

Summary
Chromatin immunoprecipitation followed by sequencing (ChIP-seq) has been instrumental to our current view of chromatin structure and function. It allows genome-wide mapping of histone marks, which demarcate biologically relevant domains. However, ChIP-seq is an ensemble measurement reporting the average occupancy of individual marks in a cell population. Consequently, our understanding of the combinatorial nature of chromatin states relies almost exclusively on correlation between the genomic distributions of individual marks. Here, we report the development of combinatorial-iChIP to determine the genome-wide co-occurrence of histone marks at single-nucleosome resolution. By comparing to a null model, we show that certain combinations of overlapping marks (H3K36me3 and H3K79me3) co-occur more frequently than would be expected by chance, while others (H3K4me3 and H3K36me3) do not, reflecting differences in the underlying chromatin pathways. We further use combinatorial-iChIP to illuminate aspects of the Set2-RPD3S pathway. This approach promises to improve our understanding of the combinatorial complexity of chromatin.
Keywords: chromatin, ChIP, histone modifications, nucleosomes, Set2, Rpd3
Graphical Abstract
Highlights
-
•
High-throughput method for mapping the genome-wide co-occurrence of histone marks
-
•
H3K36me3 and H3K79me3 co-occur more frequently than would be expected by chance
-
•
Initiating and elongating Pol II-deposited marks co-occur on the same nucleosomes
-
•
Disruption of RPD3 increases the co-occurrence of H3K36me3 and H3 acetylation
Standard chromatin immunoprecipitation followed by sequencing reports on the average occupancy of individual histone marks in a population of cells. As such, it cannot ascertain whether two marks co-exist on the same nucleosome. Sadeh et al. describe methodology to determine the genome-wide co-occurrence of histone marks at single-nucleosome resolution.
Introduction
Nucleosomal histones, the fundamental packaging units of DNA, are extensively decorated by a large number of posttranslational modifications (PTMs) or “marks.” These marks are highly conserved and play key roles in all genomic transactions (Rivera and Ren 2013). Enzymes that deposit, remove, or bind histone marks, as well as modified residues in histones, are frequently mutated in diseases such as cancer (Baylin and Jones, 2011, Maze et al., 2014, Chi et al., 2010, Lewis et al., 2013, Lu et al., 2016). Chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq) has been widely used to determine the genome-wide location of nucleosomes bearing specific histone marks and has been instrumental to our understanding of chromatin architecture, structure, and function in many cell types (ENCODE Project Consortium, 2012, Weiner et al., 2015). ChIP-seq studies in a variety of organisms identify combinations of spatially correlated histone marks; these combinatorial patterns demarcate biologically relevant domains such as actively transcribed or polycomb repressed genes, heterochromatin, paused and active regulatory elements, and enhancers. These patterns can be used to annotate the genome and predict unknown genomic functionalities (Guttman et al., 2009, Heintzman et al., 2007, Ernst and Kellis, 2012). Moreover, several chromatin-binding proteins that contain multiple recognition domains were shown to bind to specific combinations of histone marks (Ruthenburg et al., 2011), suggesting that mark co-occurrence can have functional implications.
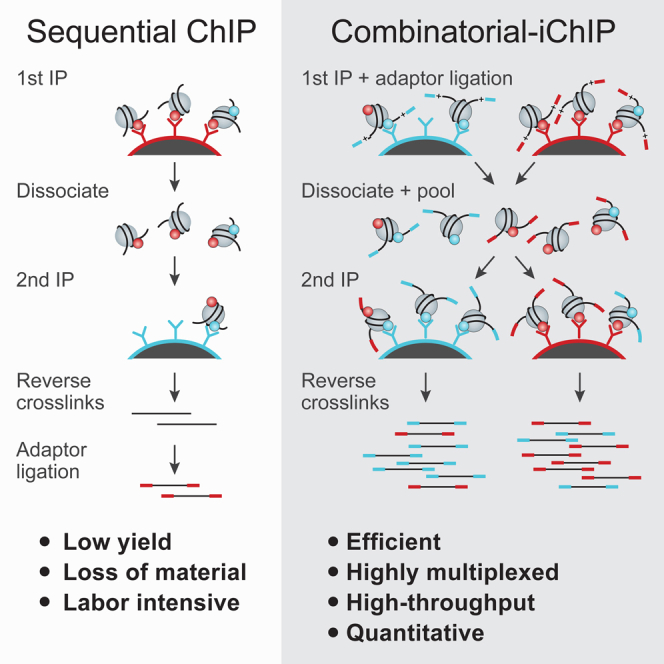
A typical ChIP-seq experiment reports on the average position and occupancy of a single modification averaged over a large population of potentially heterogeneous cells. As a result, our current understanding of the combinatorial nature of chromatin states relies almost exclusively on genomic correlations between chromatin features that may or may not occur in the same cell. Biochemical studies have identified dozens of different histone marks, as well as multiple proteins that deposit, erase, and bind them. However, efforts to probe the complexity of chromatin in various cell types have identified a limited number of combinations of histone marks that specify defined genomic regions (ENCODE Project Consortium, 2012, Weiner et al., 2015). Mass spectrometry has proven to be a powerful tool for identification of histone mark complexity; however, it lacks genomic localization and is limited to marks co-residing on a single, relatively short, peptide (Garcia et al., 2007, Young et al., 2009). Recently, single-molecule imaging allowed visualization of combinations of histone modifications; however, it has limited genomic context (Shema et al., 2016). Importantly, it is generally unknown if genomically correlated histone marks coexist or, alternatively, if they represent different chromatin states occurring in different subsets of a population of cells (Figure 1A). Sequential ChIP, where histone marks are sequentially immunoprecipitated, can report on the actual combinatorial nature of histone marks (Bernstein et al., 2006), yet such experiments are surprisingly rare and fraught with technical challenges. One factor that may impair the robustness and reliability of such experiments is the large amount of biological material required to provide sufficient input to the second ChIP, which can result in low signal-to-background ratio. Here, we report the development of a combinatorial indexed ChIP method (combinatorial-iChIP) to map the genome-wide co-occurrence of histone marks at single-nucleosome resolution (Figure 1B) and provide experimental and analytical tools for efficient, reliable, and reproducible detection of combinations of histone marks.
Figure 1.

Combinatorial-iChIP Protocol for Assaying Combinations of Histone Modifications
(A) Overlapping signal of standard ChIP (red and cyan) can be due to co-occurrence of the two marks on the same nucleosomes (left peak, co-occurring), but can also be due to disjoint occurrence in the same location in different cells (right peak, disjointed). Combinatorial signal (purple) allows us to distinguish the two scenarios.
(B) Outline of the combinatorial-iChIP protocol: MNase-digested chromatin is immobilized on magnetic beads coated with antibodies of interest (first IP). Immobilized nucleosomes are ligated to barcoded adaptors to specify the 1st antibody. Following antibody inactivation and nucleosome release, samples are pooled, re-divided, and subjected to a second ChIP. Nucleosomes are reverse crosslinked and NGS sequences are added by PCR to barcoded DNA to generate NGS-compatible libraries. At this stage a second barcode denoting the second ChIP pool is added to the fragments.
(C) The signal from the first ChIP step of the MNase combinatorial-iChIP protocol (first ChIP, solid colors) is in close agreement to standard MNase-ChIP (faded colors) (Weiner et al., 2015). Shown are coverage tracks for a representative genomic region.
(D) Reciprocal combinatorial-iChIP signals are in good agreement. Same locus as (C).
(E) Combinatorial-iChIP uncovers co-occurrences and disjoint occurrences that are not available from individual ChIP. Shown is a representative genomic region. The gray boxes highlight locations that are similar in terms of individual ChIP but different in combinatorial-iChIP for these individual marks (black arrows). See also Figures S1 and S2.
Design
To investigate combinations of histone marks on individual nucleosomes, we need to address several concerns:
-
(1)
To obtain single-nucleosome resolution, we need to be able to determine the chromatin fragments that undergo IP.
-
(2)
Once we consider successive IP stages, we need to deal with the issue of low amounts of material.
-
(3)
We need to deal with multiple samples, each probed by a large number of IP pair combinations.
-
(4)
We must ensure that the method is quantitative and reproducible.
-
(5)
We would like the experimental design to be as compatible as possible with high-throughput practices.
Following our experience with MNase-ChIP (Liu et al., 2005, Weiner et al., 2015), we chose to deal with the first challenge by using MNase digestion to break chromatin into nucleosomal particles. The DNA fragment size allows us to distinguish mono-nucleosomal particles from multi-nucleosomal ones. In addition, unlike sonication, MNase digestion can be readily adapted to high-throughput practices.
Performing library construction on small amounts material following IP has been the primary challenge in applying ChIP-seq to small populations of cells. Several recent works in the literature avoid this problem by performing early adaptor ligation on chromatin before the IP step (van Galen et al., 2016, Lara-Astiaso et al., 2014, Rhee and Pugh, 2011, Singh et al., 2014). This implies that, following IP, the material can be amplified and sequenced without additional processing. The use of early adaptor ligation enables barcoding of samples. Once samples are barcoded, they can be pooled together and demultiplexed after sequencing (Lara-Astiaso et al., 2014). Specifically, the iChIP method (Lara-Astiaso et al., 2014) combines these ideas to multiplex ChIP from multiple samples with limited numbers of cells.
Sample pooling can multiplex many samples, which allows us to use small amounts of input material per sample and to process a combinatorial number of samples. Second, by performing IP for multiple samples in a single tube, we reduce technical variability between samples.
Results
Combinatorial-iChIP Can Detect Co-occurrence of Histone Marks
To test the feasibility of chromatin barcoding for detecting co-occurrence of two histone marks on the same nucleosome, we selected well-established antibodies against promoter and gene 5′ region (H3K4me3, and H3K18ac) and gene-body (H3K36me3, and H3K79me3) histone marks. We used exponentially growing S. cerevisiae (budding yeast) as a model in studying the interactions of these marks (Rando and Winston, 2012).
In combinatorial-iChIP the first IP and barcoding step provide the first layer of specificity (Figure 1B). To test the specificity provided by this step, we sequenced material obtained from our first IP and barcoding steps. The resulting coverage maps are in good agreement with previously published MNase-ChIP-seq datasets (Weiner et al., 2015) at both local (Figure 1C) and genomic (Figure S1) scales.
Following the first IP, we pooled the barcoded chromatin and used it for the second IP step with the same battery of antibodies (Figure 1B), thus reading out pairwise co-occurrence of histone modifications. The use of MNase-digested chromatin ensures mononucleosome resolution for this assay. Combinatorial-iChIP produced clear signal that was distinguishable from the parental ChIP experiments. Independent combinatorial-iChIP experiments showed highly similar enrichment patterns (Figure S1B). Importantly, combinatorial-iChIP signal was highly similar between reciprocal experiments in which the order of the two antibodies was reversed (Figures 1D and S1B) and exhibited different patterns compared to either of the relevant individual ChIPs (Figures 1E and S2), implying that combinatorial-iChIP captures genomic location where the tested histone marks co-reside on a single nucleosome.
To further determine whether combinatorial- iChIP signal is specific to its target modification, we repeated these assays in cells that express histones mutated at the antibody target residue (e.g., histone 3 lysine 18 replaced by an arginine, H3K18R). This experiment allows one to measure specificity of both IP steps (Figure 2A). After the first IP step, we sequenced the pooled material and subjected all samples to the same amplification and sampling depth. Comparing the number of unique reads, we observed a dramatic reduction in yield when the target residue is mutated compared to WT sample (Figure 2B). The effect was somewhat smaller in H3K36R, potentially due to cross-reactivity of the specific batch of antibody used and the lack of competition with high-affinity target of the antibody in IP reaction. The dramatic loss of material in the mutant backgrounds confirms that the barcoding step during the first IP is highly specific to its target residue. To measure yield in the second IP stage, we compared the number of unique reads from a specific first IP (e.g., H3K4R-H3K36me3) before and after the second IP. This yield was dramatically and significantly reduced when the target residue of the second IP was mutated (Figure 2C and Table S1). We stress that, unlike canonical ChIP, our second IP was carried in the same tube for all samples, thus eliminating any noise due to sample handling. Together, these observations suggest that the signal obtained by combinatorial-iChIP is specific and is not the result of nonspecific background interactions during the first and second IPs.
Figure 2.

Combinatorial-iChIP Signal Is Specific
(A) Schematic diagram of the flow of material in our histone KO experiment. Samples from four strains (first column from the left) were each subjected to first IP step with four different antibodies (listed on the second column) and each barcoded with a different index. We pooled the resulting material. A fraction was directly sequenced (first IP), and the rest was divided into the second IP step with four antibodies.
(B) Number of unique sequenced reads in different first IP steps shown relative to the same IP in the WT sample. Observed numbers are 0.5%, 3.7%, and 17.8% for H3K4me3, H3K18ac, and H3K36me3, respectively.
(C) Number of unique sequenced reads in different second IP libraries. For each index, we calculate the yield as the ratio between the output (number reads after the second IP) and input (number of reads in the pooled input). We report yield normalized to the yield of the same sample from the WT strain. Results are shown as boxplots (25%, 50%, and 75% percentile) with individual indexes (strain/first IP combinations) as dots. p values are computed using t test. Median yield is 2.5%, 9.2%, and 8.2% for H3K4me3, H3K18ac, and H3K36me3, respectively. See also Table S1.
Combinatorial-iChIP Is a Quantitative Assay
We next turned to examine the quantitative nature of the combinatorial-iChIP signal. At any genomic locus, we define the co-abundance of nucleosomes with a combination of two marks (e.g., H3K36me3 and H3K79me3) as the fraction of cells in the population with a nucleosome with both marks at this locus. This abundance cannot be larger than the abundance of individual marks at the same locus (Figure 3A). Specifically, the co-abundance of two marks cannot exceed the abundance of each of the individual marks, leading to two constraints (Figures 3B and 3C) on the co-abundance. On the other hand, if both the individual marks are highly abundant, then the co-abundance must be above a linear constraint (Figure 3D). These constraints hold for absolute co-abundances. However, the actual read counts in each combinatorial-iChIP library depend not only on the co-abundance but also on other factors, such as antibody yield (fraction of targets retained in the IP steps) and the sequencing depth.
Figure 3.

Combinatorial-iChIP Signal Is a Quantitative Measure
(A) Schematic description of the constraints relating the abundances of individual marks at a location to the co-abundance of the dual marks at the same location. If combinatorial-iChIP signal is quantitative, then it should obey these constraints (up to a multiplicative constant).
(B–D) Comparison of the individual ChIP of H3K36me3 and H3K79me3 to their combinatorial signal. Each panel interrogates one constraint (dashed lines). Occupancy levels are as assigned by the model’s transformation of the data (Experimental Procedures).
(E) Comparison of observed combinatorial signal against the value expected based on independence between the two individual marks. See also Figure S3.
We reasoned that if the signal is quantitative, we would expect to observe these constraints in the data up to an (unknown) amplification and measurement noise. In other words, the signal in each library should have a linear relation with the true abundances. This hypothesis leads to a testable prediction: there is a multiplicative scaling coefficient that would make the combinatorial-iChIP signal obey the underlying constraints. To test this prediction, we searched for a scaling coefficient that minimizes the violations of these constraints in each (first-second) IP pair (Figures 3B–3D, Experimental Procedures). Indeed, for each pair we find a scaling coefficient (one parameter) that agrees with the constraints (less than 2.5% constraint violations). Additionally, the complementary set (>97.5% of nucleosomes) spans the range of allowed interactions (Figures 3B–3D and S3). Finding simple scaling rules for our samples indicates that the combinatorial-iChIP signal is approximately linear in the actual co-abundances. The scaled signal, for any given location in the genome, provides a quantitative estimate of the abundance of single or combinatorial states at this locus. This allows us to distinguish nucleosomes whose combinatorial-iChIP signal is higher (or lower) than the expected levels given the single ChIP signal and an independent modification model (Figure 3E).
Co-occurrence of Gene-Body-Associated Marks
We observe that, for most nucleosomes, the co-occurrence of H3K36me3 and H3K79me3 is higher than we would expect from an independent model (Figure 3E). Both marks are known to accumulate in gene bodies in a pattern anticorrelated with nucleosome turnover rates (Venkatesh et al., 2012, Weiner et al., 2015, Dion et al., 2007). H3K36me3 is deposited by Set2, which is recruited by elongating RNA Pol II (Li et al., 2003), and its presence is currently viewed as protecting gene-body nucleosomes from eviction and thus reducing nucleosome turnover rates (Venkatesh et al., 2012). H3K79me3 is deposited by Dot1 in a manner that is dependent on H2B ubiquitylation (Ng et al., 2002). Moreover, there are no known histone demethylases that erase H3K79 methylation (unlike other lysine methylations). Thus, it is currently assumed that removal of H3K79me3 is only through nucleosome eviction. The combinatorial-iChIP signal shows large co-occurrence of H3K36me3 and H3K79me3, even in nucleosomes with moderate levels of each individual mark. This observation is in agreement with the evidence that H3K36me3 slows nucleosome turnover, which will result in accumulation of H3K79me3 on nucleosomes marked with H3K36me3.
Co-occurrence of Transcription-Associated Marks
We next used this approach to gain insight into the relationship between H3K4me3 and H3K36me3 (Figure 4A). These marks are deposited by enzymes recruited by initiating (H3K4me3) and elongating (H3K36me3) forms of RNA Pol II that are differentially phosphorylated at the C-terminal domain (CTD) (Ng et al., 2002, Ng et al., 2003). Additionally, H3K36me3 has been implicated in suppressing transcription initiation within gene bodies (Carrozza et al., 2005, Venkatesh et al., 2012). These data suggest that H3K36me3 nucleosomes should be depleted of the initiation mark H3K4me3. This prediction is supported by the sparse overlap of the individual ChIP signals (Figure 4B). Examining the H3K4me3 and H3K36me3 combinatorial signal (Figures 4C and 4D), we see that a majority of the nucleosomes with noticeable signal for one of the marks do not display a combinatorial signal (12,735/23,832 and 22,165/30,721 of H3K4me3 and H3K36me3 nucleosomes, respectively). Focusing on the nucleosomes where both marks are present at the population level (Figure 4B, red outline), we observe a combinatorial signal that is proportional to the expected values from a multiplicative model that assumes independence between the marks (Figure 4E). This is in contrast to the behavior of H3K36me3 and H3K79me3 (Figure 3E). Examining the location of nucleosomes with combinatorial signal for these marks, we see that most of the combinatorial signal is in nucleosomes +3 to +5, which are in the overlap zone between the individual marks (Figures 4A and 4F). Moreover, this signal scales with expression level (Figure 4G). These results support a model where gene-body nucleosomes have a low chance to be modified at both lysines during passage of any individual RNA Pol II molecule through the nucleosomes. However, the border between the two modifications is fuzzy, either due to variation in timing of Pol II CTD modification or due to a large diameter of action of CTD-bound enzymes. Hence, the accumulation of both H3K4me3 and H3K36me3 on the same nucleosome is more likely to occur at highly expressed genes that experience repeated cycles of Pol II passages (Figure 4G).
Figure 4.

Co-occurrence of H3K4me3 and H3K36me3 Scales with Expression
(A) Polymerase-deposited marks H3K4me3 and H3K36me3 appear at gene start and body, respectively. Shown is a representative genomic region. Overlapping regions of the individual signals show combinatorial-iChIP signal (gray area).
(B) Scatter of normalized H3K4me3 and H3K36me3 individual ChIP levels on all nucleosomes. Most nucleosomes have strong signal for one or the other marks. A subpopulation of nucleosomes show co-enrichment for both marks (red box).
(C and D) Comparison of individual ChIP to combinatorial-iChIP (as in Figures 3B and 3C). Red lines denote population of nucleosomes with high levels of the individual ChIP signal and either low or high levels of combinatorial-iChIP signal.
(E) Comparison of expected co-occurrence signal by chance versus combinatorial-iChIP signal (as in Figure 3E) for the subpopulation marked in (B).
(F) Meta genes of ChIP signal of H3K4me3, H3K36me3, and their combinatorial-iChIP. “High” and “low” denote averages on genes in the 80%–100% and the 20%–40% quantiles of expression, respectively. Combinatorial-iChIP signal is highest in nucleosomes +3 to +5 (gray background).
(G) Comparison of expression levels of genes to the average combinatorial-iChIP signal on nucleosomes +3 to +5 (area marked in gray in F). Red line marks the smoothed mean (gray area, confidence interval in the mean). The location of genes shown in (A) are marked on the scatterplot.
Dissecting the Set2-RPD3S Pathway
One of the best-studied cases of chromatin regulation and crosstalk in yeast is the repression of “cryptic” transcription by the histone deacetylase Rpd3 small (RPD3S) complex (Figure S4A). RPD3S is recruited to active gene bodies by RNA Pol II and likely gets activated by binding of its Eaf3 subunit to H3K36me3 (Drouin et al., 2010, Govind et al., 2010). This, in turn, leads to hypoacetylation at gene-body nucleosomes of active genes. Thus, interference with H3K36 methylation or RPD3S activity results in hyperacetylation of these nucleosomes, which in turn increases transcription initiation from “cryptic” promoters found in gene bodies (Carrozza et al., 2005, Venkatesh et al., 2012). These previous findings predict that Eaf3 knockout strains will have increased co-occurrence of H3K36me3 and H3K18ac at gene-body nucleosomes.
We carried out a combinatorial-iChIP experiment in set2 and eaf3 deletion strains (Figure S4B). We reproduce previous reports (Carrozza et al., 2005, Venkatesh et al., 2012) showing an increase in gene-body H3K18 acetylation in cells lacking Eaf3 or Set2, with no apparent change in H3K36me3 in Eaf3 knockout cells (Figures 5A and 5B). We next tested the co-occurrence of H3K18ac and H3K36me3. As predicted, combinatorial signal of these marks increases specifically at gene bodies in Eaf3 knockout cells (Figure 5C), thus directly demonstrating this co-occurrence on individual nucleosomes.
Figure 5.

Gain of Co-occurrence of Marks as a Result of Genetic Perturbation
(A–C) Metagene profiles over long genes (ORFs of 2,000 bp or longer). Average occupancy (arbitrary units) versus location relative to TSS in different strains. Median increase in gene body signal in Eaf3 knockout (relative to the increase in 5′ signal for each gene) is reported in red. See also Figure S4.
Discussion
The experimental and analytic framework presented here represents an important step toward determining the genome-wide co-occurrence of histone marks at a single-nucleosome resolution. We demonstrate the power of combinatorial-iChIP in resolving population correlations of histone marks into a functional understanding of chromatin states. Using combinatorial-iChIP we were able to demonstrate that a certain combination of histone marks (H3K36me3 and H3K79me3) tends to co-occur while another (H3K4me3 and H3K36me3) appears to be deposited independently during transcription. These differences likely reflect underlying mechanisms that drive the co-accumulation of these histone marks. We further used combinatorial-iChIP to shed light on longstanding questions in chromatin biology. By applying combinatorial-iChIP to cells compromised on the Set2-Rpd3 pathway, we found that a mark for H3 acetylation (H3K18ac) coexists with H3K36me3, directly supporting the current theory about the working of this pathway.
Combinatorial-iChIP provides a powerful extension of the widely used ChIP-seq assays. It is not limited to histone marks and can be readily adapted for detecting the co-occurrence of transcription factors as well as other chromatin-associated molecules. Combinatorial-iChIP is amenable to simultaneous processing of many samples. It requires much less biological material compared to canonical ChIP and re-ChIP. This is mainly achieved by early barcoding of chromatin in the first IP step and pooling samples prior to the second IP step. Another advantage of early barcoding and sample pooling is that the second IP step is done in a single tube, simultaneously for all samples, which reduces handling noise and contributes to the quantitative nature of combinatorial-iChIP (Figure 3). By distinguishing co-occurring from merely correlating histone marks, combinatorial-iChIP can help to pinpoint marks that co-specify distinct chromatin states (Ruthenburg et al., 2011).
Limitations
As with ChIP-seq, combinatorial-iChIP relies on antibodies, with the known caveats of antibody specificity and sensitivity. In addition, due to differences in abundance of modifications and in antibody yield efficiencies, the read counts can vary dramatically between different antibodies. As result, much of the sequenced reads can be dominated by few samples. To avoid this, calibration of antibody concentrations and the amount of chromatin is required prior to experiments (Experimental Procedures). We noticed that the barcoding efficiency can differ between different antibodies used during the first IP step. This could be a result of steric interference and/or the binding constants of the antibody. This problem can be overcome either by barcoding more material when using “problematic” antibodies or by barcoding nucleosomes in solution (van Galen et al., 2016) prior to the first IP.
In summary, MNase combinatorial-iChIP is a robust, straightforward methodology that can be easily adjusted to robotic frameworks to probe the combinatorial nature of chromatin. We believe that combinatorial-iChIP can greatly improve our understanding of the compositional and functional complexity of chromatin.
Experimental Procedures
See Supplemental Information for detailed experimental steps.
Yeast Strains and Growth
Yeast strains were obtained from the yeast KO collection (BY4741 with KanMX cassette replacing the deleted gene) and the histone substitution and deletion library (Dai et al., 2008). As “WT” strains we used Bar1 knockout from the yeast KO collection and the H3 WT from the histone substitution and deletion library. Yeast cells were grown in YPD media at 30°C with constant shaking to OD 0.6–0.8.
Cell Growth, Fixation, and MNase Digestion
Cells were fixed with 1% formaldehyde and treated with zymolyase to prepare spheroplasts. Spheroplasts were treated with MNase to digest chromatin into nucleosomes. In all cases MNase pattern showed ∼80% mononucleosomal fragments.
Chromatin Immobilization
MNase-digested chromatin was allowed to bind to antibodies for 2 hr. Protein G dynabeads were added for an additional hour, and the beads were extensively washed.
Chromatin Barcoding and Release
Chromatin barcoding and release was performed as previously reported by Lara-Astiaso et al. (2014), with minor modifications.
Second ChIP and Next-Generation Sequencing
The barcoded chromatin was pooled and divided into fresh tubes as the number of antibodies used for the second IP step. Chromatin immobilization and washing was done essentially as for the first ChIP. Chromatin elution and library preparation were performed as reported by Lara-Astiaso et al. (2014). DNA libraries were paired-end sequenced by Illumina NextSeq 500.
Read Mapping
Pair-end reads were mapped to the yeast genome (sacCer3) using bowtie2 with maximal fragment size of 2,000 bp. We removed duplicate fragments, as they are potential PCR artifacts. We defined mononucleosome fragments as these shorter than 220 bp.
Nucleosome Coverage
We used the nucleosome location atlas defined by Weiner et al. (2015). We measured nucleosome coverage by counting the number of fragments overlapping a window of size 150 bp around the center of the nucleosome.
Model Normalization
Consider two modifications, X and Y. Let Xl and Yl denote the event that a nucleosome at location l has either mark. Thus, the abundance in the population of each mark, or the combination, is P(Xl), P(Yl), and P(Xl,Yl). From the laws of probabilities, we have three constraints:
-
(1)
P(Xl) ≥ P(Xl,Yl)
-
(2)
P(Yl) ≥ P(Xl,Yl)
-
(3)
P(Xl,Yl) ≥ P(Xl) + P(Yl) − 1
These three constraints define the boundaries of the allowed region shown in Figure 2A. We assume that the number of reads, , , and , in our libraries is related to the abundance of each of the combination. The simplest assumption is that
Similarly, such equations can be used for other marks, each with its own multiplicative scaling coefficient.
To test whether we can assign such scaling coefficients, we did the following steps:
-
(1)
We assigned ChIP and combinatorial-iChIP coverage (raw counts) per nucleosome (as described above).
-
(2)
The coverage counts were divided by nucleosome occupancy levels, as measured by Weiner et al. (2015).
-
(3)
Each single ChIP nucleosome coverage vector (value per nucleosome position) was transformed to the range [0,1] by dividing by its 99.5% quantile value. This provides the single ChIP scaling coefficient. The scaled value defines our estimate of occupancy for mark X.
-
(4)
For each combinatorial-iChIP, we performed a line search for the scaling coefficient that minimizes the sum of the deviations from the allowed region constraints (as shown in Figures 2B–2D). Formally, we define the loss of a coefficient as
where is the normalized value of co-occurrence counts for X and Y from Step 2, and S(z) = max(z, 0). The value of α that minimizes this loss is chosen for normalizing the counts for the combinatorial-iChIP of X and Y. In all cases, the fraction of nucleosome violations did not exceed 2.5%.
We repeated this procedure with different values of quantiles in Step 2. While the actual values were somewhat different, the relative conclusions, including differences from expected value (Figure 2E), were fairly robust to this choice.
Density Plots and Smoothing
All plots were generated using the ggplot2 library of R (version 3.2.3). Density scatters were generated using the geom_bin2d(), contours by geom_density2d(), and smoothed averages by geom_smooth().
Author Contributions
R.S. and N.F. conceived and designed this study. R.S., R.L.-W., and H.W. conducted the experiments with support from A.R. N.F. performed data analysis. R.S. and N.F. wrote the manuscript with contributions from all authors.
Acknowledgments
We thank D. Lara-Astiaso and I. Amit for providing guidance and support setting up the iChIP protocol. We thank A. Appleboim, C. de Boer, J. Engreitz, S. Grossman, N. Habib, S. Myers, O.J. Rando, K. Shekhar, and I. Tirosh for critical comments on the manuscript and L. Friedman and L. Gaffney for help with the graphics. This work was supported by ERC Grant 340712 and by the ISF I-Core on Chromatin and RNA in Gene Regulation.
Published: August 2, 2016
Footnotes
Supplemental Information includes four figures, one table, and a Detailed Combinatorial-iChIP Protocol and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2016.07.023.
Contributor Information
Ronen Sadeh, Email: ronensadeh@gmail.com.
Nir Friedman, Email: nir@cs.huji.ac.il.
Accession Numbers
The data in this paper are deposited at Gene Expression Omnibus (GEO) under accession number GEO: GSE84240.
Supplemental Information
For each sample, the number of reads and the number of unique reads in two size ranges, 50,220 bp and 2,201,000 bp, is listed.
References
- Baylin S.B., Jones P.A. A decade of exploring the cancer epigenome - biological and translational implications. Nat. Rev. Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Carrozza M.J., Li B., Florens L., Suganuma T., Swanson S.K., Lee K.K., Shia W.-J., Anderson S., Yates J., Washburn M.P., Workman J.L. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Chi P., Allis C.D., Wang G.G. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J., Hyland E.M., Yuan D.S., Huang H., Bader J.S., Boeke J.D. Probing nucleosome function: a highly versatile library of synthetic histone H3 and H4 mutants. Cell. 2008;134:1066–1078. doi: 10.1016/j.cell.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion M.F., Kaplan T., Kim M., Buratowski S., Friedman N., Rando O.J. Dynamics of replication-independent histone turnover in budding yeast. Science. 2007;315:1405–1408. doi: 10.1126/science.1134053. [DOI] [PubMed] [Google Scholar]
- Drouin S., Laramée L., Jacques P.-É., Forest A., Bergeron M., Robert F. DSIF and RNA polymerase II CTD phosphorylation coordinate the recruitment of Rpd3S to actively transcribed genes. PLoS Genet. 2010;6:e1001173. doi: 10.1371/journal.pgen.1001173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia B.A., Pesavento J.J., Mizzen C.A., Kelleher N.L. Pervasive combinatorial modification of histone H3 in human cells. Nat. Methods. 2007;4:487–489. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- Govind C.K., Qiu H., Ginsburg D.S., Ruan C., Hofmeyer K., Hu C., Swaminathan V., Workman J.L., Li B., Hinnebusch A.G. Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol. Cell. 2010;39:234–246. doi: 10.1016/j.molcel.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M., Amit I., Garber M., French C., Lin M.F., Feldser D., Huarte M., Zuk O., Carey B.W., Cassady J.P. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Lara-Astiaso D., Weiner A., Lorenzo-Vivas E., Zaretsky I., Jaitin D.A., David E., Keren-Shaul H., Mildner A., Winter D., Jung S. Immunogenetics. Chromatin state dynamics during blood formation. Science. 2014;345:943–949. doi: 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis P.W., Müller M.M., Koletsky M.S., Cordero F., Lin S., Banaszynski L.A., Garcia B.A., Muir T.W., Becher O.J., Allis C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Howe L., Anderson S., Yates J.R., 3rd, Workman J.L. The Set2 histone methyltransferase functions through the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem. 2003;278:8897–8903. doi: 10.1074/jbc.M212134200. [DOI] [PubMed] [Google Scholar]
- Liu C.L., Kaplan T., Kim M., Buratowski S., Schreiber S.L., Friedman N., Rando O.J. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 2005;3:e328. doi: 10.1371/journal.pbio.0030328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Jain S.U., Hoelper D., Bechet D., Molden R.C., Ran L., Murphy D., Venneti S., Hameed M., Pawel B.R. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. 2016;352:844–849. doi: 10.1126/science.aac7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I., Noh K.-M., Soshnev A.A., Allis C.D. Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet. 2014;15:259–271. doi: 10.1038/nrg3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng H.H., Xu R.-M., Zhang Y., Struhl K. Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J. Biol. Chem. 2002;277:34655–34657. doi: 10.1074/jbc.C200433200. [DOI] [PubMed] [Google Scholar]
- Ng H.H., Robert F., Young R.A., Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- Rando O.J., Winston F. Chromatin and transcription in yeast. Genetics. 2012;190:351–387. doi: 10.1534/genetics.111.132266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee H.S., Pugh B.F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011;147:1408–1419. doi: 10.1016/j.cell.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C.M., Ren B. Mapping human epigenomes. Cell. 2013;155:39–55. doi: 10.1016/j.cell.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg A.J., Li H., Milne T.A., Dewell S., McGinty R.K., Yuen M., Ueberheide B., Dou Y., Muir T.W., Patel D.J., Allis C.D. Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell. 2011;145:692–706. doi: 10.1016/j.cell.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema E., Jones D., Shoresh N., Donohue L., Ram O., Bernstein B.E. Single-molecule decoding of combinatorially modified nucleosomes. Science. 2016;352:717–721. doi: 10.1126/science.aad7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.S., Singh N., Bonocora R.P., Fitzgerald D.M., Wade J.T., Grainger D.C. Widespread suppression of intragenic transcription initiation by H-NS. Genes Dev. 2014;28:214–219. doi: 10.1101/gad.234336.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Galen P., Viny A.D., Ram O., Ryan R.J.H., Cotton M.J., Donohue L., Sievers C., Drier Y., Liau B.B., Gillespie S.M. A Multiplexed System for Quantitative Comparisons of Chromatin Landscapes. Mol. Cell. 2016;61:170–180. doi: 10.1016/j.molcel.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S., Smolle M., Li H., Gogol M.M., Saint M., Kumar S., Natarajan K., Workman J.L. Set2 methylation of histone H3 lysinec36 suppresses histone exchange on transcribed genes. Nature. 2012;489:452–455. doi: 10.1038/nature11326. [DOI] [PubMed] [Google Scholar]
- Weiner A., Hsieh T.-H.S., Appleboim A., Chen H.V., Rahat A., Amit I., Rando O.J., Friedman N. High-resolution chromatin dynamics during a yeast stress response. Mol. Cell. 2015;58:371–386. doi: 10.1016/j.molcel.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N.L., DiMaggio P.A., Plazas-Mayorca M.D., Baliban R.C., Floudas C.A., Garcia B.A. High throughput characterization of combinatorial histone codes. Mol. Cell. Proteomics. 2009;8:2266–2284. doi: 10.1074/mcp.M900238-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For each sample, the number of reads and the number of unique reads in two size ranges, 50,220 bp and 2,201,000 bp, is listed.