

Abstract
Accurate determination of elements in various kinds of samples is essential for many areas, including environmental science, medicine, as well as industry. Inductively coupled plasma mass spectrometry (ICP-MS) is a powerful tool enabling multi-elemental analysis of numerous matrices with high sensitivity and good precision. Various calibration approaches can be used to perform accurate quantitative measurements by ICP-MS. They include the use of pure standards, matrix-matched standards, or relevant certified reference materials, assuring traceability of the reported results. This review critically evaluates the advantages and limitations of different calibration approaches, which are used in quantitative analyses by ICP-MS. Examples of such analyses are provided.
This article is part of the themed issue ‘Quantitative mass spectrometry’.
Keywords: ICP-MS, quantitative measurements, calibration, isotopic dilution, standards and reference materials
1. Introduction
Analytical chemistry is an information-oriented discipline of science. It focuses on detection of various physico-chemical phenomena, and collects relevant information on the composition of the investigated materials. According to the definition given in the IUPAC Gold Book [1], quantitative analysis refers to procedures during which the amount or concentration of an analyte can be determined (estimated) and expressed as a numerical value in appropriate units. Undoubtedly, obtaining reliable and accurate results from quantitative chemical measurements is of common interest. However, it is only possible when all the relevant processes involved in the signal detection and quantification are understood and taken into account. Highest priority is always given to the so-called ‘definitive methods’ of chemical analysis. The commonly used instrumental methods require calibration using appropriate standards. Adequate calibration of instrumental methods allows for reliable quantification, even when analysing ‘difficult samples’ with complex chemical composition [2,3]. Thus, proper calibration of analytical instruments is a critical step of every instrumental analysis. Such calibration must be based on the most suitable chemical standards, which are selected specifically for the given purpose. Apart from the reliable calibration, it is also essential to perform an analytical risk assessment and to verify analytical correctness of the conversion of the recorded values into meaningful quantitative results. A good indicator of analytical correctness is the so-called uncertainty, a numerical value that should accompany every final result.
According to the definition of a ‘definite method’ [1] is ‘a method of exceptional scientific status which is sufficiently accurate to stand-alone in the determination of a given property for the certification of a reference material. Such a method must have a firm theoretical foundation so that systematic error is negligible relative to the intended use. Analyte masses (amounts) or concentrations must be measured directly in terms of the base units of measurements, or indirectly related through sound theoretical equations. Definitive methods, together with certified reference materials, are primary means for transferring accuracy, i.e. establishing traceability’. The base units of measurements are as follows: m (metre), kg (kilogram), s (second), A (ampere), K (kelvin), mol (mole), cd (candela). The definitive methods are considered as those of highest metrological status and are used for the reference measurements, e.g. for the certification of standard reference materials.
The scope of any measurement is to collect accurate data, according to the recognized context, in which the obtained results are intended to be used. Measurements of chemical properties involve assigning numbers reflecting dimensions of the measured properties, which can be compared to well-characterized standards. In principle, the measured values are only approximations of the true values. Acquiring accurate results is essential for many applications. Therefore, a significant effort should be taken to make chemical measurements as accurate as possible, according to the specified requirements.
The accuracy of analytical results can be defined by the use of the well-defined standards, which are accepted by all interested parties. Traceability is an intrinsic feature of a measurement result that allows one to evaluate the quality of that result. The commonly accepted definitions of accuracy and traceability are given by the Vocabulaire international de métrologie (VIM) 3 (clause 2.12 and 2.41, respectively): (i) ‘measurement accuracy—closeness of agreement between a measured quantity value and a true quantity value of a measurand’; (ii) ‘metrological traceability—property of a measurement result whereby the result can be related to a reference through a documented unbroken chain of calibrations, each contributing to the measurement uncertainty’ [4].
Other important terms, which are frequently used to characterize the quantitative features of the results, are ‘measurement precision—closeness of agreement between indications or measured quantity values obtained by replicate measurements on the same or similar objects under specific conditions' (VIM 3, clause 2.15), and ‘measurement uncertainty—non-negative parameter characterizing the dispersion of the quantity values being attributed to a measurand, based on the information used’ (VIM 3, clause 2.26). These terms are essential for reflecting about quantitative measurements. Referring to these terms is also useful when discussing quantification by instrumental approaches such as inductively coupled plasma mass spectrometry (ICP-MS). Chemical standards are basic reference materials they are used to test and calibrate analytical devices.
ICP-MS is a technique that enables measurements of elements and their isotopes over a very wide dynamic range. It is very useful in a large number of applications [5–8]. ICP-MS is a comparative technique: it requires a set of well-defined standards and/or reference materials for accurate calibration. A variety of reference materials can be used in chemical measurements, namely (i) pure substances (either essentially pure chemicals or well-characterized substances containing trace amounts of impurities); (ii) standard solutions and gas mixtures prepared from precursory pure substances; (iii) matrix reference materials (mimicking the chemical composition of the investigated objects). It should also be noted that the measurement methods are often standardized: the operational standards are defined by an exact procedure. Interlaboratory comparison can be additionally used to assure the accuracy of quantitative measurements [9].
The aim of this overview is to describe the principles of measurements performed by ICP-MS with special emphasis on acquiring quantitative results with high accuracy and low uncertainty. The detailed characteristics of ICP discharge and MS measurements have been extensively discussed in monographs and review papers; thus, in this article, only selected representative publications are referred to.
2. Principles of analysis by ICP-MS
ICP-MS has become a well-established technique for very sensitive, trace and ultra-trace multi-elemental analysis, as well as for determinations of isotopic ratios—for those elements that possess more than one isotope. The utility of ICP-MS was proven during the last decade. The advantages of ICP-MS include very low detection limits, speed and a large number of possible applications. Nowadays, ICP-MS can be considered as a mature technique. Thus, its newest applications are technically advanced. This also imposes the requirement for stringent quality control of results.
ICP-MS is often equipped with a liquid-phase sample introduction system (nebulizer). It can be coupled to chromatographic separation techniques. Laser ablation is a very useful tool for sampling solids prior to ICP-MS detection. The analytical features of ICP-MS are related to the efficient generation of ions in the inductively coupled plasma zone as well as the sensitivity of the ion detector [10,11]. A practical feature of ICP-MS is that the ICP discharge operates at atmospheric pressure. Thus, the technique is highly versatile, and can readily be connected to different sample introduction systems, including solution nebulization, laser ablation, electrothermal vaporization, chemical gas generation or high performance liquid chromatography (HPLC) (figure 1).
Figure 1.

Schematic overview of different systems of sample introduction to ICP-MS.
The ion intensity ICP mass spectra reflect the number of ions detected in every second (counts per second or cps), and are obtained in the course of a series of successive analytical steps [12]. These steps involve (i) conversion of a given sample (liquid or solid) into an aerosol or its direct introduction in gaseous form, (ii) transport of the aerosol/gas into plasma, (iii) atomization of the introduced compounds, followed by ionization of atoms in the hot plasma environment, (iv) transport of the generated ions from the plasma operated at atmospheric pressure to the mass separator (analyser) operating under high vacuum, involving ion extraction through ion optics, (v) separation of the ions according to their mass to charge ratios (m/z), and (vi) detection of the separated ions by detector, and conversion of ion flux intensity to an electronic signal (cps).
Considering the introduction of liquids, transport of the sample aerosol through the spray chamber can be influenced by the sample uptake rate, as well as by the density of the aliquots, e.g. the concentration of acids, or by the particle size distribution—in the case of dry aerosol produced for example by laser ablation or electrothermal vaporization [12–16]. Once the aerosol enters the hot plasma zone, some of the particles vaporize before gaseous species undergo atomization and ionization. The effectiveness of those processes depends on the nature of chemical species, as well as on the plasma conditions. It should be stressed that not all particles vaporize completely, but some of the largest particles and/or droplets are able to pass through the plasma without complete vaporization. In most of the commercially available instruments, argon gas is used. Thus, the resulting plasma consists mainly of argon atoms accompanied by numerous argon ions, electrons and species originating from the sample. Once formed in the hot plasma, all those species (particles, vapours, atoms and ions) disperse along as they travel through the plasma zone. The ions formed in plasma are transported into the mass analyser, separated according to their mass-to-charge ratios (m/z). Eventually, the ions with the appropriate mass-to-charge-ratio reach the detector, where they are counted [17–19]. Elemental mass spectrometers are most typically cover the mass range from 6 to 254 a.m.u. Hence, they enable detection of isotope ions of the majority of elements from the periodic table.
Intensities of electronic signals are related to the numbers of ions formed in the plasma and transported through the ICP-MS ion optics. The efficiency of ion formation is influenced by the ionization energy of particular elements, as well as by the plasma temperature. By using the principle of Saha–Eggert equation [20], the degree of ionization for a chemical element depends on the plasma temperature. In general, ICP is considered to be an efficient ion source. Assuming that the plasma temperature is ca 6000 K and electron density is 1015 cm−3, one can estimate the ionization efficiency to vary between 0.99 for sodium to 10−6 for chlorine. Certainly, the efficiency of ionization of given element influences the (signal to noise) S/N ratio. It is worth noting that signal intensities, and thus sensitivities, depend not only on the ionization efficiency but also on the sample introduction rate, efficiency of sample transport/delivery, and—last but not least—on the extraction and transmission efficiency of the spectrometer's ion channel. The matrix effects, both of chemical and physical origin, can occur at each step of any of the above-described processes. These effects are influenced by sample composition, occurrence of isobaric interferences, as well as spectral interferences. Such interferences may result from double-charged or cluster ions. The former only slightly but the latter strongly depend on the plasma parameters [19,21–23].
3. Quantification by ICP-MS
Most instrumental analytical techniques use direct comparison of the signals generated by a sample of unknown composition with the signals generated by a (series of) standard(s) of exactly known isotopic composition with associated uncertainties that are much smaller than the expected uncertainty of the results. Thus, an instrument has to be thoroughly calibrated before performing any quantitative analysis.
Calibration approaches, which can be applied, include
— external calibration, with or without internal standard (IS);
— standard addition;
— isotope dilution (ID).
Most state-of-the-art instruments are computerized. Therefore, mathematical processing of the data into analytically relevant terms, such as concentration of the analyte, is an integral part of the analytical system.
4. Data collection in ICP-MS
ICP-MS enables performing measurements with various quantification strategies. Depending on the general concept of data collection, analyses can be semi-quantitative or quantitative. The first step should always be the routine maintenance and daily tuning, which is obviously needed for the proper work and calibration of the spectrometer. The mass scale should normally be calibrated across the entire accessible mass range. For some applications, when the analytical goal is focused on the determination of a limited number of elements within a narrow mass range, calibration of the corresponding mass range may be sufficient.
Peak hopping mode or scanning mode are commonly used for data acquisition. Peak hopping mode enables collection of data for pre-set masses of selected isotopes of interest. In most of the instrument set-ups, a dwell time allocated to each isotope can vary according to the isotope abundance and concentration of the element in the analysed sample. Varying dwell time allows one to improve the counting statistics. An important advantage of the peak hopping mode is that time is not spent on collecting data for those isotopes that are not of interest. Of course, this feature can also be regarded as a limitation because further on no records would be available for quantification of other elements if the corresponding signals are not recorded. It is also possible to collect data for a large number of points (scanning mode) to construct the full spectrum for the pre-set mass range. In this mode, the data are collected not only for the defined isotopes but also over a wider mass range, mainly for archival purposes. Moreover, the scanning mode facilitates identification of possible interfering signals.
5. Semi-quantitative analysis by ICP-MS
All commercially available ICP-MS instruments enable quick semi-quantitative multi-elemental analysis following a single analysis of a sample, using a blank and standards. Standard solutions can contain species of one element or can be mixtures of species of several elements. The elements in the standard solutions can be the same elements as those to be quantified. Alternatively, the standard solution can include selected elements across the mass range involved in the measurement. Default algorithms are then used to compute approximate concentrations, with automatic correction for isobaric and some of the polyatomic interferences. This method of analysis is commonly used for quick fingerprinting of the elemental composition, if the target elements have more than one isotope [24]. Then, an assumption is made that relative signal intensities correspond to the relative natural abundances of the isotopes of the elements of interest—in the absence of spectroscopic interferences. If all signals are monitored for the elements present in the concentration above their detection limits, such a semi-quantitative analysis can provide information on potential sources of spectroscopic interferences from the matrix. The approximate concentrations obtained in such preliminary measurements—for example, for matrix components—facilitate subsequent quantitative analysis. An example of the use of such an approach in the semi-quantitative analysis of gaseous samples was given by Gerdes & Carter [25]. The instrument response to the multi-elements standard solution was used to establish empirical factors relating the detector response for those elements introduced as liquid or gaseous sample. These empirical factors were then used to obtain semi-quantitative results for gaseous samples.
A semi-quantitative analysis while scanning the entire m/z range allows the analyst to predict interferences, and to select appropriate ISs. It is worth noting that semi-quantitative data can also be used to estimate the content of the elements to be quantified. This helps to select the concentrations of the standard solutions to be prepared for the following quantitative analyses [26].
6. Calibration of ICP-MS for quantitative analysis
Various calibration approaches can be implemented to determine accurate concentrations of analytes in samples. They include external calibration with pure standard solution or matrix-matched standards; internal calibration; as well as the use of isotopic dilution. Calibration standards are required for constructing multi-point standard curves, covering the anticipated range of analyte concentrations in samples. The role of a standard is to determine the mathematical relationship between the selected signal intensities and the analyte concentration within the concentration range involved in the analysis. Certified reference materials (CRMs) can also be used to direct quantitative analysis. The available CRMs include certified solutions, alloys and even pure metals. CRMs are primarily used to verify the accuracy of the results obtained by conducting the entire analytical procedure, and to demonstrate traceability of the results to the fundamental units pertaining to the International System of Units (SI). A general overview of most commonly used quantifications strategies in ICP-MS is described in table 1, summarizing their advantages and limitations.
Table 1.
Features of the main quantification approaches used in ICP-MS.
| quantification mode | advantages | limitations |
|---|---|---|
| semi-quantitative |
|
|
| external calibration |
|
|
| external calibration with internal standard (IS) |
|
|
| isotopic ratio measurements |
|
|
| isotopic dilution |
|
7. External calibration in ICP-MS
The external calibration method relies on preparation of synthetic samples containing known quantities of the element to be measured. These synthetic samples are then introduced to the mass spectrometer, and the ion signal intensities are recorded. The signal intensities can be linked with the known concentrations of the elemental standards. The obtained signal-concentration dependences are basic for further quantification. External calibration is the most popular calibration approach. It is widely used for analysis of samples for which the assumption can be made on the negligible effects of the matrix on the instrumental response. However, one should be aware that in the case of severe interferences, the matrix could significantly influence the slope of the calibration, thus hampering the accuracy of the results (figure 2). This calibration approach demands the use of a series of standard solutions (often prepared in 2% HNO3) and the calibration blank. The calibration blank consists of the same concentration(s) of the same acid(s) as those used to prepare the final dilution of the calibration standards. The calibration blank solution must also contain the same ISs as those added to the calibration standard solutions. The equation of the line of best fit, obtained through linear regression (signal versus concentration of the standards) is then used to convert analyte signal intensities, recorded for the samples, to concentration values. Blank subtraction is conducted using the spectral data recorded for the blank solution. The detection limit for the particular element depends mostly on the noise, both the white noise (i.e. fundamental or Gaussian noise) and the shot noise (due to the detector used) [27,28].
Figure 2.
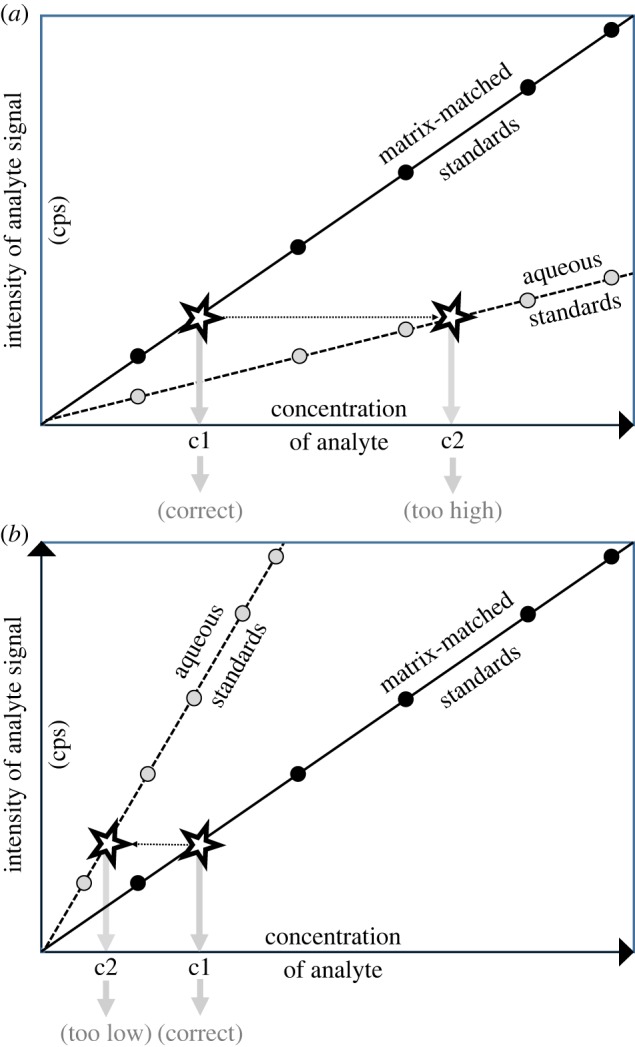
Schematic representation of the use of external calibration when matrix matching is required. The matrix effect is shown for: (a) increase of the signal (due to e.g. spectral interferences); (b) decrease of the signal (due to e.g. reduced recovery during sample pre-treatment).
The linear dynamic range of the calibration depends on the instrument performance and is typically based on the response of the instrument (signal intensity in cps units) versus concentration, covering the linear range over six to eight orders of magnitude. When the ion detector operates in the pulse-counting mode with the dead-time correction, this range is restricted to about six orders of magnitude. The range is greater when the detector is switched to the analogue mode. In the analogue mode, signal range in the calibration plot is one to two orders of magnitude wider than in the pulse mode. The calibration line is usually drawn by applying the least-squares regression procedure, to relate signal intensities (cps value) with concentrations of the analyte standard solutions. The correlation coefficient is used to evaluate quality of the calibration characteristics.
Another issue related to the calibration of ICP-MS instruments is the measurement of standard and procedural blanks. The standard blank refers to the sample prepared in the same way as a regular standard sample, which does not contain the compound of interest. However, it contains all the reagents that are used during the preparation of standard solutions. This blank could be used to correct the results for contamination arising from both chemicals and instrument, as well as to establish the ‘zero point’ for the calibration plot. The procedural blank refers to the standard blank sample subjected to all preparatory steps, thus reflects not only the contribution from all reagents used but also that of all preparatory steps.
The instrumental calibration of ICP-MS requires the use of standard chemical compounds with defined purity accompanying its uncertainty. In the case of use of liquid samples, the standard solutions may be prepared by dissolving pure substances in an acid-containing matrix. Direct analysis of solids, by means of electrothermal vaporization or laser ablation, requires careful selection of matrix-matched solid standards. Pure aqueous standards are typically used for the calibration of an instrument, but they do not provide adequate matching with respect to the viscosity and the matrix composition of the samples. Variation in these physical properties could be corrected for by using matrix-matched standards. It should be pointed out that the use of standard reference matrix-matched materials is not recommended for primary calibration. However, such materials are certainly recommended to be used as quality control samples to asses both accuracy and precision.
8. External calibration with internal standard
Internal calibration is widely used in ICP-MS analyses mostly to compensate for physical interferences, to correct for variations in the instrument response as the analysis proceeds (signal drift), and to calculate the analyte concentrations of the samples. ISs are the selected isotope of the element added to the blank, standards and samples in known concentrations. The concentration of a given IS should be the same in all the solutions involved in the analytical process (i.e. blank, standards and samples) to enable effective correction of matrix effects and monitoring, to enable correction of short and long-term fluctuations of signals.
The element which could be selected as IS should fulfil several requirements: it should not exhibit isobaric interferences with the analyte(s); the content of the element considered to serve as IS in the samples and standards should be negligible; the primary ionization potential of the element used as IS should be as close as possible to that of the analyte. When analysing a group of elements with a wide range of masses, several ISs should be used with a similarly wide range of masses (e.g. Be, In and Bi). The commonly used ISs are 6Li, 45Sc, 89Y, 103Rh, 115In, 159Tb, 165Ho and 209Bi. The study by Vanhaecke et al. [29] provides an example of the use of an IS with the mass number close to that of the analyte, what improves analytical precision.
9. Standard addition
In this method, calibration is performed by adding (spiking) each of the sub-samples with increasing amounts of a solution containing the element(s) of interest. In this case, quantification is carried out based on the analysis of the original sample and a number of spiked samples, all of them having the same matrix. The intercept of the calibration line on the ‘concentration’ axis gives the concentration of the element of interest in the unspiked sample. Although this approach can offer good accuracy of data, it is usually too time-consuming to be performed in most laboratories.
10. Isotope dilution
Mass spectrometry, especially high-resolution mass spectrometry, allows for monitoring several isotopes of elements of interest with good precision. It is also possible to calculate the isotope ratio of the selected pairs of isotopes. Those values can be used as such, e.g. for the monitoring of isotopic profiles of the elements [30]. The most important use of the isotope ratio measurements is the isotope dilution (ID) mode of quantification. Previously, ID was used in conjunction with thermal ionization. Later on, it was also implemented with ICP-MS. ID provides advantages over traditional calibration because the calculated isotope ratios, and hence the analytical results, obtained by means of ID-ICP-MS do not depend on instrumental instabilities and possible matrix effects [31–33]. Thus, highly accurate measurements can be performed, and the ID-MS technique may be regarded as one of definitive analytical methods. In this mode of analysis, known amounts of selected isotopically enriched standards are added to each sample, most preferably before sample preparation steps, in order to account for any possible losses of analytes during the entire procedure. As isotopically enriched compound exhibits the same chemical properties as a compound present in the original sample, both will proceed in the same way. This was exemplified by the determination of Cu in two groundwater candidate reference materials (BCR CRM 609 and BCR CRM 610) [34].
The detailed comparison of the external and ID calibration was described for the multi-elemental analysis of coal slurries [35]. For this purpose, the standard containing isotopes 201Hg, 206Pb, 77Se and 119Sn was added to the slurry to achieve an altered isotopic ratio close to 1. The proposed approach was validated by the analysis of four certified coal reference materials. Similar methodology was applied for the determination of Cd, Hg, Pb and Se in sediments reference materials by adding the standard of enriched isotopes (198Hg, 206Pb, 111Cd and 77Sn) to the sediment slurry followed by slurry sampling chemical vapour generation (CVG) with ID calibration and detection by ICP-MS [36]. The multi-elemental analysis of soil prepared using HNO3 leaching or pseudo-total digestion with HNO3, HCl and HF in a microwave oven was performed. In this case, two calibration approaches were evaluated: external calibration, combined with internal standardization; and ID after appropriate spiking of the soils with stable isotopes mixture prior to sample preparation. Although both methods of quantification yield results of essentially equivalent accuracy and precision, external calibration allows quantifying a greater number of elements [37].
The accuracy of ICP-ID-MS was demonstrated recently by examples of its use for the certification of the content of mercury and methyl mercury (MeHg) in several environmental candidates for CRMs [38,39]. For both analytes, mercury and methyl mercury, ID-MS was applied as a primary method of measurement, assuring the traceability of obtained values to the SI units: the mole, the kilogram and the second. The determination of total mercury in environmental samples (the candidates for CRMs) was then performed with the use of cold vapour atomic absorption spectrometry (CV-AAS), ICP-MS (with external calibration or standard addition method), and finally validated by ID-MS as a primary method of measurement. The ERM-AE640 isotopically enriched standard (95.7% abundance for 202Hg) was used for this purpose [38]. The certification of the mass fraction of the total Hg and MeHg, in marine biota samples, candidates for CRMs (oyster and clam Gafrarium tumidum) was also performed with the use of reference measurements. Two modes of ID-ICP-MS, namely direct ID and species-specific ID analysis were compared. The mass fraction values of total Hg and MeHg, determined in this study, were then used by the International Atomic Energy Agency (IAEA) Environment Laboratories for characterization of the IAEA 461 and IAEA 470 certified reference materials [39].
Although the ID-MS has proved to be unvanquishable in many applications, unfortunately many elements are monoisotopic (9Be, 23Na, 27Al, 45Sc, 55Mn, 75As, 89Y, 103Rh, 127I, 133Cs, 141Pr, 159Tb, 165Ho, 169Tm, 197Au, and 232Th), which makes ID-ICP-MS useless in analysis of these elements.
11. Quantitative measurements by ICP-MS with different sample introduction modes
Analysis of elemental composition by ICP-MS can be performed during continuous delivery of a liquid sample to the plasma zone via nebulization, so a steady signal is obtained. Calibration of the MS detector can be performed with a standard solution of given element(s) with the addition of properly selected IS(s). Analysis can also be performed when the solution is delivered via liquid chromatography and a transient signal is obtained with a unique concentration profile reflecting the performance of chromatographic separation. Then, calibration can be performed either by using mono-compound standard solutions, or by using a set of the standard solutions containing the analysed species, which elute from the column at different times. The analysis can also be also performed using laser ablation as the microsampling method, and with transient signal registration and laser parameters selected depending on the type of solid [40]. These measurements are usually influenced by the fractionation effects [41]. The calibration can be done either by using the same solid matrix as that of the sample (e.g. glass, alloys), by using home-made solid standards that mimic, to the best extent, the structure and the composition of the analysed solids, or by preparing pellets from CRM powders.
12. Introduction of liquid samples
ICP-MS is well suited for analysis of liquid samples introduced into the plasma via nebulization. Pure chemical solutions are used in calibration [42], and to verify the analytical performance of the method, e.g. its detection limit, the linear range of concentration, as well as precision. In order to evaluate the possible interferences, matrix-matched standards should be applied. A well-established method to assure high accuracy and traceability of the results is the use of CRMs. CRMs are often available as solids containing the relevant matrix, e.g. of environmental origin. When the solids undergo digestion, it is expected that the element of interest will be present in the form of one compound. Thus, the calibration solution should also contain known concentrations of this compound [43].
The most commonly used practice in a routine laboratory is to perform instrumental calibration with the use of single compound standard solutions for a given element. With this approach, one can perform analysis of the aliquots derived from the original sample solutions (e.g. from drinking water), or after dilution (when analysing liquids containing high content of dissolved solids; e.g. seawater, waste water, extracts or digestion solution). Alternatively, the effluent of the liquid chromatography column can be directed to the plasma zone. It should be emphasized that in most cases, besides the chemical environment, the elements of interest can be present in the form of various chemical species. The sensitivities of the response of ICP-MS may be different for every species. Thus, it is often necessary to digest the sample (liquid or solid) in order to bring the element of interest to the same chemical form.
Appropriately selected CRMs were successfully used in the research on biologically relevant processes taking place in plants tissues. They were focused on determination of Se [44], Cd [15,45], Zn, Fe and Cd [46,47]. Moreover, species of noble metals such as Pt and Pd were analysed in environmental samples [48].
External calibration with the use of ISs has been used in various applications. An interesting example is elemental analysis of petroleum-related samples, e.g. determination of Cu in crude oil distillation products, in which case sample preparation by digestion of organic matter is required [49], or direct determination of Fe and Ca in alumina-based based nickel-molybdenum hydrodesulfurization catalysts [50].
13. Species-specific calibration: single versus multiple species calibration
A main advantage of ICP-MS is that it can be coupled with the chromatographic separations; for example, with HPLC, which is widely used in studies of chemical speciation. In most cases, coupling HPLC to ICP-MS is technically simple. The exit of the chromatographic column is directly connected to a nebulizer. In the course of chromatographic separation, the species are eluted consecutively, with the order determined by their retention in the column, reaching the plasma zone and the MS system. This means that, in every time interval, different compounds of the element of interest undergo atomization, ionization and are transported to the mass analyser zone. When using gradient elution, precautions need to be made to reduce the biases caused by an unequal response of the detector at the beginning and the end of the separation.
Coupling chromatography to ICP-MS has been used in the speciation analysis of selenium. In all cases, the MS system responded to different chemical forms of the element of interest [51–53]. As shown in figure 3, four standard solutions of selenium were introduced into the ICP-MS via HPLC. Interestingly, the slope of the calibration depends on the type of selenium species. The influence of species type is apparent not only in the case of organic compounds but also for both inorganic compounds in which the metal appears at various oxidation states. Therefore, in order to obtain accurate results, it is essential to perform separate calibration with respective mono-compound solutions of an element of interest (here, selenium).
Figure 3.
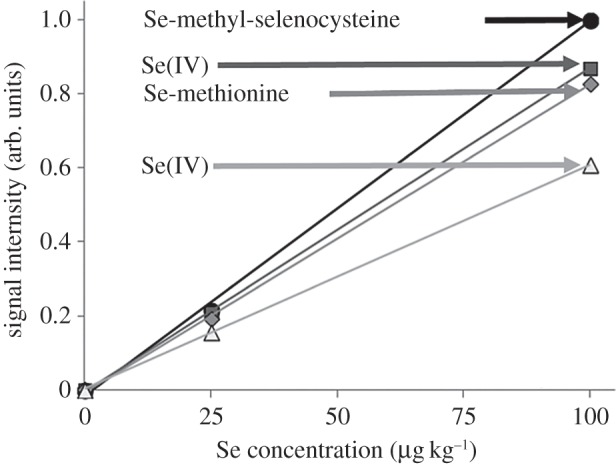
Calibration lines for a selected standard of selenium. Analysed inorganic species: Se(IV) nitrate and Se(VI) nitrate.
14. Sampling analytes from solid matrices
Laser ablation (LA) systems can also be coupled to ICP-MS allowing for micro sampling of solid materials. This way of sample introduction to ICP-MS has been constantly gaining popularity within the last few years because it requires almost no sample preparation and permits direct analysis of solid samples [40,41]. Minor depletion of the sample material by the ablation processes is the only side-effect, which has to be taken into account while using LA-ICP-MS. However, it cannot be regarded as a serious limitation, and it is acceptable in many applications.
An important feature of LA-ICP-MS is the need to refer to appropriate matrix-matched CRMs. Probably owing to this feature, the literature reports many custom-developed matrix-matched calibration approaches, which should fit to various applications of LA-ICP-MS in quantitative elemental analysis [54–58]. The most often reported calibration approach is based on the use of Standard Reference Material (SRM) NIST 610 glass, which is available from the National Institute of Standards and Technology (NIST). It nominally contains ca 500 µg g−1 of 61 elements present in the silicate matrix (Si-Na-Ca-Al) [58,59]. Interestingly, this homogeneous SRM glass material is commonly used as the external standard, even if no matrix matching is assured [60]. Successful applications of NIST 610 as the external standard for multi-elemental analysis of other glass samples are reported widely in the literature devoted to investigations of historic objects [61–65]. It should be stressed that the composition of historic glasses usually differs to some extent from the major elemental composition of NIST 610. Thus, the quality of the results can be then assessed with Corning Archeological Reference Glasses: A, B, C and D, which have been produced by the Corning Museum of Glass (USA), and are used during the evaluation of the quantitative data obtained with NIST 610. The Corning Glasses reflect three different technologies of historic glass production and are usually used while investigating archaeological beads, as well as other historic glass samples [66–68]. Estimation of the precision and accuracy of measurements can be done according to the chemometric protocols [68].
The advantages of laser ablation are fully appreciated during analysis of environmental samples, which are regarded as difficult, and are often delivered to laboratories as heterogeneous powders [69,70]. They can be analysed either after digestion or in the solid state. A digestion can be difficult and time-consuming. Moreover, it may increase the risk of loss of volatile elements, or can even contribute to contamination. LA-ICP-MS can help to avoid the above-mentioned problems. However, it requires immobilization of powdered samples, and selection of a reliable calibration approach [70]. Various calibration strategies for LA-ICP-MS have been proposed in the literature. Pakieła et al. [70] proposed a multi-point calibration for the LA-ICP-MS analysis based on CRMs, available in the form of powders e.g. soils, sediments or ashes. Powdered samples were prepared by mixing the original material with zinc oxide, and adding 2-methoxy-4-(2-propenyl)phenol in order to form zinc complex. Stable, homogeneous and mechanically resistant targets, containing increasing concentrations of selected elements (Al, Ba, Co, Cr, Fe, Mg, Mn, Pb, Sb, U and V), were subjected to laser ablation to enable calibration of multi-elemental ICP-MS measurements. The calculated linear correlation coefficients between the mass of the CRM and the intensity of the signal exceeded the value of 0.99. This calibration approach was evaluated as fit-for-purpose for investigations of environmental powders. Then the validation of the proposed approach was done by analysing soil and sediment certified reference materials (river sediment, RM 8704 and bone ash, RM 1400) [70].
A numerous creative approaches to calibrate LA-ICP-MS for quantitative analysis exist. Some of them enable improving the quality of the final results in different ways. Analysis of environmental samples containing natural carbonate compounds can be mentioned here as a good example [71,72]. Craig et al. [72] compared calibration using a standard glass material, calcium carbonate powders spiked with liquid standards of selected elements and natural geological reference materials, and the last approach was preferred over the first two approaches. Bellotto & Miekeley [71] proposed co-precipitation of multi-element liquid standards into a CaCO3 matrix. All approaches are regarded as beneficial for precise and accurate LA-ICP-MS quantification of the final results versus matrix-matched standards. In some cases, the accurate calibration of LA-ICP-MS was possible using liquids without assessment of the similarity between calibration solutions and analysed samples [73]. The proposed method reduced the limitations of the currently available reference materials, and offered a possibility to quantify analytes in solids with variable chemical composition.
In other work, a method for quantitative analysis of trace elements in biological samples by LA-ICP-ID-MS has been developed. The accuracy of this approach was verified while analysing several elements (Cr, Fe, Cu, Zn, Sr, Cd and Pb) in selected CRMs [74]. Another sound example was provided by Thieleke & Vogt [75]. In their work, 204Pb-enriched lead was used for performing ID-MS calibration, while bismuth served as IS. In order to assure high accuracy of the results, a standard sample containing lead as well as a lead-containing polymer CRM (BAM H010) has been used. The mass fractions for lead, calculated after single and double ID experiments in the test samples, showed a deviation of less than 1% (relative to the certified values) [75].
15. Validation of qualitative results with the use of various measurement techniques
Mercury can be analysed by various techniques, thus direct comparison of the accuracies can be made [30,76]. The comparison of the accuracy of measurements for multi-elemental analysis of peat cores with ICP-MS and X-ray fluorescence (XRF) is a very interesting example. In this case, cross-calibration was used to assure high quality of the obtained results. A digestion procedure was used to prepare samples for ICP-MS analyses [77].
Yet other interesting approach is to use chemometric modelling to develop an accurate one-step method for quantitative analysis, where the results obtained by electrochemical analysis were used to validate independent quantitative ICP-MS data, without the need for standard additions [78].
In other work, analytical performance of ICP-MS for determination of lanthanides in biological materials was investigated and compared with the performances of neutron activation analysis (NAA), as well as ion chromatography (IC) with UV-VIS detection [79]. It should be nevertheless emphasized that the biggest advantage of ICP-MS is that it provides very low limits of detection. Two sample preparation protocols were tested: (i) microwave-assisted digestion by concentrated nitric acid; (ii) microwave digestion involving silica and fluoride removal, followed by selective and quantitative lanthanide group separation from the plant matrix. Several CRMs of plant origin were used for the evaluation of the accuracy of the applied analytical procedures. Thus, by comparing the data obtained with ICP-MS with those obtained by NAA reference measurements, the provisional (named as ‘tentative’) values can be obtained [79].
LA-ICP-MS was also compared with LA-ICP-ID-MS during determination of Cd in coral skeletons. Following careful optimization of the instrumental parameters, and subsequent analyses, the results obtained by LA-ICP-MS and ID-ICP-MS were found to be highly correlated [80].
16. Conclusion
Accurate determination of elements in a variety of matrices has become essential for many areas, including environmental science, medicine and industry. In order to achieve high quality chemical results, it is essential to consider all the important measures related to the accuracy and traceability of the results. It is necessary to choose an adequate calibration method, and to use individually selected chemical standard or reference materials. In the above, we aimed to review the possible approaches. The main approaches are summarized in table 1.
Thus, it may be concluded that semi-quantitative measurements are used in the preliminary screening of the analysed samples, and to learn about possible spectral interferences. When no matrix effects are expected, quantitative measurements are often performed with the use of external calibration, using standards of pure substances (as solid or in the solution). When matrix effects are expected, it is recommended to implement matrix-matched standards. In addition, it can be used to compensate for instrumental fluctuations and possible analyte losses during sample pre-treatment. Analytical robustness with respect to the above-mentioned effects can be avoided by determining ratios of the selected isotopes of the element of interest. Finally, the use of isotopic dilution, followed by ICP-MS analysis, is highly beneficial for the measurements with the highest possible accuracy and very low uncertainty. It should be pointed out that the main advantage of ID-ICP-MS is that as soon as isotopic equilibrium is established, any losses of the analyte do not affect the analytical results. This is especially important for the investigations of the elements that have been isolated by means of chromatographic separation, extraction, co-precipitation, electrolytic deposition or hydride generation prior to their determination by ICP-MS. In this case, ‘non-quantitative’ variations of recoveries are automatically corrected for. Although various quantification approaches are available, ICP-ID-MS can be regarded as accurate calibration procedure among all of them. However, it is not applicable to analysis of mono-isotope elements.
Authors' contributions
Both authors, E.B. and B.W., have equally contributed to this article, which involved the following tasks: (i) substantial contributions to conception and design of this review; (ii) drafting the article and contributions to the important intellectual content; and (iii) final approval of the version to be published.
Competing interests
We declare we have no competing interests.
Funding
The authors acknowledge the financial support from the National Science Centre (NCN), Poland (project number 2012/05/B/ST4/01219). This study was carried out in the Biological and Chemical Research Centre, University of Warsaw, established within the project co-financed by the European Union—European Regional Development Fund under the Operational Programme Innovative Economy (2007–2013).
References
- 1.IUPAC Compendium of Chemical Terminology. 1997. IUPAC Gold Book, 2nd edn (eds AD McNaught, A Wilkinson) Oxford, UK: Blackwell Scientific Publications. See; XML corrected version http://goldbook.iupac.org (2006) created by Nic M, Jirat J, Kosata B, updates compiled by Jenkins A (accessed July 2016). [Google Scholar]
- 2.Danzer K, Currie LA. 1998. Guidelines for calibration. Part I. Fundamentals and single component calibration (IUPAC Recommendations 1998). Anal. Chem. Pure Appl. Chem. 70, 993–1014. ( 10.1351/pac199870040993) [DOI] [Google Scholar]
- 3.Matschat R, Czerwensky M, Pattberg S, Heinrich H-J, Tutschku S. 2002. High purity metals as primary calibration materials for elemental analysis—their importance and their certification. Mat. Trans. 43, 90–97. ( 10.2320/matertrans.43.90) [DOI] [Google Scholar]
- 4.ISO/IEC Guide 99. 2007. International vocabulary of metrology—basic and general concepts and associated terms (VIM). See http://www.iso.org/iso/catalogue_detail?csnumber=45324 (accessed July 2016).
- 5.Beauchemin D. 2010. Environmental analysis by inductively coupled plasma mass spectrometry. Mass Spectrom. Rev. 29, 560–592. ( 10.1002/mas.20257) [DOI] [PubMed] [Google Scholar]
- 6.Krachler M, Alvarez-Sarandes R, Van Winckel S. 2015. Challenges in the quality assurance of elemental and isotopic analyses in the nuclear domain benefitting from high resolution ICP-OES and sector field ICP-MS. J. Radioanal. Nucl. Chem. 304, 1201–1209. ( 10.1007/s10967-015-3952-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hann S, Dernovics M, Koellensperger G. 2015. Elemental analysis in biotechnology. Curr. Opin. Biotech. 31, 93–100. ( 10.1016/j.copbio.2014.08.008) [DOI] [PubMed] [Google Scholar]
- 8.Pröfrock D, Prange A. 2012. Inductively coupled plasma—mass spectrometry for quantitative analysis in environmental and life science: a review of challenges, solutions, and trends. Appl. Spectr. 66, 843–868. ( 10.1366/12-06681) [DOI] [PubMed] [Google Scholar]
- 9.Foster GL, et al. 2013. Interlaboratory comparison of boron isotope analysis of boric acid, seawater and marine CaCO3 by MC-ICPMS and NTIMS. Chem. Geol. 358, 1–14. ( 10.1016/j.chemgeo.2013.08.027) [DOI] [Google Scholar]
- 10.Holland G, Tanner SD. 1999. Plasma source mass spectrometry, new developments and applications. London, UK: The Royal Society of Chemistry. [Google Scholar]
- 11.Broekaert JAC. 2005. Analytical atomic spectrometry with flames and plasmas. New York, NY: Wiley. [Google Scholar]
- 12.Gray AL. 1985. The ICP as an ion source—origins, achievements and prospects. Spectrochim. Acta B. 40, 1525–1537. ( 10.1016/0584-8547(85)80176-2) [DOI] [Google Scholar]
- 13.Kántor T, Maestre S, de Loos-Vollebregt MTC. 2005. Studies on transport phenomena in electrothermal vaporization sample introduction applied to inductively coupled plasma for optical emission and mass spectrometry. Spectrochim. Acta B. 60, 1323–1333. ( 10.1016/j.sab.2005.06.011) [DOI] [Google Scholar]
- 14.Paredes E, Bosque J, Mermet JM, Todoli JL. 2010. Influence of the nebulizer design and aerosol impact bead on the analytical sebsitivities of inductively coupled plasma mass spectrometry. Spectrochim. Acta B. 65 908–917. ( 10.1016/j.sab.2010.08.006) [DOI] [Google Scholar]
- 15.Xu JQ, Balik D, Agnes GR. 2001. Aerosol static electrification and its effects in inductively coupled plasma spectrometry. J. Anal. At. Spectrom. 16, 715–723. ( 10.1039/B009014J) [DOI] [Google Scholar]
- 16.Fraser MM, Beauchemin D. 2009. Evidence supporting the occurrence of coulomb fission during conventional sample introduction in inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 24, 469–475. ( 10.1039/B819816K) [DOI] [Google Scholar]
- 17.Faser MM, Beauchemin D. 2000. Effect of concomitant elements on the distribution of ions in inductively coupled plasma mass spectrometry. Spectrochim. Acta B. 55, 1705–1731. ( 10.1016/S0584-8547(00)00273-1) [DOI] [Google Scholar]
- 18.Niu H, Houk RS. 1996. Fundamental aspects of ion extraction in inductively coupled plasma mass spectrometry. Spectrochim. Acta B. 51, 779–815. ( 10.1016/0584-8547(96)01506-6) [DOI] [Google Scholar]
- 19.Tanner SD. 1995. Characterisation of ionization and matrix suppression in inductively coupled ‘cold’ plasma mass spectrometry. J. Anal. At. Spectrom. 10, 905–921. ( 10.1039/JA9951000905) [DOI] [Google Scholar]
- 20.Blades MW. 1988. Fundamental reference data and the inductively coupled plasma. Spectrochim. Acta B. 43, 35–43. ( 10.1016/0584-8547(88)80035-1) [DOI] [Google Scholar]
- 21.Garcia-Poyo MC, Grindlay G, Gras L, de Loos-Vollebregt MTC, Mora J. 2015. Non-spectral interferences due to the presence of sulfuric acid in Inductively coupled mass spectrometry. Spectrochim. Acta B. 105, 71–76. ( 10.1016/j.sab.2014.11.003) [DOI] [Google Scholar]
- 22.Agatemor C, Beauchemin D. 2011. Towards the reduction of matrix effects in inductively coupled plasma mass spectrometry without compromising detection limits. Spectrochim. Acta B. 66, 1–11. ( 10.1016/j.sab.2010.11.011) [DOI] [PubMed] [Google Scholar]
- 23.Agatemor C, Beauchemin D. 2011. Matrix effects in inductively coupled plasma mass spectrometry: a review. Anal. Chim. Acta. 706, 66–830. ( 10.1016/j.aca.2011.08.027) [DOI] [PubMed] [Google Scholar]
- 24.Krzciuk K. 2016. Intelligent analysis of samples by semiquantitative inductively coupled plasma—mass spectrometry technique; a review. Crit. Rev. Anal. Chem. 46, 284–290. ( 10.1080/10408347.2015.1053106) [DOI] [PubMed] [Google Scholar]
- 25.Gerdes K, Carter KE. 2011. Calibration strategy for semi-quantitative direct gas analysing using inductively coupled plasma mass spectrometry. Spectrochim. Acta B. 66 712–725. ( 10.1016/j.sab.2011.09.007) [DOI] [Google Scholar]
- 26.Chen H, Dąbek-Złotorzyńska E, Rasmussen PE, Hassan N, Lanouette M. 2008. Evaluation of semiquantitative analysis mode in ICP-MS. Talanta 74, 1547–1555. ( 10.1016/j.talanta.2007.09.037) [DOI] [PubMed] [Google Scholar]
- 27.Sesi NN, Borer MW, Starn TK, Hieftje GM. 1998. A standard approach to collecting and calculation noise amplitude spectra. J. Chem. Ed. 75, 788–792. ( 10.1021/ed075p788) [DOI] [Google Scholar]
- 28.Mermet JM, Granier G, Fichet P. 2012. A logical way through the limit of quantitation in inductively coupled plasma mass spectrometry. Spectrochim. Acta B. 76 221–225. ( 10.1016/j.sab.2012.06.002) [DOI] [Google Scholar]
- 29.Vanhaecke F, Vanhoe H, Dams R, Vandecasteele C. 1992. The use of internal standards in ICP-MS. Talanta 39, 737–742. ( 10.1016/0039-9140(92)80088-U) [DOI] [PubMed] [Google Scholar]
- 30.Wojciechowski M, Krata A, Bulska E. 2008. Determination of mercury isotopic profile by inductively coupled plasma mass spectrometry: possibilities and limitations. Chem. Anal. (Warsaw) 53, 797–808. [Google Scholar]
- 31.Shiel AE, Barling KJ, Orlans KJ, Weis D. 2009. Matrix effects on the multicollector inductively coupled plasma mass spectrometric analysis of high-precision cadmium and zinc isotope ratios. Anal. Chim. Acta 633, 29–37. ( 10.1016/j.aca.2008.11.026) [DOI] [PubMed] [Google Scholar]
- 32.Barling J, Weis D. 2008. Influence of non-spectral matrix effects on the accuracy of Pb isotope ration measurement by MC-ICP-MS: implication for the external normalization method of instrumental mass bias correction. J. Anal. At. Spectrom. 23, 1017–1025. ( 10.1039/B717418G) [DOI] [Google Scholar]
- 33.Teran-Baamonde J, Andrade JM, Soto-Ferreiro RM, Carlosena A, Prada D. 2015. A simple procedure to select a model for mass discrimination correction in isotope dilution inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 30, 1197–1206. ( 10.1039/C4JA00475B) [DOI] [Google Scholar]
- 34.Vanhaecke F, Moens L, Dams R. 1998. The accurate determination of copper in two groundwater candidate reference materials by means of high resolution inductively coupled plasma mass spectrometry using isotope dilution for calibration. J. Anal. At. Spectrom. 13, 1189–1192. ( 10.1039/A803782E) [DOI] [Google Scholar]
- 35.Vieira MA, Ribeiro AS, Curtius AJ. 2006. Determination of As, Ge, Hg, Pb, Sb, Se and Sn in coal slurries by CVG-ETV-ICP-MS using external or isotopic dilution calibration. Microchem. J. 82, 127–136. ( 10.1016/j.microc.2006.01.002) [DOI] [Google Scholar]
- 36.Vieira MA, Ribeiro AS, Dias LF, Curtius AJ. 2005. Determination of Cd, Hg, Pb and Sn in sediment slurries by isotopic dilution calibration ICP-MS after chemical vapour generation using an on-line system or retention in an electrothermal vaporizer treated with iridium. Spectrochim. Acta B. 60, 643–652. ( 10.1016/j.sab.2003.12.016) [DOI] [Google Scholar]
- 37.Engström E, Stenberg A, Baxter DC, Malinovsky D, Mäkinen I, Pönni S, Rodushkin I. 2004. Effects of sample preparation and calibration strategy on accuracy and precision in the multi-elemental analysis of soil by sector-field ICP-MS. J. Anal. At. Spectrom. 19, 858–866. ( 10.1039/B315283A) [DOI] [Google Scholar]
- 38.Bulska E, Krata A, Kałabun M, Wojciechowski M. In press On the use of certified reference materials for assuring the quality of results for the determination of mercury in environmental samples. Environ. Sci. Poll. Res. ( 10.1007/s11356-016-7262-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krata A, Vassileva E, Bulska E. 2016. Reference measurements for total mercury and methyl mercury content in marine biota samples using direct or species-specific isotope dilution inductively coupled plasma. Talanta. 160, 562–569. ( 10.1016/j.talanta.2016.07.033) [DOI] [PubMed] [Google Scholar]
- 40.Longerich HP, Jackson SE, Gunther D. 1996. Laser ablation inductively coupled plasma mass spectrometric transient signal data acquisition and analyte concentration calculation. J. Anal. At. Spectrom. 11/9, 899–904. ( 10.1039/JA9961100899) [DOI] [Google Scholar]
- 41.Longerich HP, Gunther D, Jackson SE. 1996. Elemental fractionation in laser ablation inductively coupled plasma mass spectrometry. Fres. J. Anal. Chem. 355/5-6, 538–542. [DOI] [PubMed] [Google Scholar]
- 42.Carré S, Excoffier S, Mermet JM. 2012. A study of the relation between the limit of detection and the limit of quantification in inductively coupled plasma spectrochemistry. Spectrochim. Acta B. 76, 221–225. ( 10.1016/S0584-8547(97)00114-6) [DOI] [Google Scholar]
- 43.Mermet JM, Granier G. 2012. Potencial of accuracy profile for method validation in inductively coupled plasma spectrochemistry. Spectrochim. Acta B. 76, 214–220. ( 10.1016/j.sab.2012.06.003) [DOI] [Google Scholar]
- 44.Wysocka IA, Bulska E, Wróbel K, Wróbel K. 2003. A comparison of electrothermal atomic absorption spectrometry and inductively coupled plasma mass spectrometry for the determination of selenium in garlic. Chem. Anal. (Warsaw) 48, 919–930. [Google Scholar]
- 45.Siemianowski O, Barabasz A, Kendziorek M, Ruszczynska A, Bulska E, Williams LE, Antosiewicz DM. 2014. AtHMA4-expression in tobacco reduces Cd accumulation due to the induction of the apoplastic barrier. J. Experim. Bot. 65, 1125–1139. ( 10.1093/jxb/ert471) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barabasz A, Wilkowska A, Tracz K, Ruszczyńska A, Bulska E, Mills RF, Williams LE, Antosiewicz DM. 2013. Expression of HvHMA2 in tobacco modifies Zn-Fe-Cd homeostasis. J. Plant Phys. 170, 1176–1186. ( 10.1016/j.jplph.2013.03.018) [DOI] [PubMed] [Google Scholar]
- 47.Siemianowski O, Barabasz A, Weremczuk A, Ruszczynska A, Bulska E, Williams L, Antosiewicz DM. 2013. Development of Zn-related necrosis in tobacco is enhanced by expressing AtHMA4 and depends on the apoplastic Zn levels. Plant Cell Environ. 36, 1093–1104. ( 10.1111/pce.12041) [DOI] [PubMed] [Google Scholar]
- 48.Leśniewska BA, Godlewska-Żyłkiewicz B, Ruszczyńska A, Bulska E, Hulanicki A. 2006. Elimination of interferences in determination of platinum and palladium in environmental samples by inductively coupled plasma mass spectrometry. Anal. Chim. Acta 564, 236–242. ( 10.1016/j.aca.2006.01.066) [DOI] [Google Scholar]
- 49.Kowalewska Z, Ruszczyńska A, Bulska E. 2005. Copper determination in crude oil distillation products by atomic absorption and inductively coupled plasma mass spectrometry after analyte transfer to aqueous solution. Spectrochim. Acta B 60, 351–359. ( 10.1016/j.sab.2005.02.022) [DOI] [Google Scholar]
- 50.Kowalewska Z, Ruszczynska A, Jaron I, Bulska E. 2012. Determination of Fe and Ca in alumina-based nickel-molybdenum hydrodesulphurization catalysts using inductively coupled plasma optical emission spectrometry and inductively coupled plasma dynamic reaction cell mass spectrometry. Atom. Spectr. 33, 196–204. [Google Scholar]
- 51.Michalska-–Kacymirow M, Kurek E, Smolis A, Wierzbicka M, Bulska E. 2014. The biological and chemical investigation of Allium cepa L. response to the selenium inorganic compounds. Anal. Bioanal. Chem. 406, 3717–3722. ( 10.1007/s00216-014-7742-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kurek E, Ruszczyńska A, Wojciechowski M, Czauderna M, Bulska E. 2009. Study on speciation of selenium in animal tissues using high performance liquid chromatography with on-line detection by inductively coupled plasma mass spectrometry. Chem. Anal. (Warsaw) 54, 43–57. [Google Scholar]
- 53.Wróbel K, Wróbel K, Kannamkumarath SS, Caruso JA, Wysocka IA, Bulska E, Świątek J, Wierzbicka M. 2004. HPLC-ICP-MS speciation of selenium in enriched onion leaves—a potencial dietary sources of Se-methyloselenocysteine. Food Chem. 86, 617–623. ( 10.1016/j.foodchem.2003.11.005) [DOI] [Google Scholar]
- 54.Hoesl S, Neumann B, Techritz S, Lindscheid M, Theuring F, Scheler C, Jakubowski N, Mueller L. 2014. Development of calibration and standardization procedure for LA-ICP-MS using a conventional ink-jet printer for quantification of proteins in electro- and Western-blot assays. J. Anal. At. Spectrom. 29, 1282–1291. ( 10.1039/C4JA00060A) [DOI] [Google Scholar]
- 55.Horner NS, Beauchemin D. 2014. The use of sol-gel as solid calibration standards for the analysis of soil samples by laser ablation inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 29, 715–720. ( 10.1039/C3JA50374G) [DOI] [Google Scholar]
- 56.Kovacs R, Schlosser S, Staub SP, Schmiderer A, Pernicka E, Guenther D. 2009. Characterization of calibration materials for trace element analysis and fingerprint studies of gold using LA-ICP-MS. J. Anal. At. Spectrom. 24, 476–483. ( 10.1039/B819685K) [DOI] [Google Scholar]
- 57.O'Reilly J, Douglas D, Braybrook J, So P-W, Vergucht E, Garrevoet J, Vekemans B, Vincze L, Goenaga-Infante H. 2014. A novel calibration strategy for the quantitative imaging of iron in biological tissues by LA-ICP-MS using matrix-matched standards and internal standardisation. J. Anal. At. Spectrom. 29, 1378–1384. ( 10.1039/C4JA00002A) [DOI] [Google Scholar]
- 58.Miliszkiewicz N, Walas S, Tobiasz A. 2015. Current approaches to calibration of LA-ICP-MS analysis. J. Anal. At. Spectrom. 30, 327–338. ( 10.1039/C4JA00325J) [DOI] [Google Scholar]
- 59.Jochum KP, et al. 2011. Determination of reference values for NIST SRM 610–617 glasses following ISO guidelines. Geos. Geoanal. Res. 35/4, 397–429. ( 10.1111/j.1751-908X.2011.00120.x) [DOI] [Google Scholar]
- 60.Velasquez G, Borisova AY, Salvi S, Béziat D. 2012. In situ determination of au and cu in natural pyrite by near-infrared femtosecond laser ablation-inductively coupled plasma-quadrupole mass spectrometry: no evidence for matrix effects. Geos. Geoanal. Res. 36/3, 315–324. ( 10.1111/j.1751-908X.2012.00152.x) [DOI] [Google Scholar]
- 61.Van Der Linden V, Cagno S, Cosysns P, Janssens K, Nowak A, Wagner B, Bulska E. 2009. Deeply coloured and black glass in the northern provinces of the Roman Empire: differences and similarities in chemical composition before and after 150 AD. Archaeometry 51, 822–844. ( 10.1111/j.1475-4754.2008.00434.x) [DOI] [Google Scholar]
- 62.Wagner B, Nowak A, Bulska E, Kunicki-Goldfinger J, Schalm O, Janssens K. 2008. Complementary analysis of historical glass by SEM/EDS and LAICPMS. Microchim. Acta 162, 405–424. ( 10.1007/s00604-007-0835-7) [DOI] [Google Scholar]
- 63.Purowski T, Wagner B, Bulska E, Syta O, Dzierżanowski P. 2014. Glassy faience from the Hallstatt C period in Poland: a chemico-physical study. J. Archaeol. Sci. 50, 288–304. ( 10.1016/j.jas.2014.06.022) [DOI] [Google Scholar]
- 64.Purowski T, Dzierżanowski P, Bulska E, Wagner B, Nowak A. 2012. A study of glass beads from the hallstatt C-D from southwestern Poland: implications for glass technology and provenance. Archaeometry 54, 144–166. ( 10.1111/j.1475-4754.2011.00619.x) [DOI] [Google Scholar]
- 65.Cagno S, et al. 2013. Composition data of a large collection of black-appearing Roman glass. Open J. Archaeom. 1, 104–108. ( 10.4081/arc.2013.e22) [DOI] [Google Scholar]
- 66.Dussubieux L, Gratuze B, Blet-Lemarquand M. 2010. Mineral soda alumina glass: occurrence and meaning. J. Archaeol. Sci. 37, 1646–1655. ( 10.1016/j.jas.2010.01.025) [DOI] [Google Scholar]
- 67.Dussubieux L, Robertshaw P, Glascock MD. 2009. LA-ICP-MS analysis of African glass beads: laboratory inter-comparison with an emphasis on the impact of corrosion on data interpretation, Intern. J. Mass Spectrom. 284, 152–161. ( 10.1016/j.ijms.2008.11.003) [DOI] [Google Scholar]
- 68.Wagner B, Nowak A, Bulska E, Hametner K, Günther D. 2012. Critical assessment of the elemental composition of corning archeological reference glasses by LA-ICP-MS. Anal. Bioanal. Chem. 402, 1667–1677. ( 10.1007/s00216-011-5597-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cousin H, Magyar B. 1994. Precision and accuracy of laser ablation-ICP-MS analysis of rare earth elements with external calibration. Microchim. Acta 113, 313–323. ( 10.1007/BF01243621) [DOI] [Google Scholar]
- 70.Pakieła M, Wojciechowski M, Wagner B, Bulska E. 2011. Immobilization of powder for elemental analysis by laser-ablation inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 26, 1539–1543. ( 10.1039/C0JA00201A) [DOI] [Google Scholar]
- 71.Bellotto VR, Miekeley N. 2000. Improvements in calibration procedures for the quantitative determination of trace elements in carbonate material (mussel shells) by laser ablation ICP-MS. Fres. J. Anal. Chem. 367, 635–640. ( 10.1007/s002160000421) [DOI] [PubMed] [Google Scholar]
- 72.Craig C-A, Jarvis K, Clarke LJ. 2000. An assessment of calibration strategies for the quantitative and semi-quantitative analysis of calcium carbonate matrices by laser ablation inductively coupled plasma—mass spectrometry (LA-ICP-MS). J. Anal. At. Spectrom. 15, 1001–1008. ( 10.1039/B002097O) [DOI] [Google Scholar]
- 73.Halicz L, Gunther D. 2004. Quantitative analysis of silicates using LA-ICP-MS with liquid calibration. J. Anal. At. Spectrom. 19, 1539–1545. ( 10.1039/B410132D) [DOI] [Google Scholar]
- 74.Tibi M, Heumann G. 2003. Isotope dilution mass spectrometry as a calibration method for the analysis of trace elements in powder samples by LA-ICP-MS. J. Anal. At. Spectrom. 18, 1076–1081. ( 10.1039/B301835K) [DOI] [Google Scholar]
- 75.Thieleke J, Vogt C. 2016. Calibration stragtegy for LA-ICP-MS using isotope dilution for solid reference materials. J. Anal. At. Spectrom. 31, 1198–1205. ( 10.1039/C6JA00042H) [DOI] [Google Scholar]
- 76.Krata A, Bulska E. 2005. Critical evaluation of analytical performance of atomic absorption spectrometry and inductively coupled plasma mass spectrometry for mercury determination. Spectrochim. Acta B 60, 345–350. ( 10.1016/j.sab.2004.11.011) [DOI] [Google Scholar]
- 77.Poto L, Gabrieli J, Crowhurst S, Agostinelli C, Spolaor A, Cairns WRL, Cozzi G, Barbante C. 2015. Cross calibration between XRF and ICP MS for high spatial resolution analysis of ombrotrophic cores for palaeoclimatoc studies. Anal. Bioanal. Chem. 407, 379–385. ( 10.1007/s00216-014-8289-3) [DOI] [PubMed] [Google Scholar]
- 78.Donachie A, Walmsley AD, Haswell SJ. 1999. Application and comparison of chemometric techniques for calibration modelling using electrochemical/ICP MS data for trace elements in UHQ water and humic acid matrices. Anal. Chim. Acta 378, 235–243. ( 10.1016/S0003-2670(98)00609-6) [DOI] [Google Scholar]
- 79.Bulska E, Danko B, Dybczyński RS, Krata A, Kulisa K, Samczyński Z, Wojciechowski M. 2012. Inductively coupled plasma mass spectrometry in comparison with neutron activation and ion chromatography with UV/VIS detector for the determination of lanthanides in plant materials. Talanta 97, 303–311. ( 10.1016/j.talanta.2012.04.035) [DOI] [PubMed] [Google Scholar]
- 80.Grottoli AG, Matthews KA, Palardy JE, McDonough WF. 2013. High resolution coral Cd measurements using LA-ICP-MS and ID-ICP-MS: calibration and interpretation. Chem. Geol. 356, 151–159. ( 10.1016/j.chemgeo.2013.08.024) [DOI] [Google Scholar]