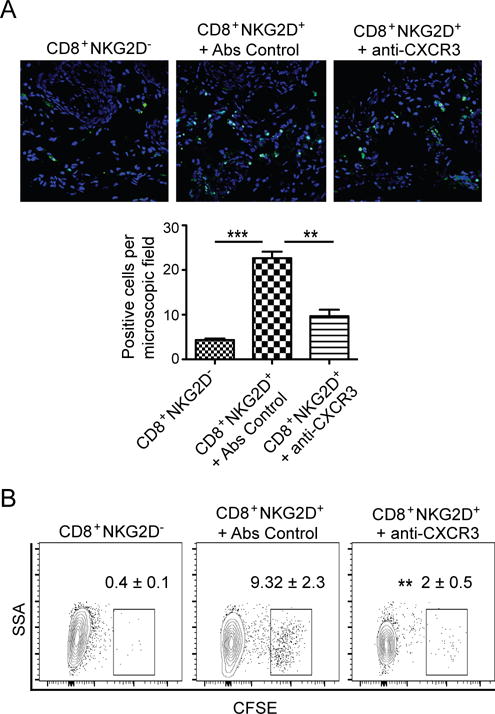
Migration of CD8+NKG2D+ T cells into alopecic skin. CD8+NKG2D− T cells or CD8+NKG2D+ T cells were purified, CFSE-labeled and injected i.v. together with anti-CXCR3 mAb or control mAb. (A) Migration of CFSE-labeled T cells into AA skin grafts was evaluated by immunofluorescent microscopy. Data are the average of three mice, with at least three fields counted per mouse. (B) Representative FACS plots of single cell suspension of mouse skin. Shown are the percentages of the CFSE positive cells gated on CD45+CD3+CD8+. The relative frequency of CD8+CFSE positive cells was reduced by anti-CXCR3 mAb treatment. Data are representative of three independent experiments. ** indicates p<0.01, and *** indicates p<0.001.
Treatment with neutralizing anti-CXCR3 mAb prevents onset of AA
We have previously shown that NKG2D+CD8+ T cells are both necessary and sufficient to induce AA (5), and that CXCR3 blockade significantly inhibited NKG2D+CD8+ T cells migration to the skin (Fig. 4). We next tested the potential effect of CXCR3 blockade on disease development in skin grafted mice. At the start of the study, C3H/HeJ mice were grafted with AA-affected skin from C3H/HeJ AA donor and injected with 200 ug anti-CXCR3 mAb or Ab isotype control twice a week for 12 weeks. In the control group, all mice developed AA by 12 weeks after grafting. In contrast, only 30% of mice receiving anti-CXCR3 antibodies developed AA by 12 weeks (Fig. 5A, 5B).
FIGURE 5.
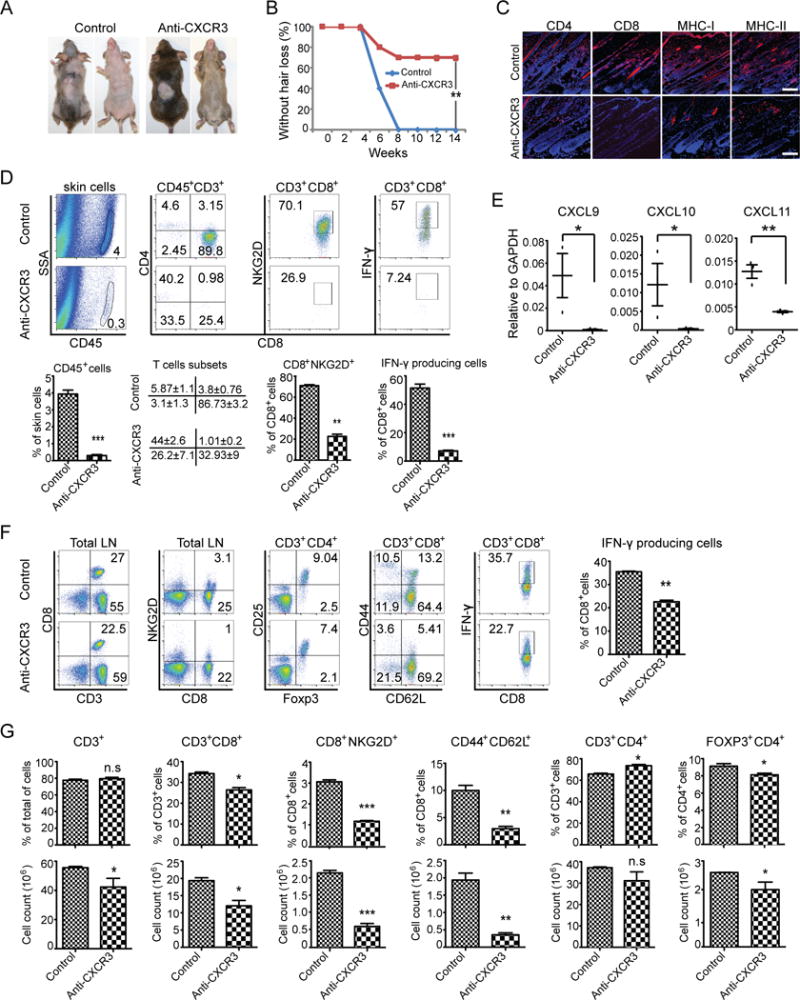
CXCR3 blockade prevents the onset of alopecia in AA skin grafted C3H/HeJ mice. Mice were treated beginning the day of grafting (n=5–10 mice per group). anti-CXCR3 mAb or isotype control IgG was administrated by i.p. injection (200 μg) two times weekly for 12 weeks. (A) The onset of alopecia was inhibited by administration of anti-CXCR3 mAb. (B) Time course of onset of AA in control mice and anti-CXCR3 treated mice was shown as weeks after grafting. (C) The expression of CD4, CD8, MHC class I and MHC class II in skin was significantly decreased in anti-CXCR3 treated mice compared to control mice. (D) Representative FACS plots of cell suspension of mouse skin. The frequencies of infiltrating CD45+ leukocytes and IFN-γ-producing CD8+NKG2D+ T cell in skin of anti-CXCR3 treated mice were significantly decreased compared to control mice. (E) CXCR3 ligand expression in skin from anti-CXCR3 treated mice and controls was analyzed using quantitative PCR. The expression of CXCR3 ligands in skin are significantly decreased in anti-CXCR3 treated mice compared to control mice. (F) Representative FACS plots of the SDLNs. Shown are the percentages of lymphocyte subsets. (G) The absolute numbers and the percentages of lymphocyte subsets in SDLNs are compared between the anti-CXCR3 treated group and control. Data are representative of two experiments with 10 mice total each group. Scale bar = 200μM. * indicates p<0.05, ** indicates p<0.01, and *** indicates p<0.001
We next performed immunofluorescence staining of skin at the end of the treatment period. Affected skin was heavily infiltrated by CD8+ T cells in control IgG-treated mice, but to a lesser extent in mice treated with the anti-CXCR3 antibody (Fig. 5C). Staining of additional histological markers of disease revealed that control Ab-treated animals showed extensive upregulation of MHC class I and MHC class II on HF epithelium, indicative of inflammation, whereas, anti-CXCR3 mAb-treated mice showed reduced staining of MHC class I and MHC class II on HF epithelium, indicative of scarce or absent inflammation (Fig. 5C). We found that anti-CXCR3 treatment blocked the accumulation of IFN-γ producing CD8+NKG2D+ T cells in the skin in the majority of grafted recipients (Fig. 5D). Correspondingly, the expression of the IFN-inducible CXCL9, CXCL10 and CXCL11 mRNAs was markedly decreased in the skin of anti-CXCR3 mAb-treated mice in comparison to controls (Fig. 5E).
We next compared the lymphocyte subset numbers and frequencies between these two groups. Although no difference was detected in the frequencies of CD3+ T cells between these two groups, because of the overall reduction in the total SDLNs cell number, anti-CXCR3 antibody treatment significantly decreased the absolute numbers of total CD3+ T cells in SDLNs. CD8+ T cells are the major pathogenic cells in AA (5). Mice treated with anti-CXCR3 antibody had a significant reduction in the number and frequency of CD8+ T cells, and especially CD8+NKG2D+ T cells and CD8+CD44+CD62L− effector memory T cells known to be associated with the pathogenesis of AA (5). Interestingly, the relative frequencies of CD4+ T cells were increased anti-CXCR3 mAb-treated mice. However, no statistical difference was detected in the absolute number of CD4+ T cells between these two groups due to the overall reduction in SDLNs cell number in anti-CXCR3 mAb-treated mice. The frequencies and the absolute number of CD4+FOXP3+ Tregs were decreased in anti-CXCR3 mAb-treated mice. Taken together, our data shows that blockade of CXCR3 by administration of a neutralizing antibody decreases accumulation of CD8+NKG2D+ T cells infiltration in the skin, which in turn prevents the development of AA.
CXCR3 blockade prevents accumulation of autoreactive T cells in SDLNs from C3H/HeJ skin grafted mice
We next turned to investigate whether CXCR3 and its ligands are also involved in autoreactive T cell accumulation in the lymph node. Robust expression of CXCL9 and CXCL10 were observed in SDLNs from C3H/HeJ mice with AA (Fig. 6A, 6B). Although CXCR3 ligands are induced in variety of cell types including DCs, macrophages, fibroblasts and epithelial cells (34, 35), we observed that CD11c+ MHC IIhigh DCs in SDLNs strongly expressed CXCL9 and CXCL10 (Fig. 6C).
FIGURE 6.
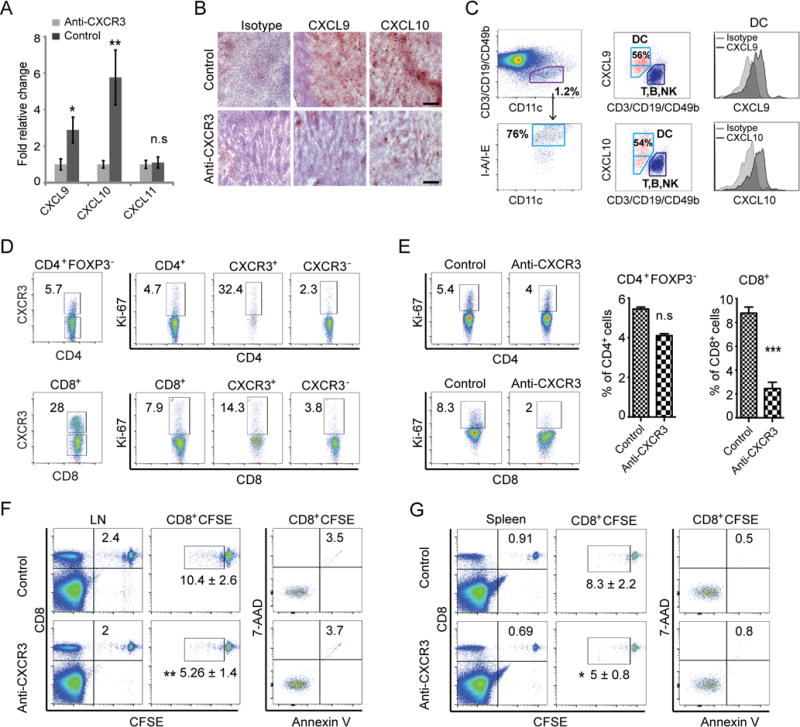
CXCR3 blockade inhibits the proliferation of CD8+NKG2D+ T cells in SDLNs. C3H/HeJ mice skin grafted mice were treated as described in Methods. (A) Expression of CXCR3 ligands in SDLNs from anti-CXCR3 treated mice and controls was determined by quantitative RT-PCR. Data are indicated as the mean ± SEM of four mice in each group. (B) Frozen sections of cutaneous lymph nodes from each mouse in anti-CXCR3 treated or control groups were stained with anti-CXCL9 or anti-CXCL10 Abs. The expression of CXCR3 ligands in SDLNs were significantly decreased in anti-CXCR3 treated mice compared to control mice. Scale bar = 200μM. (C) The CD11c+MHC-II+ DCs in SDLNs were identified by gating on the T-cell-negative, B-cell-negative and NK-cell-negative population. The dot plots show that CD11c+MHC-II+ DCs within SDLNs strongly expressed CXCL9 and CXCL10. (D) Representative FACS plots showed the expression of CXCR3 was strongly associated with the expression of Ki-67 in either CD4+ or CD8+ T cell subsets within SDLNs of control skin grafted mice. Treatment with anti-CXCR3 reduced Ki67 expression in CD8 T cells. (E) CD8+ T cell proliferation was significantly inhibited by anti-CXCR3 treatment. CD8+ T cells purified from SDLN from C3H/HeJ mice with AA were CFSE stained and injected i.v. into C3H/HeJ AA mice recipients. Proliferation of the CFSE-stained CD8+ T cells within LNs (F) and spleen (G) was assessed by CFSE dilution. Data are representative of three independent experiments. * indicates p<0.05, ** indicates p<0.01, *** indicates p<0.001, n.s. indicates not significant.
In addition to chemotaxis, CXCR3 is known to be required for the optimal generation of CD8+ effector T cells, by facilitating T cell-dendritic cell (DC) interactions during T cell priming because CD8+ T cells in CXCR3 receptor knockout or ligand knockout mice have reduced proliferative and cytotoxic ability (36–38). We next investigated if the expression of CXCR3 receptor on T cell was associated with T cell proliferation. We assessed the expression of Ki-67, a cellular marker for proliferation in CXCR3+ and CXCR3− T cell subsets. The expression of CXCR3 receptor was strongly associated with the expression of Ki-67 in both CD4+ and CD8+ T cells from SDLNs in C3H/HeJ mice with AA (Fig. 6D), indicating that CXCR3+ effector T cells are proliferative. Interestingly, the proliferation of CD8+ T cells, but to a lesser extent the proliferation of CD4+ T cells, was significantly inhibited in anti-CXCR3 mAb-treated mice (Fig. 6E), and this was in concordance with the decrease in the absolute number of CD8+ T cells, but not CD4+ T cells in anti-CXCR3 mAb-treated mice (Fig. 5G). To further test the effects of CXCR3 on CD8+ T cell proliferation, we analyzed the proliferation of adoptively transferred CFSE-labelled CD8+ T cells in vivo. The proliferation of the CFSE-stained CD8+ T cells in SDLN and spleen was significantly inhibited by anti-CXCR3 antibody treatment (Fig. 6F, 6 G). Collectively, these results suggest that anti-CXCR3 antibody treatment may prevent trafficking of T cells, and disrupt T cell-DC interactions in SDLN for autoreactive T cell activation during AA pathogenesis.
Discussion
AA is a common autoimmune disease resulting in an immune attack on the HF. AA is thought to be the result of loss of local immune privilege in the HF with local T cell-mediated cytotoxicity induced by CD8+ T cells with the help of CD4+ T cells (4, 6). Under a permissive genetic background, AA may be induced by environmental factors, such as infection, and potentially emotional and physical stresses (39–41). These factors may contribute to immune privilege collapse by inducing IFN-γ production in and around the HF immune privilege site (4, 6, 26). The role of IFN-γ in AA was previously established using both knockout studies as well as pharmacological administration of cytokine which precipitated disease (10). We previously showed that neutralizing IFN-γ in C3H/HeJ mice model of AA prevented the onset of hair loss (5), suggesting a key role of the IFN pathway in AA pathogenesis.
Our prior studies identified cytolytic CD8+NKG2D+ T cells in the SDLNs as the predominant immune effectors responsible for autoimmune attack of the HF (5). In AA mice, the immunophenotype of the skin-infiltrating CD8+ T cells was similar to the CD8+NKG2D+ T population found in the SDLNs (5). The signals that recruit autoreactive T cell migration into skin and HF during AA are unknown. Our global transcriptional profiling of mouse and human AA skin revealed striking signatures indicative of a robust IFN-γ response, including the IFN-inducible chemokines CXCL9/10/11 (5). IFN-inducible chemokines and their common receptor, CXCR3, have been implicated as playing a central role in the maintenance and amplification of autoimmunity-related inflammatory processes (42). Inhibition of CXCR3 or the chemokine CXCL10 by specific blocking mAbs have been successfully tested in other models of autoimmune diseases, such as RA and vitiligo (19, 23).
In this study, we found that expression of CXCR3 ligands was upregulated in the HF in C3H/HeJ AA mice, and that CD8+ T cells infiltrating the HFs expressed CXCR3 receptor, underscoring the importance of CXCR3 ligands/CXCR3 function in CD8+NKG2D+ T cells in AA progression. To demonstrate the effect of CXCR3 ligands/CXCR3 in disease pathogenesis, we showed that therapeutic anti-CXCR3 treatment delayed or prevented the development of AA in C3H/HeJ mice (Fig. 5). Anti-CXCR3 mAb-treated mice had fewer autoreactive CD8+NKG2D+ T cells in the skin and peripheral lymphoid tissues (Fig. 5). Our studies suggest that CXCR3 is a key contributor to the pathogenesis of AA by mediating the infiltration of autoreactive CD8+NKG2D+ T cells into the skin during the development of adoptively grafted AA.
Although CXCL9, CXCL10 and CXCL11 all signal through the CXCR3 receptor, it has been reported that CXCR3 ligands may have overlapping, nonredundant or dominant functions depending on the model of disease studied (42). For example, in the MRL/lpr lupus mouse model, kidney disease was suppressed in both CXCR3−/− and CXCL9−/− mice, but not in CXCL10−/− mice, suggesting CXCL9 dominance in that system (43). In a mouse model of vitiligo, CXCL10−/− hosts were protected for disease, whereas CXCL9−/− hosts developed depigmentation, indicating that CXCL10 is the single chemokine responsible for vitiligo (19). In humans, the therapeutic activity of a blocking CXCL10 antibody (MDX-1100) was reported in a phase 2 clinical trial for RA, and emphasized the crucial role of CXCL10 in this disease (23). We have shown that the expression of CXCL9, CXCL10 and CXCL11 were all upregulated in mice with AA. Further studies will be needed to address which ligands of CXCR3 play the potential dominant role in the pathogenesis of AA, or whether anti-chemokine monotherapy could offer therapeutic benefit as shown in RA, vitiligo and other autoimmune diseases.
The C3H/HeJ model is a well-established graft-transfer model of AA. Skin from an AA affected mouse can initiate the entire autoimmune process in C3H/HeJ recipients. HF antigens, activated APCs and/or T cells present in the graft may initiate and/or perpetuate this process (5, 24, 44). Our data shows that CXCR3 blockade prevents alopecic T cell accumulation in the end organ, lesional skin. This could be due to depletion, impaired recruitment, proliferation, survival and/or retention. Previous studies have shown that the antibody used here, mAb CXCR3-173, is not a depleting antibody (30), and our studies confirmed that CD8+NKG2D+ T cells were not depleted by administration of 1mg CXCR3-173 (Supplemental figure 1). Here, using cell transfer of CFSE labeled T cells, we found that blockade of CXCR3 is not only able to prevent the recruitment of CD8+NKG2D+ T cells to skin, but moreover, inhibits the proliferation of effector T cells in the SDLN. Previously, CXCR3 was shown to be required for the optimal generation of CD8+ effector T cells (36–38) (Fig. 5 and Supplemental figure 2). We found that CXCL9 and CXCL10 were upregulated in SDLNs form AA mice, and that most of the CXCL9 and CXCL10 producing cells were DCs. In mouse viral infection models, CXCR3 interaction with CXCR3 ligands produced by DCs and accessory cells in draining LNs, can promote T cell differentiation (34). A CXCR3-dependent inflammatory loop may not only increase recruitment of T cells into peripheral tissues, but also could enhance the generation of effector T cells and promote increased effector responses (34, 35). If so, it is possible that activated APCs and/or T cells present in the graft can migrate to draining LNs, producing IFN that upregulates expression of CXCR3 ligands by DCs or accessory cells, leading to intra-nodal chemokine gradients (34, 35). These, in turn, may promote CXCR3+ T cell interaction with DCs or other APCs for increased antigen stimulation and/or unique cytokine signals for T cell activation and proliferation. However, we cannot exclude the possibility of a different signal leading to CXCR3+ T cell interaction with APCs and LN retention.
Although the IFN-CXCR3-chemokine-dependent inflammatory loop has been implicated in some inflammatory diseases (19–23, 43), including AA (24–26), the cellular source of the ligands in different types of inflammation remains unanswered. CXCR3 ligands have been reported to be expressed by different cell types, such as macrophages, DCs, fibroblasts and epithelial cells (16–18). Ito et al previously reported that in human AA, CXCL10 was highly expressed in the HFs and surrounding interstitial cells, while normal HFs showed weak CXCL10 staining in the outer root sheath (26). We present evidence that in human AA, CXCL9 and CXCL11 were also expressed in HFs in addition to the upregulation of CXCL10. This is consistent with our gene expression microarray data that CXCL9, CXCL10 and CXCL11 mRNAs are upregulated in human AA alopecic skin (5, 45). Further, we found that normal human HFs upregulate the expression of CXCL9, CXCL10 and CXCL11 mRNA and protein levels when treated with IFN-γ. In vivo, we also found that intradermal injection of IFN-γ induced CXCL9, CXCL10 and CXCL11 mRNA and protein expression in the HFs of normal hair C3H mice. Beyond the HF, infiltrating leukocytes such as DCs and macrophages may also be important source of CXCR3 ligands in AA.
Our finding unexpectedly revealed that the expression of CXCR3 on some HFs infiltrating CD8+ T cells from mice with AA was downregulated or absent when examined by immunofluorescence microscopy. Meiser et al showed that cell surface CXCR3 was internalized and degraded following incubation with its ligands, and CXCL11 was the most potent ligand to downregulate CXCR3 expression (31). CXCR3-dependent T cell recruitment permits the entry of CXCR3 negative T cells into HF immune privilege sites, and it is also postulated that in early stage of AA, the first waves of CXCR3+ T cells were recruited and migrated to inflamed skin containing high levels of CXCR3 ligands, producing proinflammatory cytokines or chemokines, leading to the recruitment of CXCR3 negative T cells expressing other chemokine receptors (35, 42). Taken together, it is possible that ligand-mediated cell surface CXCR3 receptor downregulation and recruitment of CXCR3 negative T cells may explain CXCR3 negative T cells in AA alopecic skin from mice, and the therapeutic activity of a blocking CXCR3 antibody in prevention of AA. Further studies are needed to ascertain whether recruitment and retention of alopecic T cells in both initiation and maintenance of disease remains CXCR3 dependent, by establishing whether CXCR3 blockade can reverse established disease.
Although neutralizing antibodies to CXCR3 or its ligands have proven successful in blocking several inflammatory diseases, their efficacy is often challenged by limited penetration and accelerated elimination after repetitive administration. Small molecule CXCR3 antagonists could overcome some of these challenges, and have shown efficacy in reducing inflammation in the collagen-induced arthritis (CIA) and experimental autoimmune encephalomyelitis (EAE) models, as well as prolonged allograft survival (46). Topical delivery of a selective CXCR3 antagonist could provide an improved therapeutic index, by limiting systemic exposure to immuomodulatory agents.
Collectively, our data suggest that CXCR3 is a key contributor to the pathogenesis of AA by reducing accumulation of T cells in the skin during the development of AA in the grafted C3H/HeJ mouse model of AA. Our data suggests that CXCR3 blockade could impact disease progression by impacting both recruitment to the end organ and the priming and expansion of autoaggressive T cells in the lymph node. In the future, studies targeting CXCR3 and its ligands may prove useful therapeutic targets for the treatment of human AA.
Supplementary Material
Acknowledgments
We thank Emily Chang, Jade Huang and Ming Zhang for expert assistance in the laboratory. We appreciate the support of the Skin Disease Research Center in the Department of Dermatology at Columbia University.
This work was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health Grant P30AR44535 and R01AR64014 (to A.M.C.). Additional supports were obtained from Locks of Love. Z.D. was supported by a Research Award from the National Alopecia Areata Foundation.
Abbreviations in this article
- AA
alopecia areata
- SDLN
skin-draining lymph nodes
- HF
hair follicle
- ORS
outer root sheath
- IRS
inner root sheath
- DC
dendritic cell
- qRT-PCR
quantitative RT-PCR
References
- 1.Safavi KH, Muller SA, Suman VJ, Moshell AN, Melton LJ., 3rd Incidence of alopecia areata in Olmsted County, Minnesota, 1975 through 1989. Mayo Clin Proc. 1995;70:628–633. doi: 10.4065/70.7.628. [DOI] [PubMed] [Google Scholar]
- 2.Paus R, Nickoloff BJ, Ito T. A ‘hairy’ privilege. Trends Immunol. 2005;26:32–40. doi: 10.1016/j.it.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 3.Sun J, Silva KA, McElwee KJ, King LE, Jr, Sundberg JP. The C3H/HeJ mouse and DEBR rat models for alopecia areata: review of preclinical drug screening approaches and results. Exp Dermatol. 2008;17:793–805. doi: 10.1111/j.1600-0625.2008.00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McElwee KJ, Gilhar A, Tobin DJ, Ramot Y, Sundberg JP, Nakamura M, Bertolini M, Inui S, Tokura Y, King LE, Jr, Duque-Estrada B, Tosti A, Keren A, Itami S, Shoenfeld Y, Zlotogorski A, Paus R. What causes alopecia areata? Exp Dermatol. 2013;22:609–626. doi: 10.1111/exd.12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, de Jong A, Harel S, DeStefano GM, Rothman L, Singh P, Petukhova L, Mackay-Wiggan J, Christiano AM, Clynes R. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. 2014;20:1043–1049. doi: 10.1038/nm.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilhar A, Paus R, Kalish RS. Lymphocytes, neuropeptides, and genes involved in alopecia areata. J Clin Invest. 2007;117:2019–2027. doi: 10.1172/JCI31942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruckert R, Hofmann U, van der Veen C, Bulfone-Paus S, Paus R. MHC class I expression in murine skin: developmentally controlled and strikingly restricted intraepithelial expression during hair follicle morphogenesis and cycling, and response to cytokine treatment in vivo. J Invest Dermatol. 1998;111:25–30. doi: 10.1046/j.1523-1747.1998.00228.x. [DOI] [PubMed] [Google Scholar]
- 8.Paus R, Slominski A, Czarnetzki BM. Is alopecia areata an autoimmune-response against melanogenesis-related proteins, exposed by abnormal MHC class I expression in the anagen hair bulb? Yale J Biol Med. 1993;66:541–554. [PMC free article] [PubMed] [Google Scholar]
- 9.Gilhar A, Kam Y, Assy B, Kalish RS. Alopecia areata induced in C3H/HeJ mice by interferon-gamma: evidence for loss of immune privilege. J Invest Dermatol. 2005;124:288–289. doi: 10.1111/j.0022-202X.2004.23580.x. [DOI] [PubMed] [Google Scholar]
- 10.Freyschmidt-Paul P, McElwee KJ, Hoffmann R, Sundberg JP, Vitacolonna M, Kissling S, Zoller M. Interferon-gamma-deficient mice are resistant to the development of alopecia areata. Br J Dermatol. 2006;155:515–521. doi: 10.1111/j.1365-2133.2006.07377.x. [DOI] [PubMed] [Google Scholar]
- 11.Petukhova L, Duvic M, Hordinsky M, Norris D, Price V, Shimomura Y, Kim H, Singh P, Lee A, Chen WV, Meyer KC, Paus R, Jahoda CA, Amos CI, Gregersen PK, Christiano AM. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466:113–117. doi: 10.1038/nature09114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rollins BJ. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- 13.Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol. 1997;15:675–705. doi: 10.1146/annurev.immunol.15.1.675. [DOI] [PubMed] [Google Scholar]
- 14.Schall TJ, Bacon KB. Chemokines, leukocyte trafficking, and inflammation. Curr Opin Immunol. 1994;6:865–873. doi: 10.1016/0952-7915(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 15.Loetscher M, Loetscher P, Brass N, Meese E, Moser B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol. 1998;28:3696–3705. doi: 10.1002/(SICI)1521-4141(199811)28:11<3696::AID-IMMU3696>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 16.Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, Baggiolini M, Moser B. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP, Lin W, Boyd JG, Moser B, Wood DE, Sahagan BG, Neote K. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med. 1998;187:2009–2021. doi: 10.1084/jem.187.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao F, Rabin RL, Yannelli JR, Koniaris LG, Vanguri P, Farber JM. Human Mig chemokine: biochemical and functional characterization. J Exp Med. 1995;182:1301–1314. doi: 10.1084/jem.182.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rashighi M, Agarwal P, Richmond JM, Harris TH, Dresser K, Su MW, Zhou Y, Deng A, Hunter CA, Luster AD, Harris JE. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci Transl Med. 2014;6:223ra223. doi: 10.1126/scitranslmed.3007811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motoki Y, Tani K, Shimizu T, Tamiya H, Hase K, Ohmoto Y, Matsushima K, Sone S. The expression of chemokine receptor CXCR3: relevance to disease activity of rheumatoid arthritis. Mod Rheumatol. 2003;13:114–120. doi: 10.3109/s10165-002-0209-2. [DOI] [PubMed] [Google Scholar]
- 21.Flier J, Boorsma DM, van Beek PJ, Nieboer C, Stoof TJ, Willemze R, Tensen CP. Differential expression of CXCR3 targeting chemokines CXCL10, CXCL9, and CXCL11 in different types of skin inflammation. J Pathol. 2001;194:398–405. doi: 10.1002/1096-9896(200108)194:4<397::aid-path899>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 22.Goebeler M, Toksoy A, Spandau U, Engelhardt E, Brocker EB, Gillitzer R. The C-X-C chemokine Mig is highly expressed in the papillae of psoriatic lesions. J Pathol. 1998;184:89–95. doi: 10.1002/(SICI)1096-9896(199801)184:1<89::AID-PATH975>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 23.Yellin M, Paliienko I, Balanescu A, Ter-Vartanian S, Tseluyko V, Xu LA, Tao X, Cardarelli PM, Leblanc H, Nichol G, Ancuta C, Chirieac R, Luo A. A phase II, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of MDX-1100, a fully human anti-CXCL10 monoclonal antibody, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 2012;64:1730–1739. doi: 10.1002/art.34330. [DOI] [PubMed] [Google Scholar]
- 24.McPhee CG, Duncan FJ, Silva KA, King LE, Jr, Hogenesch H, Roopenian DC, Everts HB, Sundberg JP. Increased expression of Cxcr3 and its ligands, Cxcl9 and Cxcl10, during the development of alopecia areata in the mouse. J Invest Dermatol. 2012;132:1736–1738. doi: 10.1038/jid.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zainodini N, Hassanshahi G, Arababadi MK, Khorramdelazad H, Mirzaei A. Differential expression of CXCL1, CXCL9, CXCL10 and CXCL12 chemokines in alopecia areata. Iran J Immunol. 2013;10:40–46. [PubMed] [Google Scholar]
- 26.Ito T, Hashizume H, Shimauchi T, Funakoshi A, Ito N, Fukamizu H, Takigawa M, Tokura Y. CXCL10 produced from hair follicles induces Th1 and Tc1 cell infiltration in the acute phase of alopecia areata followed by sustained Tc1 accumulation in the chronic phase. J Dermatol Sci. 2013;69:140–147. doi: 10.1016/j.jdermsci.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Nakae S, Komiyama Y, Narumi S, Sudo K, Horai R, Tagawa Y, Sekikawa K, Matsushima K, Asano M, Iwakura Y. IL-1-induced tumor necrosis factor-alpha elicits inflammatory cell infiltration in the skin by inducing IFN-gamma-inducible protein 10 in the elicitation phase of the contact hypersensitivity response. Int Immunol. 2003;15:251–260. doi: 10.1093/intimm/dxg028. [DOI] [PubMed] [Google Scholar]
- 28.Ito T, Ito N, Saathoff M, Bettermann A, Takigawa M, Paus R. Interferon-gamma is a potent inducer of catagen-like changes in cultured human anagen hair follicles. Br J Dermatol. 2005;152:623–631. doi: 10.1111/j.1365-2133.2005.06453.x. [DOI] [PubMed] [Google Scholar]
- 29.Glotzer DJ, Zelzer E, Olsen BR. Impaired skin and hair follicle development in Runx2 deficient mice. Dev Biol. 2008;315:459–473. doi: 10.1016/j.ydbio.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uppaluri R, Sheehan KC, Wang L, Bui JD, Brotman JJ, Lu B, Gerard C, Hancock WW, Schreiber RD. Prolongation of cardiac and islet allograft survival by a blocking hamster anti-mouse CXCR3 monoclonal antibody. Transplantation. 2008;86:137–147. doi: 10.1097/TP.0b013e31817b8e4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meiser A, Mueller A, Wise EL, McDonagh EM, Petit SJ, Saran N, Clark PC, Williams TJ, Pease JE. The chemokine receptor CXCR3 is degraded following internalization and is replenished at the cell surface by de novo synthesis of receptor. J Immunol. 2008;180:6713–6724. doi: 10.4049/jimmunol.180.10.6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanda N, Shimizu T, Tada Y, Watanabe S. IL-18 enhances IFN-gamma-induced production of CXCL9, CXCL10, and CXCL11 in human keratinocytes. Eur J Immunol. 2007;37:338–350. doi: 10.1002/eji.200636420. [DOI] [PubMed] [Google Scholar]
- 33.Kraft M, Riedel S, Maaser C, Kucharzik T, Steinbuechel A, Domschke W, Luegering N. IFN-gamma synergizes with TNF-alpha but not with viable H. pylori in up-regulating CXC chemokine secretion in gastric epithelial cells. Clin Exp Immunol. 2001;126:474–481. doi: 10.1046/j.1365-2249.2001.01634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, von Andrian UH, Moon JJ, Mempel TR, Luster AD. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity. 2012;37:1091–1103. doi: 10.1016/j.immuni.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res. 2011;317:620–631. doi: 10.1016/j.yexcr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168:3195–3204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- 37.Whiting D, Hsieh G, Yun JJ, Banerji A, Yao W, Fishbein MC, Belperio J, Strieter RM, Bonavida B, Ardehali A. Chemokine monokine induced by IFN-gamma/CXC chemokine ligand 9 stimulates T lymphocyte proliferation and effector cytokine production. J Immunol. 2004;172:7417–7424. doi: 10.4049/jimmunol.172.12.7417. [DOI] [PubMed] [Google Scholar]
- 38.Thapa M, Carr DJ. CXCR3 deficiency increases susceptibility to genital herpes simplex virus type 2 infection: Uncoupling of CD8+ T-cell effector function but not migration. J Virol. 2009;83:9486–9501. doi: 10.1128/JVI.00854-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodriguez TA, Duvic M, R. National Alopecia Areata Onset of alopecia areata after Epstein-Barr virus infectious mononucleosis. J Am Acad Dermatol. 2008;59:137–139. doi: 10.1016/j.jaad.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 40.Gupta MA, Gupta AK, Watteel GN. Stress and alopecia areata: a psychodermatologic study. Acta Derm Venereol. 1997;77:296–298. doi: 10.2340/0001555577296298. [DOI] [PubMed] [Google Scholar]
- 41.de Andrade M, Jackow CM, Dahm N, Hordinsky M, Reveille JD, Duvic M. Alopecia areata in families: association with the HLA locus. The journal of investigative dermatology Symposium proceedings / the Society for Investigative Dermatology, Inc [and] European Society for Dermatological Research. 1999;4:220–223. doi: 10.1038/sj.jidsp.5640215. [DOI] [PubMed] [Google Scholar]
- 42.Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci. 2009;1173:310–317. doi: 10.1111/j.1749-6632.2009.04813.x. [DOI] [PubMed] [Google Scholar]
- 43.Menke J, Zeller GC, Kikawada E, Means TK, Huang XR, Lan HY, Lu B, Farber J, Luster AD, Kelley VR. CXCL9, but not CXCL10, promotes CXCR3-dependent immune-mediated kidney disease. J Am Soc Nephrol. 2008;19:1177–1189. doi: 10.1681/ASN.2007111179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McElwee KJ, Boggess D, King LE, Jr, Sundberg JP. Experimental induction of alopecia areata-like hair loss in C3H/HeJ mice using full-thickness skin grafts. J Invest Dermatol. 1998;111:797–803. doi: 10.1046/j.1523-1747.1998.00380.x. [DOI] [PubMed] [Google Scholar]
- 45.Jabbari A, Nguyen N, Cerise JE, Ulerio G, de Jong A, Clynes R, Christiano AM, Mackay-Wiggan J. Treatment of an alopecia areata patient with tofacitinib results in regrowth of hair and changes in serum and skin biomarkers. Exp Dermatol. 2016 doi: 10.1111/exd.13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jenh CH, Cox MA, Cui L, Reich EP, Sullivan L, Chen SC, Kinsley D, Qian S, Kim SH, Rosenblum S, Kozlowski J, Fine JS, Zavodny PJ, Lundell D. A selective and potent CXCR3 antagonist SCH 546738 attenuates the development of autoimmune diseases and delays graft rejection. BMC Immunol. 2012;13:2. doi: 10.1186/10.1186/1471-2172-13-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.