

Abstract
Metabolomics is a research field used to acquire comprehensive information on the composition of a metabolite pool to provide a functional screen of the cellular state. Studies of the plant metabolome include the analysis of a wide range of chemical species with very diverse physico-chemical properties, and therefore powerful analytical tools are required for the separation, characterization and quantification of this vast compound diversity present in plant matrices. In this review, challenges in the use of mass spectrometry (MS) as a quantitative tool in plant metabolomics experiments are discussed, and important criteria for the development and validation of MS-based analytical methods provided.
This article is part of the themed issue ‘Quantitative mass spectrometry’.
Keywords: mass spectrometry, plant metabolomics, metabolite quantification, method validation, matrix effects, analytical recoveries
1. Introduction
Quantitative plant metabolomics is a tool that helps to improve our understanding of plant biochemistry and metabolism by delivering the accurate measurement, prior to statistical and bioinformatics analysis, of the concentrations of known metabolites that occur in different levels in plant samples [1,2]. The two most commonly used analytical technologies driving quantitative plant metabolomics studies are mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy. However, due to its high sensitivity relative to NMR, MS is by far the technology of choice in most plant metabolomics studies, and, when coupled to powerful chromatographic techniques (e.g. liquid chromatography–mass spectrometry (LC-MS), capillary electrophoresis–mass spectrometry (CE-MS) and gas chromatography–mass spectrometry (GC-MS)), allows the separation and characterization of the extremely high compound diversity present in the plant metabolome [3–5]. Nonetheless, NMR methods are still increasingly being applied as a particular approach for structure elucidation in plant metabolomics studies [6]. Indeed, both approaches provide complementary as well as supplementary information on the concentrations of metabolites, their ranges and changes in complex plant matrices, and, when combined with new experimental and computational methods, enable routine quantitative analysis of hundreds of plant metabolites[7–10].
Despite the state-of-the-art technology employed, the need for standardizing key steps in plant metabolomics experiments, from experimental design to instrument performance, is yet considered a critical issue. In line with this, back in 2007, the Metabolomics journal dedicated an entire issue to the Metabolomics Standards Initiative (MSI), an initiative of the Metabolomics Society developed to ensure that clear information on the biological system and workflow of the metabolomics experiment is delivered to the community [11,12]. As part of the MSI, the Chemical Analysis Working Group (CAWG) proposed minimum community-agreed reporting standards for chemical analysis that also included minimum reporting metadata related to metabolite identification and quantification [13]. Since then, great interest in standardizing data associated with large-scale metabolomics experiments has become a general consensus, and data sharing, data standards and workflows for metabolomics are progressively becoming more FAIR-compliant (i.e. findable, accessible, interoperable and reusable) within the metabolomics community [14–19].
Quantitative MS-based plant metabolomics has established itself as a powerful tool to address interesting biological questions related to plant environment and agriculture. Because plants are sessile organisms, they cannot escape from changing environmental conditions and/or plant–attacker interactions that adversely affect their growth and development (figure 1). Therefore, plant survival mostly depends on the initiation of complex adaptive responses that involve stress sensing, signal transduction and the activation of a number of stress-related genes and metabolites [20]. Central metabolism is involved in the regulation of the various developmental processes that allow plants to survive such environmental threats, and the measurement of known primary metabolites (e.g. carbohydrates, amino and organic acids) has largely contributed to elucidate how and to what extent plant metabolism readjusts to a changing environment [5,21,22]. On the other hand, the measurement of specific secondary metabolites such as phytohormones (e.g. auxins, cytokinins, gibberellins, jasmonates, abscisic and salicylic acids), which are key metabolites of signalling and communication between an organism and the abiotic/biotic environment, has contributed to improve our current understanding of the plant growth–defence system [23–27] (figure 1).
Figure 1.

Mass spectrometry-based plant metabolomics is a powerful tool to study the metabolic mechanisms underlying plant responses to adverse environmental conditions (abiotic stress) and/or plant–attacker interactions (biotic stress) that adversely affect plant growth and development.
In this review, we first present an overview of the most critical steps in quantitative plant metabolomics experiments (§2), from experimental design and sample handling procedures to comprehensive analytical method validation (§3). We further discuss applications of MS as a quantitative tool in the field of plant metabolomics (§4), with special focus on the analysis of key primary metabolites and some classes of phytohormones.
2. Experimental design for plant metabolomics
A good experimental design in plant metabolomics heavily depends on the starting point of the experiment and must include (i) the selection of appropriate growth conditions (e.g. photoperiod, relative humidity, temperature), (ii) a suitable number of biological replicates and controls, and (iii) the use of randomization practices throughout the entire experimental workflow [11–13,28–30]. It is also advisable to monitor environmental variations during the progress of the experiment, such as small differences in temperature or light intensity that might occur in the greenhouse or growth chamber; minor variations can greatly affect the biochemical status of the plant [31] (figure 2).
Figure 2.

Typical plant metabolomics workflow: from experimental design and sample preparation to MS-based metabolite analysis.
(a). Sample preparation
Sample preparation is another critical step in the design of plant metabolomics experiments and must be carefully planned, from plant tissue harvest and quenching to metabolite extraction [32] (figure 2).
(i). Harvesting and quenching
Tissue harvest should be carried out as fast as possible by immediately freezing the plant tissue in liquid nitrogen (i.e. shock freezing) and storing it at −80°C. Quick inactivation of enzymatic reactions and metabolic processes during tissue harvest is particularly important to avoid fluctuations in the levels of fast turnover metabolites (e.g. glycolytic intermediates) [33–36]. Fresh-frozen plant tissues should be then finely homogenized, a process often performed using a pre-cooled pestle and mortar filled with liquid nitrogen. Finally, aliquots of fine powdered plant tissue must be rapidly weighed in pre-cooled polypropylene microfuge tubes to ensure that the plant material does not thaw [5,37,38].
(ii). Extraction of metabolites
The extraction of metabolites from plant tissues should be as comprehensive as possible while making sure that (i) the chemical stability of metabolites is not affected, (ii) the analytical recoveries of metabolites throughout the extraction are complete, and (iii) the variability in metabolite concentrations during the extraction is minimized [37]. The amount of tissue adopted in plant metabolomics experiments might differ between extraction methods (e.g. typically up to 100 mg for primary metabolites or even higher up to 2 g for phytohormones); however, the tissue-to-solvent ratio should be maintained. Polar organic solvents (e.g. methanol and acetonitrile) are able to extract both hydrophilic and hydrophobic compounds; therefore, solvent systems that combine both polar and non-polar organic solvents (e.g. chloroform) are often used to separate a polar phase (hydrophilic metabolites) from a non-polar phase (hydrophobic metabolites such as lipids) [37]. Solvent systems containing acetonitrile have been shown to be more efficient than methanol to precipitate proteins, thereby improving the analytical method accuracy and precision in metabolomics experiments [39–42]. Nevertheless, there is no particular extraction protocol that can cover the full metabolome, and multiple protocols are often employed in plant metabolomics studies for better metabolite coverage [5,38].
(iii). Pre-analytical requirements
Prior to metabolite analysis using MS-based technologies (e.g. LC-MS, CE-MS and GC-MS), pre-analytical strategies might be carried out to ultimately (i) concentrate the metabolite(s) of interest, (ii) prevent sample carryover in chromatographic systems, and (iii) eliminate interfering components from the plant matrix [37]. GC-MS is only capable of analysing volatile compounds and requires extensive chemical derivatization procedures to increase volatility and thermostability (e.g. most primary metabolites) [43,44]. LC and CE allow direct analysis of non-volatile metabolites. In LC-MS, the plant extract is usually re-dissolved in a solvent with the same composition of the initial conditions of the LC mobile phase. CE-MS, in turn, includes very low organic solvent consumption without the need for extensive sample pretreatment [45,46].
3. Analytical method validation for quantitative plant metabolomics
In quantitative metabolomics studies, major efforts are put into optimizing sample extraction and separation as well as instrumental conditions to measure specific plant metabolites. This is extremely important given the very diverse physico-chemical properties of the compounds that are being analysed in plant matrices. The fundamental challenge, however, is to find the best analytical conditions for validating the method (i.e. the method is precise and accurate and produces reliable quantitative data for target metabolites in a specific matrix) prior to its implementation in routine analyses [47–49]. Nonetheless, one main aspect common to most analytical validation methods is the attempt to maximize the number of known metabolites that can be quantitatively measured in a single study.
MS-based plant metabolomics experiments deal with two approaches for quantitative analyses, namely, relative and absolute quantification. Relative quantification can be defined as the analyte instrumental response relative to an internal standard, and is usually applied in metabolite profiling experiments. In CAWG-MSI-compliant MS-based experiments, relative quantification should include (i) the name and the added amount of the exogenous internal standard compound (e.g. unlabelled ribitol and/or isotopically labelled 13C-sorbitol); (ii) a description of the method used to evaluate instrument response (e.g. peak integration and deconvolution method); and (iii) the number of replicate analyses and standard error. On the other hand, absolute quantification is a more demanding process and generally includes all method validation steps for absolute metabolite quantification, namely (i) a defined range of metabolite concentration to establish calibration curves that clearly define instrument linearity, (ii) an assessment of the limit of detection (LOD) and limit of quantification (LOQ), (iii) an evaluation of method precision and accuracy, (iv) method specificity and selectivity, and (v) analytical recoveries of commercially available authentic standards during the extraction (advisable) [13]. Based on these guidelines, we next discuss key steps required for the analytical validation of MS-based methods for quantitative plant metabolomics experiments.
(a). Instrument linearity
Linearity is the study of the calibration line and its corresponding slope. For a first-order calibration function (y = a + bx), axis intercept a and slope b are characteristic of the sensitivity of the analytical method (figure 3). Calibration samples are prepared at different concentration levels, and the increasing analyte concentration is plotted against the corresponding detector response (peak height, peak area or peak intensity) [47]. The resulting graphical representation of the calibration data is then evaluated for linearity and the correlation coefficient (R2) determined. However, it has been shown that a correlation coefficient very close to one might also be obtained for a curved relationship, characteristic of a second-order calibration function [50]. In such cases, when visual inspection of the calibration line is not sufficient, Mandel's fitting test is recommended for mathematical verification of linearity [51]. The Mandel fitting test evaluates the linearity of the calibration by calculating the residual standard deviation of a first-order calibration function (Sy1) and a second-order calibration function (Sy2) adjustments. The difference between the variances (DS2) is calculated by
| 3.1 |
where N is the number of standards used in the calibration, and the equation has one degree of freedom. DS2 and the variance of the second-order calibration function are submitted to an F-test in order to examine for significant differences. The test value (PG) required for the F-test is calculated by
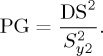 |
3.2 |
Figure 3.
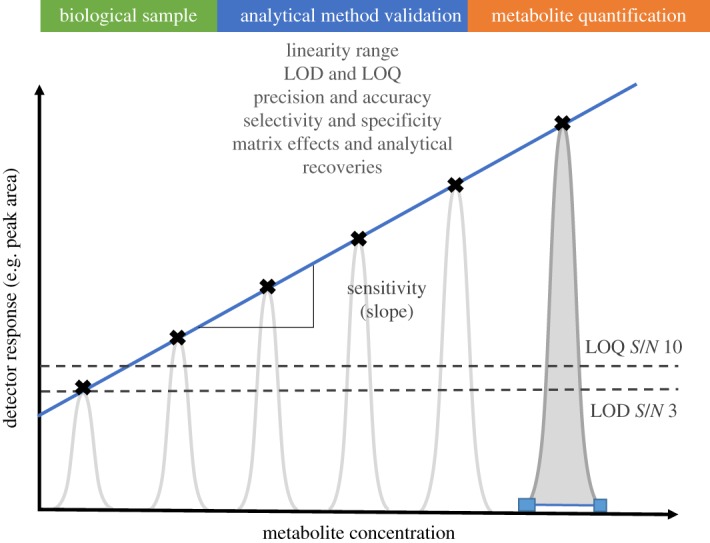
Parameters required for the analytical validation of MS-based methods for quantitative plant metabolomics experiments.
If PG ≤ F, the second-order calibration function does not lead to a significantly better adjustment and the calibration function is linear; if PG > F, the working range should be narrowed to maintain a sufficient linearity; otherwise, the obtained data must be evaluated using the second-order calibration function [52].
An appropriate number of standards is necessary to define adequately the analytical relationship between concentration and response. Although information on specific requirements for the number of replicates at each concentration level is scarce, five to eight concentration points are recommended within a calibration curve, each analysed at least in triplicate [52–55].
The choice of an appropriate calibration model is also necessary for reliable quantification. In quantitative MS-based chromatographic methods, quantification of target compounds can be achieved via: (i) external standard calibration plots, using standard solutions of the analyte; (ii) internal standard calibration plots, where an internal standard is added to the sample and is analysed together with the analyte; (iii) standard addition calibration plots, where increasing amounts of standard are added to several sample matrix aliquots and the standard addition calibration curve is calculated between the measured analytical response and the known added concentrations; and (iv) matrix-matched calibration plots, where calibration is obtained using a matrix reference material (when available) [47]. However, because there is no matrix reference material with known metabolite concentrations available for high-throughput analyses in plant metabolomics studies, this approach has not yet been adopted [56].
(b). Limit of detection and limit of quantification
The LOD represents the lowest analyte amount that can be detected in a sample, even if it cannot be quantified. In chromatographic methods, a quick estimation of the LOD can be simply defined as the concentration for which the signal-to-noise ratio (S/N) equals 3 (figure 3). The LOQ is the lowest analyte amount that can be quantitatively determined with a good degree of method precision and accuracy (§3c). In chromatographic methods, the LOQ can be defined as the concentration for which the S/N equals 10 (figure 3) [48,56].
(c). Method precision and accuracy
The precision of an analytical method is defined as the closeness of agreement between a set of independent analytical measurements of the same sample, under the optimal developed conditions, expressed as relative standard deviation (RSD, %) or coefficient of variation (CV, %). Within a laboratory, precision of the analytical method is often reported at two different levels: intra- and inter-day precision. In chromatographic methods, these two parameters are often expressed by repeatability of retention/migration times or peak areas. Intra-day precision assesses the repeatability of the analysis of the same sample under equal experimental conditions within the same day. On the other hand, inter-day precision reflects small variations in the experimental or operational conditions from repetitive analysis of the same sample within different days [48,54,56,57].
The accuracy reflects the closeness of agreement between the true value of the analyte concentration and the mean result obtained by applying the analytical method (i.e. reliable quantification) [48,54,57]. In plant metabolomics studies, where no reference materials with known metabolite concentrations are available, the accuracy of the analytical method can be assessed by determining the analytical recoveries of authentic standards or isotopically labelled metabolites spiked into the plant matrix (§3f) [56].
(d). Method selectivity and specificity
Selectivity is the ability of an analytical method to measure and discriminate the analyte(s) of interest without interference from other components in the sample, while specificity relates to how well a method is able to unequivocally measure one particular analyte in the presence of other components that might be present (e.g. endogenous or exogenous impurities, degradation products or matrix components). The presence of such interferents affects the analytical procedure, thereby leading to incorrect results [58,59]. To evaluate the selectivity of a method in terms of the contribution from interfering analytes, a standard addition method of the analyte(s) of interest might be performed using the plant matrix under study (§3a). A minimum of four standard additions within the established concentration range are advised, and the resulting calibration curve must be compared to the calibration with standard solutions [54].
(e). Evaluation of matrix effects
Matrix effects (ME) are a serious problem in LC-MS methods, which result from co-eluting matrix components that affect the ionization of the target analyte(s) (i.e. ion suppression or ion enhancement), thereby compromising the accurate quantification of metabolites in the plant matrix [60–63].
ME can be qualitatively evaluated by performing co-introduction or post-column infusion experiments as proposed by Bonfiglio and co-workers [64]. In this approach, the analyte of interest and the potentially suppressing matrix components are simultaneously introduced into the ionization source. However, the impact of ME is quite difficult to express with this approach because the risk of contamination of the ion source greatly increases when high amounts of the analyte are infused. In addition, because a high number of metabolites are usually profiled in metabolomics experiments, this approach is generally not adopted in routine analyses, as it is very time consuming.
ME can be quantitatively evaluated as first proposed by Buhrman et al. [65] and later modified by Matuszewski et al. [62]. This quantitative approach compares the signal response of a target analyte(s) in the absence of matrix (standard solution) with the signal response of the analyte(s) spiked at the same concentration in the matrix after extraction. The quantitative evaluation of the percentage ME is then calculated by
| 3.3 |
where ME = 100% indicates no matrix effect, ME < 100% indicates ion suppression, and ME > 100% indicates ion enhancement [62]. Alternatively, the presence or absence of ME can be assessed by determining the variability between the slope of a calibration curve prepared in the mobile phase and the slope of the same calibration curve prepared in the matrix, expressed as the percentage CV, where CV values below 4% indicate no ME and the analytical method is acceptable [48,66].
Overall, the evaluation of ME is an essential part of method development and validation in any LC-MS plant metabolomics experiment, and if ME are found to be relevant, their impact must be reduced or eliminated by optimizing the chromatographic conditions, by sample clean-up (if possible), or by employing corrective calibration methods such as the standard addition method (§3a).
(f). Analytical recovery
In plant metabolomics experiments, the analytical recovery determines the influence of an analytical procedure (e.g. extraction) as well as the influence of the plant matrix on the developed analytical method. The analytical recovery is expressed as the percentage of a known amount of analyte spiked into the plant matrix that remains after going through the extraction procedure, and is calculated by
| 3.4 |
It is usually assumed that if the analytical recoveries are 100% then the procedure in question is free from systematic errors (e.g. extraction). However, it is important to highlight that there is no minimum established value for analytical recoveries, as they are dependent on the analyte; low recovery values can be accepted if a high sensitivity, precision and accuracy are obtained for the analyte over the entire calibration range [48].
4. MS-based hyphenated technologies for quantitative plant metabolomics
The plant metabolome comprises a high diversity of metabolites with different physico-chemical properties, and includes carbohydrates, amino and organic acids, lipid-related compounds as well as structurally diverse phytohormones. Metabolites are also present in a large dynamic range of concentrations, from femtomolar to millimolar, and therefore a combination of MS-based hyphenated technologies is ideally applied to allow identification and quantification of as many metabolites as possible [67]. Spectral processing of raw data files (i.e. spectral pre-processing, feature detection, peak detection, spectral alignment, feature normalization and spectral deconvolution) is a key step in the overall workflow of a quantitative plant metabolomics experiment. This step is highly dependent on the MS-based platform used (e.g. LC-MS, CE-MS and GC-MS), and different open access software tools are available (e.g. XCMS, MZmine, MetAlign). More information on the impact of using different algorithms to process the acquired mass spectral data can be found in Cajka & Fiehn [42].
In this section, we discuss examples on the use of MS-based hyphenated technologies (e.g. LC-MS, CE-MS and GC-MS) for the quantitative analysis of primary metabolites (e.g. carbohydrates, amino and organic acids) and phytohormones (e.g. auxins, cytokinins, giberellins, jasmonates, abscisic and salicylic acids). Because natural products (e.g. anthocyanins and other flavonoids, glucosinolates, terpenoids and alkaloids) do not play an essential role in plant growth, this review will not provide details on these secondary metabolites; important information on their MS-based analysis can be found in recent comprehensive review articles [3,68–70] to mention just a few. Lipidomics has long been recognized as a subfield of the more comprehensive metabolomics research area, and therefore the extraction and analysis of the plant lipidome falls outside of the scope of this review. Detailed sensitive MS-based methods for the quantitative analysis of lipid-related species can also be found elsewhere [42,71–76].
(a). LC-MS
Liquid chromatography coupled to mass spectrometry (LC-MS) is particularly well suited to profile a small set of known metabolites or compound classes of the plant metabolome, an approach referred to as target metabolite analysis. Generally, in LC-MS applications, the most commonly employed ionization method is electrospray ionization (ESI), a soft-ionization technique that introduces little internal energy and gives little information on structure because few fragments are generated [77,78]. Detailed fragmentation and structural information from intact ionized species can be obtained by performing collision-induced dissociation (CID) experiments usually carried out on a tandem MS instrument that allows two (MS/MS) or more (MSn) sequential stages of mass spectrometric analysis [5,79].
One very important factor when developing LC-ESI-MS/MS methods is the selection of the column chemistry and its retention mechanism. A great number of applications have been reported for the target analysis of phytohormones (in more detail later in this section) and use reversed-phase (RP) stationary phases (i.e. C18 functional groups covalently bonded to the surface of the silica particle) with typical mobile phases composed of aqueous/organic solvent mixtures (e.g. water/acetonitrile or water/methanol). However, polar analytes (e.g. most primary metabolites) have very little interaction with the apolar C18 stationary phase, and thereby elute very close to the void volume without chromatographic retention. Consequently, alternative column chemistries (e.g. hydrophilic interaction chromatography (HILIC), porous graphitic carbon (PGC) and anion exchange chromatography (AEC)) must be applied for the target analysis of the wide range of polar metabolites typically found in the plant metabolome (in more detail later in this section).
Within the large range of MS instruments currently available for LC-MS-based plant metabolomics applications (e.g. triple quadrupole (QqQ), quadrupole ion trap (QIT), quadrupole time of flight (QTOF), quadrupole Orbitrap, triple quadrupole-linear ion trap (QTRAP)), LC-QqQ-MS methods are desirable in quantitative targeted approaches because QqQ-MS allows highly sensitive multiple reaction monitoring (MRM) experiments, and therefore reliable absolute quantification of low-abundance metabolites (in more detail later in this section).
(i). Primary metabolites
In early plant metabolomics studies, the use of HILIC-ESI-QIT-MS/MS for the quantification of a broad range of highly polar plant metabolites was first introduced by Tolstikov & Fiehn [80]. The HILIC-based 60 min gradient method was used to detect and quantify raffinose family oligosaccharides (RFOs), such as raffinose, stachyose and verbascose, as well as amino acids, amino sugars and sugar nucleotides from Cucurbita maxima phloem tissues. An LOD of 0.5 ng was reported for stachyose [80] (table 1).
Table 1.
Details of the LC-MS-based methods for quantitative plant metabolomics referenced in §4a.
| plant species | metabolites | MS-based method | method parameters | refs |
|---|---|---|---|---|
| primary metabolites | ||||
| Cucurbita maxima phloem | sucrose, raffinose, stachyose, maltoheptose, UDP-glucose, 1,4-dideoxy-1,4-imino-d-arabinitol, N-methyl-1-deoxynojirimycin, 2-amino-2-deoxy-d-glucose, N-acetyl-d-glucosamine, glucosaminic acid, l-methionine, l-alanyl-l-alanine | HILIC-ESI-QIT-MS | five-point calibration curvesLOD stachyose = 0.5 ng | [80] |
| Arabidopsis thaliana leaves | fructose, glucose, maltose, sucrose, trehalose; fructose-6-phosphate, glucose-1-phosphate, glucose-6-phosphate, sucrose-6 phosphate, trehalose-6-phosphate | PGC-ESI-QIT-MS/MS | linearity range 0–100 µMLOD range = 0.1–2 µM (sugars)LOD range = 0.3–1.5 µM (sugar-Ps)LOQ range = 0.3–6.7 µM (sugars)LOQ range = 2.5–5 µM (sugar-Ps) | [81] |
| Lupinus albus stems | galactinol, inositol, maltitol, mannitol, sorbitol, fructose, glucose, maltose, raffinose, stachyose, sucrose, verbascose | PGC-ESI-QIT-MS/MS | LOD range = 0.4–9 pmol (sugars)LOD range = 4–20 pmol (sugar alcohols)linearity range 0–100 µMLOQ range = 0.07–6.7 µmol (sugars)LOQ range = 0.25–3.33 µmol (sugar alcohols) | [82] |
| Arabidopsis thaliana leaves | glucose, raffinose, sucrose, verbascose, mannitol, maltitol, glucose-6-phosphate, trehalose-6-phosphate | HILIC-ESI-QIT-MS/MS | linearity range 0–100 µMLOD range = 0.2–1 µM (neutral sugars)LOD = 1 µM (sugar alcohols)LOD range = 1–2 µM (sugars-Ps)LOQ range = 0.7–3 µM (neutral sugars)LOQ = 3 µM (sugar alcohols)LOQ range = 3–6.7 µM (sugars-Ps) | [83] |
| Arabidopsis thaliana leaves | UDP-arabinose, UDP-galactose, UDP-galacturonic acid, UDP-glucose, UDP-glucuronic acid | PGC-LTQ-Orbitrap-MS/MS | linearity range 0.010–2.5 µmol l−1LOD = 70 nmol l−1 | [84] |
| Tritium aestivum | deoxynivalenol, DON-3-glucoside, UDP-glucose, UDP-glucuronic acid | HILIC-ESI-QqQ-IT-MS/MS | LOD range = 0.6–10 ng ml−1LOQ range = 2–33 ng ml−1recovery 85–103% | [85] |
| Arabidopsis thaliana leaves | GDP-fucose, GDP-glucose, GDP-mannose, UDP-arabinofuranose, UDP-arabinopyranose, UDP-galactosamine, UDP-galactose, UDP-galacturonate, UDP-glucosamine, UDP-glucose, UDP-glucuronate, UDP-xylose | HILIC-ESI-QqQ-MS/MS | LOD range = 2.5–5 fmolLOQ range = 5–20 fmolrecovery 84–110% | [86] |
| Arabidopsis thaliana seedlings | trehalose-6-phosphate | AEC-MS-QqQ-MS/MS | linearity range 0.18–3.4 pmolrecovery 82–86% | [87] |
| Arabidopsis thaliana seedlings | trehalose-6-phosphate | AEC-ESI-QIT-MS/MS | linearity range 80 nM–1.3 µMLOD = 40 nMLOQ = 80 nMrecovery >80% | [40] |
| Arabidopsis thaliana seedlings | trehalose-6-phosphate | HILIC-ESI-QIT-MS/MS | LOD = 3.5 nMrecovery 80–120%linearity range 25–4000 nM | [88] |
| Arabidopsis thaliana seedlings | trehalose-6-phosphate | AEC-MS-QqQ-MS/MS | LOD = 2.5 fmol | [89] |
| phytohormones | ||||
| Lactuca sativa seeds | auxins, abscisic acid, cytokinins, gibberellins | RP-ESI-QqQ-MS/MS | LOD range = 0.486–1.127 fmol | [90] |
| Arabidopsis thaliana leaves | auxins, abscisic acid, cytokinins, gibberellins, jasmonic acid, salicylic acid | RP-ESI-Qtrap-MS/MS | linearity range 1–1000 pg ml−1LOQ range = 0.011–10 pg g−1 FWrecovery 85–98% | [91] |
| Arabidopsis thaliana tissues; | auxins, abscisic acid, cytokinins, gibberellins | RP-ESI-IT-MS/MS | linearity range 1–5000 fmol | [92] |
| Nicotiana tabacum tissues | LOD range = 0.55–170 fmol | |||
| nanoflow RP-ESI-IT-MS/MS | linearity range 0.1–5000 fmolLOD range = 0.066–18 fmol | |||
| Acacia richii root nodules; Agapanthus africanus bud and root; Arabidopsis thaliana tissues; Oryza sativa tissues; Vitis vinifera leaf and root; Populus tissues; Zea mays roots | auxins, abscisic acid, cytokinins, gibberellins | RP-ESI-QIT-MS/MS | LOD range = 0.16–393 fmol recovery 22–95% | [93] |
| Coffea arabica leaves | abscisic acid, jasmonic acid, salicylic acid | RP-ESI-QqQ-MS/MS | linearity range 0,042–1,67 µg g−1LOD = 0.010 µg g−1 FW | [94] |
A similar HILIC-ESI-QIT-MS/MS approach was later developed by António and co-workers for the quantification of carbohydrate-related metabolites in Arabidopsis thaliana wild-type and its starchless phosphoglucomutase mutant (pgm1) leaves [83]. The method was rapid, and allowed the separation of a complex mixture containing several reducing and non-reducing sugars, sugar alcohols and sugar phosphates in less than 15 min [83]. In addition, a better LOD was obtained for stachyose (66 pg), which represents a considerable improvement over the previously reported HILIC method [80] (table 1). In that same study, António and co-workers also demonstrated that HILIC-ESI-QIT-MS/MS and PGC-ESI-QIT-MS/MS gave comparable quantitative results (and LODs) for the analysis of the same set of metabolites, and concluded that HILIC and PGC are two excellent alternatives to typical RP methods for the analysis of highly polar metabolites [83] (table 1). However, PGC separations have been reported to require extensive method development and strong conditions for the elution of sugar phosphates [81] and sugar nucleotides [84], which makes HILIC a more suitable approach for the quantitative analysis of these metabolites in routine plant metabolomics studies [83,85,86] (table 1). Nevertheless, less-laborious PGC-ESI-QIT-MS/MS methods have been developed, validated and reported for the analysis of neutral sugars in a wide range of plant metabolomics studies related to plant environment and agriculture, including the quantification of glucose, sucrose and raffinose from Lupinus albus stem tissues (cortex and stele) in response to drought stress and subsequent recovery treatments [82], RFOs from Haberlea rhodopensis leaf tissues in response to severe desiccation [95] (table 1), and, more recently, raffinose from Casuariana glauca tissues (nodules and branchlets) in response to several salt-stress conditions (Jorge T.F., Ribeiro-Barros A., Florêncio M.H. and António C., 2016, unpublished data).
High-performance anion exchange chromatography (HPAEC) coupled to ESI-QqQ-MS/MS in the highly selective MRM mode has been reported for the first time to offer the required low LOD for the targeted analysis of low-abundance trehalose-6-phosphate (T6P) in plant extracts [87]. In that study, a linear response ranging from 0.18 to 3.4 pmol of T6P was achieved, thereby allowing for the reliable quantification of T6P in the femto- to picomole range (table 1). T6P was found to be present in A. thaliana wild-type Col-0 tissues at very low concentrations that ranged from 23 to 298 pmol g−1 FW (fresh weight) in leaves and 18 to 482 pmol g−1 FW in seedlings [87]. This highly sensitive HPAEC-ESI-QqQ-MS/MS method has been adopted by other researchers to quantify T6P in plant tissues in a wide range of studies related to plant growth and development [89,96–100].
Delatte and co-workers reported an HPAEC-based method coupled to ESI-QIT-MS/MS to reliably quantify T6P in plant extracts in the range 80 nM to 1.3 µM, and LOD 40 nM [40]. In that study, T6P was found to be present in seedlings of A. thaliana wild-type Col-0 and mutant lines at concentrations below 0.35 nmol g−1 FW [40] (table 1). This method was later used by other authors to quantify T6P [101–103]. Another approach has been reported by Toraño and co-workers [88], who developed an HILIC-ESI-QIT-MS/MS to quantify T6P in A. thaliana wild-type Col-0 seedlings. T6P was found to be present in A. thaliana seedlings grown on sorbitol at concentrations below 0.2 nmol g−1 FW, whereas in seedlings grown on trehalose, the levels were found to be higher than 1.2 nmol g−1 FW [88]. With this method, an LOD of 3.5 nM was obtained for T6P, a considerable improvement with respect to the method previously published by the same authors using HPAEC [40] (table 1). Recently, Mata and co-workers developed an HILIC-based method using a QqQ in the highly sensitive MRM mode to obtain the low LOD required to also measure low-abundance T6P. The method was validated and its utility demonstrated by applying it to quantity the levels of this target metabolite in wild-type roots of the model legume Medicago truncatula following a water deficit treatment. Preliminary results have shown that T6P was found to be present at concentrations as low as 0.016 nmol g−1 FW and 0.09 nmol g−1 FW in M. truncatula wild-type roots, under control and water deficit conditions, respectively (Mata A.T., Jorge T.F., Bronze M.R., Branco D., Fevereiro P., Araújo S. and António C., 2016, unpublished data).
(ii). Phytohormones
Phytohormones are structurally diverse secondary metabolites that play important roles in plant growth in response to abiotic/biotic stress [104,105], and include auxins, particularly indole-3-acetic acid (IAA), cytokinins (CK), gibberellins (GA), brassinosteroids (BR), abscisic acid (ABA), jasmonates (JA) and salicylic acid (SA). However, their abundance is usually much lower when compared with primary metabolites, ranging from 0.1 to 50 ng g−1 FW [106]. RP-based LC-MS/MS methods are the most frequently reported methods to accurately estimate these phytohormone levels, which very often require the use of expensive labelled internal standards (table 1) [107,108].
A study by Chiwocha and co-workers reported the use of a sensitive and selective RP-ESI-QqQ-MS/MS method for the simultaneous analysis of four classes of phytohormones in Lactuca sativa seeds, including auxins (IAA), CK (zeatin), GA (GA1, GA3) and ABA [90]. The method was linear in the concentration ranges selected for the different classes, and LODs ranged from 0.486 fmol for zeatin and 1.127 pmol for ABA (table 1). Pan and co-workers [91] developed a rapid, sensitive RP-ESI-QTRAP-MS/MS method for the simultaneous quantification of seven major classes of phytohormones including auxins, CK, GA, ABA, JA, SA and relevant methyl ester-related metabolites in crude A. thaliana plants wounded with the fungal pathogen Botrytis cinerea [91]. Using this method, LOQ ranging from 0.01 to 10 pg g−1 FW allowed the quantification of all target phytohormones (table 1). Izumi and co-workers [92] described a comparative approach between a nanoflow RP-ESI-IT-MS method and capillary RP-ESI-IT-MS/MS for the quantification of four classes of phytohormones and their derivatives (auxins, CK, GA and ABA) in Nicotiana tabacum and A. thaliana plant tissues [92]. The authors demonstrated that the nanoflow method provided best sensitivity, with an LOD ranging from 0.066 to 18 fmol (table 1).
Another interesting approach was reported by Liu and co-workers [93], who developed an RP-ESI-QIT-MS/MS method for the simultaneous determination of around 24 phytohormones including IAA, cis- and trans-ABA, 11 CK and 10 GA in several plant tissues [93]. All phytohormones could be quantified in femtomole quantities in plant tissues, with an LOD (S/N ≥ 5) of 172.65 fmol obtained for IAA, and 13.78 and 65.84 fmol for cis- and trans-ABA, respectively. The LOD for CKs were the lowest (less than 1 fmol) and LOD for GAs were around 100 fmol (table 1).
An RP-ESI-QqQ-MS/MS method for the simultaneous quantification of ABA, JA and SA in Coffea arabica (L.) leaves was developed by de Sá and co-workers [94] to study the role of these phytohormones in coffee leaf rust. The method showed good linearity over the range 0.042–0.33 µg g−1 for JA and ABA and 0.042–1.67 µg g−1 for SA, with LOD of 0.010 µg g−1 FW obtained for all phytohormones analysed [94] (table 1).
(b). CE-MS
Capillary electrophoresis coupled to mass spectrometry (CE-MS) is a powerful analytical technique suitable for the separation of highly polar and charged low-molecular-weight metabolites. Briefly, metabolites are first separated according to their charge-to-size ratio and then selectively detected and quantified in the mass spectrometer by their specific mass-to-charge ratio (m/z). The major advantages associated with CE-MS are the non-extensive sample pretreatment procedures that require very low organic solvent consumption, and the efficient and fast separations with low LOD [45,46,109–111].
On the other hand, the CE-ESI-MS interface is more complex than that of conventional LC-ESI-MS because it operates at very low flow rates and requires thorough optimization of several parameters (e.g. capillary alignment, electrical contact for electrophoretic separation and nebulizer gas pressure) as well as MS-compatible background electrolytes in the running buffer to achieve a proper MS signal. For all these reasons, CE-ESI-MS is still considered a reasonably demanding technique that requires expert hands [112].
(i). Primary metabolites
Several CE-MS methodologies have been developed for the identification and quantification of a wide range of primary metabolites involved in different metabolic pathways (e.g. glycolysis, tricarboxylic acid cycle, pentose phosphate pathway, photorespiration and amino acid biosynthesis) (table 2).
Table 2.
Details of the CE-MS-based methods for quantitative plant metabolomics referenced in §4b.
| plant species | metabolites | MS-based method | method parameters | refs |
|---|---|---|---|---|
| primary metabolites | ||||
| Oryza sativa leaves | amino acids, amines, purine bases, organic acids, sugars and sugar phosphates, nucleotides, coenzymes | CE-MS | RSD migration times 0.1–0.9%RSD peak areas 5.1–73.2% | [113] |
| Catharanthus roseus cell cultures | sugar phosphates, organic acids, nucleotides and coenzyme A | CE-QTRAP-MS | LOD = 0.04–8.8 µmol l−1recoveries 70–100% | [114] |
| Arabidopsis thaliana seedlings | rehalose-6-phosphate | CE-TOF-MS | LOQ = 93 nM;33 pmol g−1 FWlinearity range 80 nM–8 µMrecoveries 92–102% | [115] |
| Nicotiana tabacum leaves | sugar phosphates, amino and organic acids, coenzymes and nucleotides | CE-TOF-MS | linearity range 0.02–1000 µg ml−1 | [116] |
| phytohormones | ||||
| Oryza sativa leaves | ABA, SA, GA, JA, IAA and indole-3-butyric acid (IBA) | CE-TOF-MS | linearity range 1.3–3000 ng ml−1LOD = 0.34–4.59 ng ml−1LOQ from 1.12 to 15.3 ng ml−1recoveries 84.6–112.2% | [117] |
In 2004, Sato and co-workers [113] developed an interesting CE-based platform composed of three CE-MS and one CE coupled to a diode array detector (DAD) [113]. This parallel system allowed the identification of 80 metabolites in Oryza sativa samples grouped in different compound classes ranging from amino acids, amines and purine bases (group A), organic acids and sugar phosphates (group B), nucleotides and coenzymes (group C), and sugars (group D) (table 2) [113]. All metabolites were analysed with previously validated methods reported in the literature; groups A, B and C were analysed with CE-MS [118–120] while group D was analysed with CE-DAD [121] (table 2). Therefore, in that parallel study, only the inter-day precision was assessed in rice matrices, with RSD obtained for migration times and peak areas ranging from 0.1% to 0.9% and from 5.1% to 73.2%, respectively [113].
Anionic metabolites from Catharanthus roseus cell cultures were identified and quantified with CE-QqQ-MS/MS by Harada and co-workers [114]. With specific MRM transitions, the authors could determine a total of 53 metabolites, including sugar phosphates, organic acids, nucleotides and coenzyme A. The method exhibited good linearity for almost all metabolites, with LODs that ranged from 0.04 to 8.8 µmol l−1. Intra-day precision RSD of peak areas and migration times were below 14% and 0.5%, respectively. The analytical recoveries ranged from 70% to 100%, except for those metabolites with higher molecular weight (e.g. coenzyme A) as described by the authors [114] (table 2).
Delatte and co-workers [115] developed and validated a CE-TOF-MS method for the sensitive target analysis of the low-abundance T6P in extracts of A. thaliana seedlings. This method showed good linearity over the range 80 nM to 8 µM in both aqueous standards and plant matrix. Furthermore, inter-day precision RSD of peak areas and migration times were below 4% and 6%, respectively. The lowest level of endogenous T6P measured with that method was determined to be 93 nM, which corresponds to a LOQ of 33 pmol g−1 FW [115] (table 2).
More recently, Zhao and co-workers [116] developed a CE-TOF-MS metabolite profiling method for the quantitative analysis of 154 polar metabolites in mature leaves of five tobacco accessions, including sugar phosphates, amino and organic acids, coenzymes and nucleotides [116]. The method was validated and showed good linearity over the range 0.02–1000 µg ml−1, with intra-day precision RSD of peak areas ranging from 4.7% to 8.5% and inter-day precision from 5% to 10.2% [116] (table 2).
(ii). Phytohormones
To the best of our knowledge, only one truly quantitative CE-MS method has been developed for the determination of phytohormones in plant extracts [117] (table 2). In that study, sensitive quantification of acidic phytohormones in O. sativa with CE-TOF-MS could only be achieved using a newly synthesized mass probe, namely 3-bromoacetonyltrimethylammonium bromide (BTA), followed by extensive optimization of several CE-MS parameters [117]. The use of BTA improved the ionization efficiency of the studied phytohormones and contributed to the excellent TOF-MS signal obtained. The authors were able to successfully identify and quantify 15 acidic phytohormones in rice, including ABA, SA, GA, JA, IAA and indole-3-butyric acid (IBA). The method exhibited good linearity over the range 1.3–3000 ng ml−1. Method precision was also assessed through intra- and inter-day precision RSD of peak areas below 6.7% and 9.9%, respectively [117]. LOD ranged from 0.34 to 4.59 ng ml−1 and LOQ from 1.12 to 15.3 ng ml−1. The analytical recoveries of the metabolites were also determined, and ranged between 84.6% and 112.2% [117] (table 2).
(c). GC-MS
Gas chromatography coupled to mass spectrometry (GC-MS) has established itself as one of the most important analytical techniques in plant metabolomics studies [1,43,44,67,122–124]. GC-MS methods have an advantage over LC-MS methods because they are able to profile a wide range of metabolites with different physico-chemical properties (e.g. carbohydrates, amino and organic acids) in a single plant extract, while providing structural information of the detected metabolites. In addition, GC-MS is a much more highly reproducible technique than LC-MS, due to the electron ionization (EI) method usually employed, in which gas-phase molecules interact with kinetically activated electrons at an accepted average standard energy of 70 eV [43,44,125]. However, GC-MS is only capable of analysing volatile and thermally stable metabolites and requires chemical derivatization to chemically modify non-volatile compounds (e.g. most primary metabolites) to produce volatile derivatives [43,44]. The derivatization protocol for GC-MS plant metabolomics studies is well established and includes two chemical reactions: methoxyamination followed by silylation [1,5,7,43,44,126]. Nevertheless, some thermolabile metabolites (e.g. sugar phosphates) as well as metabolites that do not become volatile even after derivatization (e.g. large oligosaccharides) are not amenable to be analysed with GC-MS, and thereby specific target approaches based on LC-MS are the best choice for their identification and quantification [125,127] (table 1).
GC-Q-MS provides sensitive and reliable analysis with an affordable cost; however, it operates at slow scan rates and exhibits low mass accuracy. GC-TOF-MS technology offers high mass accuracy, high duty cycles and fast acquisition times. Moreover, it allows accurate deconvolution of overlapping peaks such as those typically found in plant extracts [7,128]. Given the increased interest within the metabolomics community in obtaining accurate quantitative metabolite data, the use of a QqQ mass analyser coupled to GC has been recently explored in plant metabolomics [129]. This instrument configuration can simultaneously monitor a large number of MS/MS transitions, which enhances the selectivity and sensitivity of the GC-MS analysis, which constitutes a significant improvement for quantitative GC-MS metabolite profiling [129].
In GC-MS metabolite profiling approaches, metabolites are frequently quantified using relative quantification; the determination of the relative response ratio is calculated using the metabolite peak area divided by both the peak area of the internal standard (e.g. unlabelled ribitol and/or isotopically labelled 13C-sorbitol) and the sample fresh/dry weight. Absolute quantification can be obtained if calibration curves of specific metabolites are included in the GC-MS run.
(i). Primary metabolites
A wide range of GC-MS-based methods have been employed in plant metabolomics studies for the analysis of several primary metabolites in different plant tissues (table 3). In the early 2000s, Roessner and co-workers [122] pioneered the use of GC-Q-MS as a metabolite profiling platform for the quantitative and qualitative determination within a single chromatographic run of more than 150 metabolites in wild-type developing Solanum tuberosum (potato) tubers and tubers of transgenic potato plants [122]. Absolute quantification was obtained for 33 primary metabolites in wild-type developing potato tubers, including a range of amino acids, organic acids, sugars and sugar alcohols. Within these classes, the absolute levels found were 0.15–5.62 µmol g−1 FW for amino acids, 0.15–18.86 µmol g−1 FW for organic acids and 0.01–25.91 µmol g−1 FW for sugars and sugar alcohols [122] (table 3).
Table 3.
Details of the GC-MS-based methods for quantitative plant metabolomics referenced in §4c.
| plant species | metabolites | MS-based method | method parameters | refs |
|---|---|---|---|---|
| primary metabolites | ||||
| Solanum tuberosum tubers | 33 primary metabolites | GC-Q-MS | recoveries 70–140% | [122] |
| Lycopersicon esculentum leaf and fruit tissues | 32 primary metabolites | GC-Q-MS | recoveries 85–120% | [130] |
| Cicer arietinum (L.) flower and pod tissues | 48 primary metabolites | GC-QqQ-MS/MS | linearity range 0.625–320 µM | [129] |
| phytohormones | ||||
| Nicotiana tabacum rootsArabidopsis thaliana seedling | auxins, abscisic acid, cytokinins, jasmonic acid, salicylic acid | GC-Q-MS | LOD = 0.01–0.9 pmol | [131] |
| Zea mays seedsNicotiana tabacum seedsArachis hypogaea seeds | jasmonic acid, salicylic acid | GC-QIT-MS | LOD = 500 fgrecoveries 70–100% | [132] |
| Nicotiana tabacum rootsArabidopsis thaliana seedlingLycopersicon esculentum | auxins, abscisic acid, jasmonic acid, salicylic acid, phytotoxin (coronatine) | GC-Q-MS | linearity range 0–150 ngrecoveries >50% | [133] |
| Medicago truncatula leaves | auxins, abscisic acid, cytokinins, jasmonic acid, salicylic acid | GC-Q-MS | linearity range 0–150 ngLOD = 2–10 ng ml−1LOQ = 8.9–89.2 ng µl−1recoveries 55.6–109.9% | [134] |
The GC-Q-MS method optimized for potato tubers was also applied by the same authors to further study potato tuber metabolism in a metabolite profiling comprehensive assessment of four independent potato genotypes characterized by altered sucrose metabolism [123], and later, in combination with spectrophotometric and LC chromatographic assays, in a comprehensive study of primary metabolism on Lycopersicon esculentum (tomato) leaf and developing fruit tissues [130]. In that study, the application of combined platforms allowed the identification of over 70 primary metabolites that were used for the characterization of the metabolite composition of developing tomato fruit. Of these, the absolute quantification of 32 primary metabolites (e.g. amino and organic acids, sugars and sugar alcohols) were obtained in tomato leaf and green and red fruit tissue using calibration curves as previously described by Roessner et al. [122]. Absolute levels for amino acids ranged from 0.002 to 2.32 µmol g−1 FW in tomato leaves, 0.03 to 82.83 µmol g−1 FW in tomato green fruit and 0.07 to 201.3 µmol g−1 FW in tomato red fruit; those for organic acids ranged from 0.01 to 19.48 µmol g−1 FW in tomato leaves, 0.18 to 3.17 µmol g−1 FW in tomato green fruit and 0.03 to 0.17 µmol g−1 FW in tomato red fruit; and those for sugars and sugar alcohols ranged from 0.03 to 32.05 µmol g−1 FW in tomato leaves, 0.10 to 34.22 µmol g−1 FW in tomato green fruit and 0.28 to 33.33 µmol g−1 FW in tomato red fruit [130] (table 3).
A novel approach that used a GC-QqQ-MS method has been recently developed, validated and implemented by Dias and co-workers [129] for the quantitative profiling of 48 primary metabolites that ranged from sugars and sugar phosphates to organic acids in two chickpea cultivars with contrasting responses to salt stress [129] (table 3). In that study, highly selective MRM transitions and collision energy parameters for each metabolite were optimized and the GC-QqQ-MS method fully validated in terms of method precision and accuracy, linearity, LOQ and analytical recoveries. The method exhibited good linearity over the range 0.625–320 µM for organic acids and sugars [129] (table 3).
As we have seen from these pioneering examples, despite GC-MS metabolite profiling being used to determine the absolute levels of some primary metabolites after the establishment of appropriate calibration curves of authentic standards (around 30 is acceptable), this approach is most frequently used in plant metabolomics studies as a tool to determine the steady-state relative levels of all measurable metabolites in plant extracts. GC-MS metabolite profiling has therefore been widely applied to answer interesting biological questions in the field of plant environment interactions [95,135–144] to name just a few.
(ii). Phytohormones
GC-MS has long been recognized to be an important quantitative tool for the determination of low-abundance phytohormones typically found in plant tissues. Moreover, significant improvements have been made to optimize the sample extraction procedures, and, most importantly, to establish a derivatization step that can be applied to a wide range of phytohormones [107,108]. To date, vapour-phase extraction (VPE) is considered one of the most robust extraction methods, while derivatization protocols that include silylating agents have led to a broad coverage of phytohormones with GC-MS methods [131–133,145–147]. However, the use of the alkylating agent methyl chloroformate (MCF) has proved to be a good alternative mainly because it is not affected by the presence of water in the reaction bulk [148].
A number of GC-MS methods have been described for the analysis of phytohormones. One relevant study was performed by Müller and co-workers [145], who developed a GC-QIT-MS method for the quantification of IAA, ABA, JA, 12-oxo-phytodienoic acid (OPDA) and SA in A. thaliana plant tissues to create a distribution map of these phytohormones within organs at the whole-plant level [145].
Later, Birkemeyer and co-workers tested and compared different derivatization protocols for the GC-Q-MS quantification of several phytohormones [131]. The authors concluded that silylation with N-methyl-N-(tert-butyldimethylsilyl)trifluoroacetamide (MTBSTFA) was the most comprehensive derivatization protocol for phytohormone coverage, prior to GC-Q-MS analysis [131]. The optimized methodology was applied for the identification and quantification of IAA, JA, SA, ABA and two CKs (meta-topolin, trans-zeatin) in tobacco roots and seedlings of A. thaliana. In that study, LODs in the 0.01–0.9 pmol range were obtained for all target phytohormones [131] (table 3).
In the same year, Engelberth et al. [132] developed a sensitive and reproducible GC-QIT-MS method for the quantification of JA and SA in different plant tissues (e.g. Zea mays, N. tabacum, Arachis hypogaea) using a VPE and a methylation derivatization protocol. The analytical method exhibited good linearity over the range 5–1000 ng and analytical recoveries between 70% and 90% for SA and between 90% and 100% for JA [132]. According to the authors, the LOD of 500 fg indicated that the method can be applied to the quantification of both SA and JA using small amounts of plant material (5–400 mg FW) [132] (table 3).
Using a similar approach, Schmelz and co-workers [133] determined simultaneously the levels of SA, JA, IAA and ABA as well as the levels of a phytotoxin (coronatine) in different plant systems (e.g. A. thaliana, Z. mays, N. tabacum, L. esculentum) subjected to different environmental stresses (e.g. biotic and abiotic stress factors). In that study, the GC-Q-MS method showed good linearity over the range 0–150 ng, and analytical recoveries above 50% for the targeted phytohormones [133] (table 3).
Recently, Rawlinson and co-workers [134] described the development and validation of a GC-Q-MS method for the simultaneous quantification of five phytohormones, including ABA, azelaic acid (AZ), IAA, JA and SA, in the leaves of the model legume M. truncatula [134]. The method showed good linearity over four orders of magnitude for all target phytohormones, ranging from 2 to 50 ng ml−1. The LODs were also determined and were between 2 and 10 ng ml−1, and LOQs ranged from 8.9 to 89.2 ng µl−1 [134] (table 3).
5. Conclusion/perspectives
As described by the numerous examples in this review, undoubtedly, MS has proven to be a valuable quantitative tool in modern plant metabolomics studies due to its high sensitivity and selectivity. GC-MS metabolite profiling approaches have the advantage over LC-MS and CE-MS of a relatively broad coverage of compound classes, and interest in applying it will continue to grow in the field of plant metabolite responses to various genetic and/or environmental perturbations (abiotic/biotic stress factors). Despite the rapid advances in MS technology (i.e. mass resolution, mass accuracy, mass range and sensitivity), currently, no single analytical MS platform can cover the whole-plant metabolome, and our ability to identify and quantify metabolites in comprehensive plant metabolomics experiments still depends on the dynamic range of commercially available MS systems. Notably, improvements in sensitivity are always of great interest because sensitivity determines the LOD, which is particularly important for the quantification of low-abundance metabolites (e.g. T6P, phytohormones).
Competing interests
We declare we have no competing interests.
Funding
This work was supported by Fundação para a Ciência e a Tecnologia (FCT), FCT Investigator Programme (IF/00376/2012/CP0165/CT0003), Portugal. Funding of the ITQB NOVA research unit GREEN-it ‘Bioresources for sustainability’ (UID/Multi/04551/2013) is also gratefully acknowledged. T.F.J. acknowledges FCT for the PhD fellowship (PD/BD/113475/2015) and the ITQB NOVA International PhD Programme ‘Plants for Life’ (PD/00035/2013). A.T.M. acknowledges the FCT Investigator Programme for the MSc fellowship (031/BI/2015).
References
- 1.Fiehn O, Kopka J, Dormann P, Altmann T, Trethewey RN, Willmitzer L. 2000. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 18, 1157–1161. ( 10.1038/81137) [DOI] [PubMed] [Google Scholar]
- 2.Fiehn O. 2002. Metabolomics—the link between genotypes and phenotypes. Plant Mol. Biol. 48, 155–171. ( 10.1007/978-94-010-0448-0_11) [DOI] [PubMed] [Google Scholar]
- 3.Fernie AR. 2007. The future of metabolic phytochemistry: larger numbers of metabolites, higher resolution, greater understanding. Phytochemistry 68, 2861–2880. ( 10.1016/j.phytochem.2007.07.010) [DOI] [PubMed] [Google Scholar]
- 4.Saito K, Matsuda F. 2010. Metabolomics for functional genomics, systems biology, and biotechnology. Annu. Rev. Plant Biol. 61, 463–489. ( 10.1146/annurev.arplant.043008.092035) [DOI] [PubMed] [Google Scholar]
- 5.Jorge TF, Rodrigues JA, Caldana C, Schmidt R, van Dongen JT, Thomas-Oates J, António C. 2016. Mass spectrometry-based plant metabolomics: metabolite responses to abiotic stress. Mass Spectrom. Rev. 35, 620–649. ( 10.1002/mas.21449) [DOI] [PubMed] [Google Scholar]
- 6.Kim HK, Choi YH, Verpoorte R. 2011. NMR-based plant metabolomics: where do we stand, where do we go? Trends Biotechnol. 29, 267–275. ( 10.1016/j.tibtech.2011.02.001) [DOI] [PubMed] [Google Scholar]
- 7.Lisec J, Schauer N, Kopka J, Willmitzer L, Fernie AR. 2006. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 1, 387–396. ( 10.1038/nprot.2006.59) [DOI] [PubMed] [Google Scholar]
- 8.De Vos RC, Moco S, Lommen A, Keurentjes JJ, Bino RJ, Hall RD. 2007. Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2, 778–791. ( 10.1038/nprot.2007.95) [DOI] [PubMed] [Google Scholar]
- 9.Kim HK, Choi YH, Verpoorte R. 2010. NMR-based metabolomic analysis of plants. Nat. Protoc. 5, 536–549. ( 10.1038/nprot.2009.237) [DOI] [PubMed] [Google Scholar]
- 10.Tohge T, Fernie AR. 2010. Combining genetic diversity, informatics and metabolomics to facilitate annotation of plant gene function. Nat. Protoc. 5, 1210–1227. ( 10.1038/nprot.2010.82) [DOI] [PubMed] [Google Scholar]
- 11.Fiehn O, et al. 2007. The Metabolomics Standards Initiative (MSI). Metabolomics 3, 175–178. ( 10.1007/s11306-007-0070-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiehn O, et al. 2007. Minimum reporting standards for plant biology context information in metabolomic studies. Metabolomics 3, 195–201. ( 10.1007/s11306-007-0068-0) [DOI] [Google Scholar]
- 13.Sumner LW, et al. 2007. Proposed minimum reporting standards for chemical analysis. (Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI)) Metabolomics 3, 211–221. ( 10.1007/s11306-007-0082-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernie AR, et al. 2011. Recommendations for reporting metabolite data. Plant Cell 23, 2477–2482. ( 10.1105/tpc.111.086272) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salek RM, Steinbeck C, Viant MR, Goodacre R, Dunn WB. 2013. The role of reporting standards for metabolite annotation and identification in metabolomic studies. Giga-science 2, 13–15. ( 10.1186/2047-217X-2-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Creek DJ, et al. 2014. Metabolite identification: are you sure? And how do your peers gauge your confidence? Metabolomics 10, 350–353. ( 10.1007/s11306-014-0656-8) [DOI] [Google Scholar]
- 17.Sumner LW, Lei Z, Nikolau BJ, Saito K, Roessner U, Trengove R. 2014. Proposed quantitative and alphanumeric metabolite identification metrics. Metabolomics 10, 1047–1049. ( 10.1007/s11306-014-0739-6) [DOI] [Google Scholar]
- 18.Salek RM, et al. 2015. Coordination of Standards in MetabOlomicS (COSMOS): facilitating integrated metabolomics data access. Metabolomics 11, 1587–1597. ( 10.1007/s11306-015-0810-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rocca-Serra P, et al. 2016. Data standards can boost metabolomics research, and if there is a will, there is a way. Metabolomics 12, 1–13. ( 10.1007/s11306-015-0879-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirayama T, Shinozaki K. 2010. Research on plant abiotic stress response in the post-genome era: past, present and future. Plant J. 61, 1041–1052. ( 10.1111/j.1365-313X.2010.04124.x) [DOI] [PubMed] [Google Scholar]
- 21.Krasensky J, Jonak C. 2012. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 63, 1593–1608. ( 10.1093/jxb/err460) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Obata T, Fernie AR. 2012. The use of metabolomics to dissect plant responses to abiotic stresses. Cell Mol. Life Sci. 69, 3225–3243. ( 10.1007/s00018-012-1091-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bari R, Jones JDG. 2009. Role of plant hormones in plant defense responses. Plant Mol. Biol. 69, 473–488. ( 10.1007/s11103-008-9435-0) [DOI] [PubMed] [Google Scholar]
- 24.Pieterse CMJ, Leon-Reyes A, Van der Ent S, Van Wees SCM. 2009. Networking by small-molecule hormones in plant immunity. Nat. Chem. Biol. 5, 308–316. ( 10.1038/nchembio.164) [DOI] [PubMed] [Google Scholar]
- 25.Robert-Seilaniantz A, Grant M, Jones JDG. 2011. Hormone crosstalk in plant disease and defense: more than just jasmonate–salicylate antagonism. Annu. Rev. Phytopathol. 49, 317–343. ( 10.1146/annurev-phyto-073009-114447) [DOI] [PubMed] [Google Scholar]
- 26.Erb M, Meldau S, Howe GA. 2012. Role of phytohormones in insect-specific plant reactions. Trends Plant Sci. 17, 250–259. ( 10.1016/j.tplants.2012.01.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giron D, Frago E, Glevarec G, Pieterse CMJ, Dicke M. 2013. Cytokinins as key regulators in plant–microbe–insect interactions: connecting plant growth and defence. Funct. Ecol. 27, 599–609. ( 10.1111/1365-2435.12042) [DOI] [Google Scholar]
- 28.Griffin JL, et al. 2007. Standard reporting requirements for biological samples in metabolomics experiments: mammalian/in vivo experiments. Metabolomics 3, 179–188. ( 10.1007/s11306-007-0077-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morrison N, et al. 2007. Standard reporting requirements for biological samples in metabolomics experiments: environmental context. Metabolomics 3, 203–210. ( 10.1007/s11306-007-0067-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Werf MJ, et al. 2007. Standard reporting requirements for biological samples in metabolomics experiments: microbial and in vitro biology experiments. Metabolomics 3, 189–194. ( 10.1007/s11306-007-0080-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibon Y, Rolin D. 2013. Aspects of experimental design for plant metabolomics experiments and guidelines for growth of plant material. In Plant metabolomics: methods and protocols (eds NW Hardy, RD Hall), Methods in Molecular Biology, vol. 860, pp. 13–30. New York, NY: Humana Press ( 10.1007/978-1-61779-594-7_2) [DOI] [PubMed] [Google Scholar]
- 32.Gullberg J, Jonsson P, Nordström A, Sjöström M, Moritz T. 2004. Design of experiments: an efficient strategy to identify factors influencing extraction and derivatization of Arabidopsis thaliana samples in metabolomic studies with gas chromatography/mass spectrometry. Anal. Biochem. 331, 283–295. ( 10.1016/j.ab.2004.04.037) [DOI] [PubMed] [Google Scholar]
- 33.Weiner H, Stitt M, Heldt HW. 1987. Subcellular compartmentation of pyrophosphate and alkaline pyrophosphate in leaves. Biochim. Biophys. Acta 893, 13–21. ( 10.1016/0005-2728(87)90143-5) [DOI] [Google Scholar]
- 34.Jelitto T, Sonnewald U, Willmitzer L, Hajirezeai M, Stitt M. 1992. Inorganic pyrophosphate content and metabolites in potato and tobacco plants expressing E. coli pyrophosphatase in their cytosol. Planta 188, 238–244. ( 10.1007/BF00216819) [DOI] [PubMed] [Google Scholar]
- 35.De Koning W, van Dam K. 1992. A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal. Biochem. 204, 118–123. ( 10.1016/0003-2697(92)90149-2) [DOI] [PubMed] [Google Scholar]
- 36.Stitt M, Fernie AR. 2003. From measurements of metabolites to metabolomics: an ‘on the fly’ perspective illustrated by recent studies of carbon–nitrogen interactions. Curr. Opinion Biotechnol. 14, 136–144. ( 10.1016/S0958-1669(03)00023-5) [DOI] [PubMed] [Google Scholar]
- 37.Kim HK, Verpoorte R. 2010. Sample preparation for plant metabolomics. Phytochem. Anal. 21, 4–13. ( 10.1002/pca.1188) [DOI] [PubMed] [Google Scholar]
- 38.Roessner U, Dias DA. 2013. Plant tissue extraction for metabolomics. In Metabolomics tools for natural product discovery: methods and protocols (eds U Roessner, DA Dias), Methods in Molecular Biology, vol. 1055, pp. 21–28. New York, NY: Humana Press ( 10.1007/978-1-62703-577-4_2) [DOI] [PubMed] [Google Scholar]
- 39.Rabinowitz JD, Kimball E. 2007. Acidic acetonitrile for cellular metabolome extraction from Escherichia coli. Anal. Chem. 79, 6167–6173. ( 10.1021/ac070470c) [DOI] [PubMed] [Google Scholar]
- 40.Delatte TL, Selman MH, Schluepmann H, Somsen GW, Smeekens SC, de Jong GJ. 2009. Determination of trehalose-6-phosphate in Arabidopsis seedlings by successive extractions followed by anion exchange chromatography–mass spectrometry. Anal. Biochem. 389, 12–17. ( 10.1016/j.ab.2009.03.003) [DOI] [PubMed] [Google Scholar]
- 41.Raterink R-J, Lindenburg PW, Vreeken RJ, Ramautar R, Hankemeier T. 2014. Recent developments in sample-pretreatment techniques for mass spectrometry-based metabolomics. Trends Anal. Chem. 61, 157–167. ( 10.1016/j.trac.2014.06.003) [DOI] [Google Scholar]
- 42.Cajka T, Fiehn O. 2016. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Anal. Chem. 88, 524–545. ( 10.1021/acs.analchem.5b04491) [DOI] [PubMed] [Google Scholar]
- 43.Kopka J. 2006. Current challenges and developments in GC-MS based metabolite profiling technology. J. Biotechnol. 124, 312–322. ( 10.1016/j.jbiotec.2005.12.012) [DOI] [PubMed] [Google Scholar]
- 44.Kopka J. 2006. Gas chromatography mass spectrometry. In Biotechnology in agriculture and forestry: plant metabolomics (eds Saito K, Dixon RA, Willmitzer L), pp. 3–20. Berlin, Germany: Springer. [Google Scholar]
- 45.Hirayama A, Wakayama M, Soga T. 2014. Metabolome analysis based on capillary electrophoresis–mass spectrometry. Trends Anal. Chem. 61, 215–222. ( 10.1016/j.trac.2014.05.005) [DOI] [Google Scholar]
- 46.Ramautar R, Somsen GW, de Jong GJ. 2015. CE-MS for metabolomics: developments and applications in the period 2012–2014. Electrophoresis 36, 212–224. ( 10.1002/elps.201400388) [DOI] [PubMed] [Google Scholar]
- 47.Krull IS, Swartz M. 1999. Analytical method development and validation for the academic researcher. Anal. Lett. 32, 1067–1080. ( 10.1080/00032719908542878) [DOI] [Google Scholar]
- 48.González O, Blanco ME, Iriarte G, Bartolomé L, Maguregui MI, Alonso RM. 2014. Bioanalytical chromatographic method validation according to current regulations, with a special focus on the non-well defined parameters limit of quantification, robustness and matrix effect. J. Chromatogr. A 1353, 10–27. ( 10.1016/j.chroma.2014.03.077) [DOI] [PubMed] [Google Scholar]
- 49.Ruiz-Angel MJ, García-Alvarez-Coque MC, Berthod A, Carda-Broch S. 2014. Are analysts doing method validation in liquid chromatography? J. Chromatogr. A 1353, 2–9. ( 10.1016/j.chroma.2014.05.052) [DOI] [PubMed] [Google Scholar]
- 50.Loco JV, Elskens M, Croux C, Beernaert H. 2002. Linearity of calibration curves: use and misuse of the correlation coefficient. Accred. Qual. Assur. 7, 281–285. ( 10.1007/s00769-002-0487-6) [DOI] [Google Scholar]
- 51.Raposo F. 2016. Evaluation of analytical calibration based on least squares linear regression for instrumental techniques: a tutorial review. Trends Anal. Chem. 77, 167–185. ( 10.1016/j.trac.2015.12.006) [DOI] [Google Scholar]
- 52.Funk W, Dammann V, Donnevert G, Iannelli S, Iannelli E. 2007. Phase I: establishing a new analytical process. In Quality assurance in analytical chemistry: applications in environmental food, and materials analysis, biotechnology, and medical engineering (eds Funk W, Dammann V, Donnevert G), pp. 9–55. Weinheim, Germany: Wiley-VCH. [Google Scholar]
- 53.Hartmann C, Smeyers-Verbeke J, Massart DL, McDowall RD. 1998. Validation of bioanalytical chromatographic methods. J. Pharm. Biomed. Anal. 17, 193–218. ( 10.1016/S0731-7085(97)00198-2) [DOI] [PubMed] [Google Scholar]
- 54.Araujo P. 2009. Key aspects of analytical method validation and linearity evaluation. J. Chromatogr. B 877, 2224–2234. ( 10.1016/j.jchromb.2008.09.030) [DOI] [PubMed] [Google Scholar]
- 55.Stöckl D, D'Hondt H, Thienpont LM. 2009. Method validation across the disciplines—critical investigation of major validation criteria and associated experimental protocols. J. Chromatogr. B 877, 2180–2190. ( 10.1016/j.jchromb.2008.12.056) [DOI] [PubMed] [Google Scholar]
- 56.Koek MM, Jellema RH, van der Greef J, Tas AC, Hankemeier T. 2011. Quantitative metabolomics based on gas chromatography mass spectrometry: status and perspectives. Metabolomics 7, 307–328. ( 10.1007/s11306-010-0254-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Naz S, Vallejo M, García A, Barbas C. 2014. Method validation strategies involved in non-targeted metabolomics. J. Chromatogr. A 1353, 99–105. ( 10.1016/j.chroma.2014.04.071) [DOI] [PubMed] [Google Scholar]
- 58.Aboul-Enein HY. 2000. Selectivity versus specificity in chromatographic analytical methods. Accred. Qual. Assur. 5, 180–181. ( 10.1007/s007690050440) [DOI] [Google Scholar]
- 59.Danzer K. 2001. Selectivity and specificity in analytical chemistry. General considerations and attempt of a definition and quantification. Fresenius J. Anal. Chem. 369, 397–402. ( 10.1007/s002160000684) [DOI] [PubMed] [Google Scholar]
- 60.Burton L, Ivosev G, Tate S, Impey G, Wingate J, Bonner R. 2008. Instrumental and experimental effects in LC-MS-based metabolomics. J. Chromatogr. B 871, 227–235. ( 10.1016/j.jchromb.2008) [DOI] [PubMed] [Google Scholar]
- 61.Choi BK, Hercules DM, Gusev AI. 2001. Effect of liquid chromatography separation of complex matrices on liquid chromatography–tandem mass spectrometry signal suppression. J. Chromatogr. A 907, 337–342. ( 10.1016/S0021-9673(00)01052-9) [DOI] [PubMed] [Google Scholar]
- 62.Matuszewski BK, Constanzer ML, Chavez-Eng CM. 2003. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 75, 3019–3030. ( 10.1021/ac020361s) [DOI] [PubMed] [Google Scholar]
- 63.Trufelli H, Palma P, Famiglini G, Cappiello A. 2011. An overview of matrix effects in liquid chromatography–mass spectrometry. Mass Spectrom. Rev. 30, 491–509. ( 10.1002/mas.20298) [DOI] [PubMed] [Google Scholar]
- 64.Bonfiglio R, King RC, Olah TV, Merkle K. 1999. The effects of sample preparation methods on the variability of the electrospray ionization response for model drug compounds. Rapid Commun. Mass. Spectrom. 13, 1175–1185. ( 10.1002/(SICI)1097-0231(19990630)13:12%3C1175::AID-RCM639%3E3.0.CO;2-0) [DOI] [PubMed] [Google Scholar]
- 65.Buhrman DL, Price PI, Rudewiczcor PJ. 1996. Quantitation of SR 27417 in human plasma using electrospray liquid chromatography–tandem mass spectrometry: a study of ion suppression. J. Am. Soc. Mass Spectrom. 7, 1099–1105. ( 10.1016/S1044-0305(96)00072-4) [DOI] [PubMed] [Google Scholar]
- 66.Matuszewski BK. 2006. Standard line slopes as a measure of a relative matrix effect in quantitative HPLC-MS bioanalysis. J. Chromatogr. B 830, 293–300. ( 10.1016/j.jchromb.2005.11.009) [DOI] [PubMed] [Google Scholar]
- 67.Fernie A. 2003. Metabolome characterisation in plant system analysis. Funct. Plant Biol. 30, 111–120. ( 10.1071/FP02163) [DOI] [PubMed] [Google Scholar]
- 68.Saito K, Yonekura-Sakakibara K, Nakabayashi R, Higashi Y, Yamazaki M, Tohge T, Fernie AR. 2013. The flavonoid biosynthetic pathway in Arabidopsis: structural and genetic diversity. Plant Physiol. Biochem. 72, 21–34. ( 10.1016/j.plaphy.2013.02.001) [DOI] [PubMed] [Google Scholar]
- 69.Tohge T, Watanabe M, Hoefgen R, Fernie AR. 2013. The evolution of phenylpropanoid metabolism in the green lineage. Crit. Rev. Biochem. Mol. Biol. 48, 123–152. ( 10.3109/10409238.2012.758083) [DOI] [PubMed] [Google Scholar]
- 70.Sumner LW, Lei Z, Nikolau BJ, Saito K. 2015. Modern plant metabolomics: advanced natural product gene discoveries, improved technologies, and future prospects. Nat. Prod. Rep. 32, 212–229. ( 10.1039/c4np00072b) [DOI] [PubMed] [Google Scholar]
- 71.Bou Khalil M, Hou W, Zhou H, Elisma F, Swayne LA, Blanchard AP, Yao Z, Bennett SAL, Figeys D. 2010. Lipidomics era: accomplishments and challenges. Mass Spectrom. Rev. 29, 877–929. ( 10.1002/mas.20294) [DOI] [PubMed] [Google Scholar]
- 72.Giavalisco P, et al. 2011. Elemental formula annotation of polar and lipophilic metabolites using 13C, 15N and 34S isotope labelling, in combination with high-resolution mass spectrometry. Plant J. 68, 364–376. ( 10.1111/j.1365-313X.2011.04682.x) [DOI] [PubMed] [Google Scholar]
- 73.Han X, Yang K, Gross RW. 2012. Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrom. Rev. 31, 134–178. ( 10.1002/mas.20342) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Okazaki Y, Kamide Y, Hirai MY, Saito K. 2013. Plant lipidomics based on hydrophilic interaction chromatography coupled to ion trap time-of-flight mass spectrometry. Metabolomics 9, 121–131. ( 10.1007/s11306-011-0318-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li M, Yang L, Bai Y, Liu H. 2014. Analytical methods in lipidomics and their applications. Anal. Chem. 86, 161–175. ( 10.1021/ac403554h) [DOI] [PubMed] [Google Scholar]
- 76.Bromke MA, Hochmuth A, Tohge T, Fernie AR, Giavalisco P, Burgos A, Willmitzer L, Brotman Y. 2015. Liquid chromatography high-resolution mass spectrometry for fatty acid profiling. Plant J. 81, 529–536. ( 10.1111/tpj.12739) [DOI] [PubMed] [Google Scholar]
- 77.Yamashita M, Fenn JB. 1984. Electrospray ion source. Another variation on the free-jet theme. J. Phys. Chem. 88, 4451–4459. ( 10.1021/j150664a002) [DOI] [Google Scholar]
- 78.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. 1989. Electrospray ionization for mass spectrometry of large biomolecules. Science 246, 64–71. ( 10.1126/science.2675315) [DOI] [PubMed] [Google Scholar]
- 79.Shukla AK, Futrell JH. 2000. Tandem mass spectrometry: dissociation of ions by collisional activation. J. Mass Spectrom. 35, 1069–1090. ( 10.1002/1096-9888(200009)35:9%3C1069::AID-JMS54%3E3.0.CO;2-C) [DOI] [PubMed] [Google Scholar]
- 80.Tolstikov VV, Fiehn O. 2002. Analysis of highly polar compounds of plant origin: combination of hydrophilic interaction chromatography and electrospray ion trap mass spectrometry. Anal. Biochem. 301, 298–307. ( 10.1006/abio.2001.5513) [DOI] [PubMed] [Google Scholar]
- 81.António C, Larson T, Gilday A, Graham I, Bergström E, Thomas-Oates J. 2007. Quantification of sugars and sugar phosphates from Arabidopsis thaliana tissues using porous graphitic carbon liquid chromatography–electrospray ionization mass spectrometry. J. Chromatogr. A 1172, 170–178. ( 10.1016/j.chroma.2007.10.011) [DOI] [PubMed] [Google Scholar]
- 82.António C, Pinheiro C, Chaves MM, Ricardo CP, Ortuño MF, Thomas-Oates J. 2008. Analysis of carbohydrates in Lupinus albus stems on imposition of water deficit, using porous graphitic carbon liquid chromatography–electrospray ionization mass spectrometry. J. Chromatogr. A 1187, 111–118. ( 10.1016/j.chroma.2008.02.010) [DOI] [PubMed] [Google Scholar]
- 83.António C, Larson T, Gilday A, Graham I, Bergström E, Thomas-Oates J. 2008. Hydrophilic interaction chromatography/electrospray mass spectrometry analysis of carbohydrate-related metabolites from Arabidopsis thaliana leaf tissue. Rapid Commun. Mass. Spectrom. 22, 1399–1407. ( 10.1002/rcm.3519) [DOI] [PubMed] [Google Scholar]
- 84.Behmüller R, Forstenlehner IC, Tenhaken R, Huber CG. 2014. Quantitative HPLC-MS analysis of nucleotide sugars in plant cells following off-line SPE sample preparation. Anal. Bioanal. Chem. 406, 3229–3237. ( 10.1007/s00216-014-7746-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Warth B, Siegwart G, Lemmens M, Krska R, Adam G, Schuhmacher R. 2015. Hydrophilic interaction liquid chromatography coupled with tandem mass spectrometry for the quantification of uridine diphosphate-glucose, uridine diphosphate-glucuronic acid, deoxynivalenol and its glucoside: in-house validation and application to wheat. J. Chromatogr. A 1423, 183–189. ( 10.1016/j.chroma.2015.10.070) [DOI] [PubMed] [Google Scholar]
- 86.Ito J, et al. 2014. Analysis of plant nucleotide sugars by hydrophilic interaction liquid chromatography and tandem mass spectrometry. Anal. Biochem. 448, 14–22. ( 10.1016/j.ab.2013.11.026) [DOI] [PubMed] [Google Scholar]
- 87.Lunn JE, Feil R, Hendriks JH, Gibon Y, Morcuende R, Osuna D, Scheible WR, Carillo P, Hajirezaei MR. 2006. Sugar-induced increases in trehalose 6-phosphate are correlated with redox activation of ADP glucose pyrophosphorylase and higher rates of starch synthesis in Arabidopsis thaliana. Biochem. J. 397, 139–148. ( 10.1042/BJ20060083) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Toraño JS, Delatte TL, Schluepmann H, Smeekens SC, de Jong GJ, Somsen GW. 2012. Determination of trehalose-6-phosphate in Arabidopsis thaliana seedlings by hydrophilic-interaction liquid chromatography–mass spectrometry. Anal. Bioanal. Chem. 403, 1353–1360. ( 10.1007/s00216-012-5928-4) [DOI] [PubMed] [Google Scholar]
- 89.Yadav UP, et al. 2014. The sucrose–trehalose 6-phosphate (Tre6P) nexus: specificity and mechanisms of sucrose signaling by Tre6P. J. Exp. Bot. 65, 1051–1068. ( 10.1093/jxb/ert457) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chiwocha SD, Abrams SR, Ambrose SJ, Cutler AJ, Loewen M, Ross AR, Kermode AR. 2003. A method for profiling classes of plant hormones and their metabolites using liquid chromatography–electrospray ionization tandem mass spectrometry: an analysis of hormone regulation of thermodormancy of lettuce (Lactuca sativa L.) seeds. Plant J. 35, 405–417. ( 10.1046/j.1365-313X.2003.01800.x) [DOI] [PubMed] [Google Scholar]
- 91.Pan X, Welti R, Wang X. 2008. Simultaneous quantification of major phytohormones and related compounds in crude plant extracts by liquid chromatography–electrospray tandem mass spectrometry. Phytochemistry 69, 1773–1781. ( 10.1016/j.phytochem.2008.02.008) [DOI] [PubMed] [Google Scholar]
- 92.Izumi Y, Okazawa A, Bamba T, Kobayashi A, Fukusaki E. 2009. Development of a method for comprehensive and quantitative analysis of plant hormones by highly sensitive nanoflow liquid chromatography–electrospray ionization–ion trap mass spectrometry. Anal. Chim. Acta 648, 215–225. ( 10.1016/j.aca.2009.07.001) [DOI] [PubMed] [Google Scholar]
- 93.Liu S, Chen W, Qu L, Gai Y, Jiang X. 2013. Simultaneous determination of 24 or more acidic and alkaline phytohormones in femtomole quantities of plant tissues by high-performance liquid chromatography–electrospray ionization–ion trap mass spectrometry. Anal. Bioanal. Chem. 405, 1257–1266. ( 10.1007/s00216-012-6509-2) [DOI] [PubMed] [Google Scholar]
- 94.de Sá M, Ferreira JP, Queiroz VT, Vilas-Boas L, Silva MC, Almeida MH, Guerra-Guimarães L, Bronze MR. 2014. A liquid chromatography/electrospray ionisation tandem mass spectrometry method for the simultaneous quantification of salicylic, jasmonic and abscisic acids in Coffea arabica leaves. J. Sci. Food Agric. 94, 529–536. ( 10.1002/jsfa.6288) [DOI] [PubMed] [Google Scholar]
- 95.Gechev T, et al. 2013. Molecular mechanisms of desiccation tolerance in the resurrection glacial relic Haberlea rhodopensis. Cell. Mol. Life Sci. 70, 689–709. ( 10.1007/s00018-012-1155-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gomez LD, Gildaz A, Feil R, Lunn JE, Graham IA. 2010. AtTPS1 mediated trehalose-6-phosphate synthesis is essential for embryogenic and vegetative growth and responsiveness to ABA in germinating seeds and stomatal guard cells. Plant J. 64, 1–13. ( 10.1111/j.1365-313X.2010.04312.x) [DOI] [PubMed] [Google Scholar]
- 97.Vandesteene L, et al. 2012. Expansive evolution of the trehalose-6-phosphate phosphatase gene family in Arabidopsis thaliana. Plant Physiol. 160, 884–896. ( 10.1104/pp.112.201400) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Szecowka M, et al. 2013. Metabolic fluxes in an illuminated Arabidopsis rosette. Plant Cell 25, 694–714. ( 10.1105/tpc.112.106989) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wahl V, et al. 2013. Regulation of flowering by trehalose-6-phosphate signaling in Arabidopsis thaliana. Science 339, 704–707. ( 10.1126/science.1230406) [DOI] [PubMed] [Google Scholar]
- 100.Figueroa CM, et al. 2016. Trehalose 6-phosphate coordinates organic and amino acid metabolism with carbon availability. Plant J. 85, 410–423. ( 10.1111/tpj.13114) [DOI] [PubMed] [Google Scholar]
- 101.Delatte TL, et al. 2011. Growth arrest by trehalose-6-phosphate: an astonishing case of primary metabolite control over growth by way of the SnRK1 signaling pathway. Plant Physiol. 157, 160–174. ( 10.1104/pp.111.180422) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Martinez-Barajas E, et al. 2011. Wheat grain development is characterized by remarkable trehalose 6-phosphate accumulation pregrain filling: tissue distribution and relationship to SNF1-related protein kinase1 activity. Plant Physiol. 156, 373–381. ( 10.1104/pp.111.174524) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nunes C, et al. 2013. The trehalose 6-phosphate/SnRK1 signaling pathway primes growth recovery following relief of sink limitation. Plant Physiol. 162, 1720–1732. ( 10.1104/pp.113.220657) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Davies PJ. 1987. The plant hormones: their nature, occurrence, and functions. In Plant hormones and their role in plant growth and development (ed. Davies PJ.), pp. 1–11. Dordrecht, The Netherlands: Kluwer Academic. [Google Scholar]
- 105.Davies PJ. 2010. The plant hormones: their nature, occurrence, and function. In Plant hormones: biosynthesis, signal transduction, action! (ed. Davies PJ.), pp. 1–15. Dordrecht, The Netherlands: Springer. [Google Scholar]
- 106.Ljung K, Sandberg G, Moritz T. 2010. Methods of plant hormone analysis. In Plant hormones: biosynthesis, signal transduction, action! (ed. Davies PJ.), pp. 671–694. Dordrecht, The Netherlands: Springer. [Google Scholar]
- 107.Pan X, Wang X. 2009. Profiling of plant hormones by mass spectrometry. J. Chromatogr. B 877, 2806 ( 10.1016/j.jchromb.2009.04.024) [DOI] [PubMed] [Google Scholar]
- 108.Bai Y, Du F, Bai Y, Liu H. 2010. Determination strategies of phytohormones: recent advances. Anal. Methods 2, 1867–1873. ( 10.1039/C0AY00471E) [DOI] [Google Scholar]
- 109.Monton MRN, Soga T. 2007. Metabolome analysis by capillary electrophoresis–mass spectrometry. J. Chromatogr. A 1168, 237–246. ( 10.1016/j.chroma.2007.02.065) [DOI] [PubMed] [Google Scholar]
- 110.Ramautar R, Demirci A, de Jong GJ. 2006. Capillary electrophoresis in metabolomics. Trends Anal. Chem. 25, 455–466. ( 10.1016/j.trac.2006.02.004) [DOI] [Google Scholar]
- 111.Ramautar R, Somsen GW, de Jong GJ. 2009. CE-MS in metabolomics. Electrophoresis 30, 276–291. ( 10.1002/elps.200800512) [DOI] [PubMed] [Google Scholar]
- 112.Nilsson SL, Bylund D, Jornten-Karlsson M, Petersson P, Markides KW. 2004. A chemometric study of active parameters and their interaction effects in a nebulized sheath–liquid electrospray interface for CE-ESI-MS-based metabolomics. Electrophoresis 25, 2100–2107. ( 10.1002/elps.200305937) [DOI] [PubMed] [Google Scholar]
- 113.Sato S, Soga T, Nishioka T, Tomita M. 2004. Simultaneous determination of the main metabolites in rice leaves using capillary electrophoresis mass spectrometry and capillary electrophoresis diode array detection. Plant J. 40, 151–163. ( 10.1111/j.1365-313X.2004.02187.x) [DOI] [PubMed] [Google Scholar]
- 114.Harada K, Ohyama Y, Tabushi T, Kobayashi A, Fukusaki E. 2008. Quantitative analysis of anionic metabolites for Catharanthus roseus by capillary electrophoresis using sulfonated capillary coupled with electrospray ionization–tandem mass spectrometry. J. Biosci. Bioeng. 105, 249–260. ( 10.1263/jbb.105.249) [DOI] [PubMed] [Google Scholar]
- 115.Delatte TL, Schluepmann H, Smeekens SC, de Jong GJ, Somsen GW. 2011. Capillary electrophoresis–mass spectrometry analysis of trehalose-6-phosphate in Arabidopsis thaliana seedlings. Anal. Bioanal. Chem. 400, 1137–1144. ( 10.1007/s00216-011-4837-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhao J, et al. 2014. Study of polar metabolites in tobacco from different geographical origins by using capillary electrophoresis–mass spectrometry. Metabolomics 10, 805–815. ( 10.1007/s11306-014-0631-4) [DOI] [Google Scholar]
- 117.Chen M-L, Huang Y-Q, Liu J-Q, Yuan B-F, Feng Y-Q. 2011. Highly sensitive profiling assay of acidic plant hormones using a novel mass probe by capillary electrophoresis–time of flight–mass spectrometry. J. Chromatogr. B 879, 938–944. ( 10.1016/j.jchromb.2011.03.003) [DOI] [PubMed] [Google Scholar]
- 118.Soga T, Heiger DN. 2000. Amino acid analysis by capillary electrophoresis electrospray ionization mass spectrometry. Anal. Chem. 72, 1236–1241. ( 10.1021/ac990976y) [DOI] [PubMed] [Google Scholar]
- 119.Soga T, Ueno Y, Naraoka H, Ohashi Y, Tomita M, Nishioka T. 2002. Simultaneous determination of anionic intermediates for Bacillus subtilis metabolic pathway by capillary electrophoresis electrospray ionization mass spectrometry. Anal. Chem. 74, 2233–2239. ( 10.1021/ac020064n) [DOI] [PubMed] [Google Scholar]
- 120.Soga T, Ueno Y, Naraoka H, Matsuda K, Tomita M, Nishioka T. 2002. Pressure-assisted capillary electrophoresis electrospray ionization mass spectrometry for analysis of multivalent anions. Anal. Chem. 74, 6224–6229. ( 10.1021/ac0202684) [DOI] [PubMed] [Google Scholar]
- 121.Soga T, Heiger DN. 1998. Simultaneous determination of monosaccharides in glycoproteins by capillary electrophoresis. Anal. Biochem. 261, 73–78. ( 10.1006/abio.1998.2727) [DOI] [PubMed] [Google Scholar]
- 122.Roessner U, Wagner C, Kopka J, Trethewey RN, Willmitzer L. 2000. Simultaneous analysis of metabolites in potato tuber by gas chromatography–mass spectrometry. Plant J. 23, 131–142. ( 10.1046/j.1365-313x.2000.00774.x) [DOI] [PubMed] [Google Scholar]
- 123.Roessner U, Luedemann A, Brust D, Fiehn O, Linke T, Willmitzer L, Fernie AR. 2001. Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell 13, 11–29. ( 10.1105/tpc.13.1.11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fiehn O. 2006. Metabolite profiling in Arabidopsis. In Arabidopsis protocols (eds J Salinas, JJ Sanchez-Serrano). Methods in Molecular Biology, vol. 323, pp. 439–447. Totowa, NJ: Humana Press ( 10.1385/1-59745-003-0:439) [DOI] [PubMed] [Google Scholar]
- 125.Kopka J, Fernie A, Weckwerth W, Gibon Y, Stitt M. 2004. Metabolite profiling in plant biology: platforms and destinations. Genome Biol. 5, 1–8. ( 10.1186/gb-2004-5-6-109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Moritz T, Johansson AI. 2008. Plant metabolomics. In Metabolomics, metabonomics and metabolite profiling (ed. Griffiths WJ.), pp. 254–272. London, UK: The Royal Society. [Google Scholar]
- 127.Weckwerth W. 2003. Metabolomics in systems biology. Annu. Rev. Plant Biol. 5, 669–689. ( 10.1146/annurev.arplant.54.031902.135014) [DOI] [PubMed] [Google Scholar]
- 128.Lei Z, Huhman DV, Sumner LW. 2011. Mass spectrometry strategies in metabolomics. J. Biol. Chem. 286, 25 435–25 442. ( 10.1074/jbc.R111.238691) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dias AD, Hill CB, Jayasinghe NS, Atieno J, Sutton T, Roessner U. 2015. Quantitative profiling of polar primary metabolites of two chickpea cultivars with contrasting responses to salinity. J. Chromatogr. B 100, 1–13. ( 10.1016/j.jchromb.2015.07.002) [DOI] [PubMed] [Google Scholar]
- 130.Roessner-Tunali U, Hegemann B, Lytovchenko A, Carrari F, Bruedigam C, Granot D, Fernie AR. 2003. Metabolic profiling of transgenic tomato plants overexpressing hexokinase reveals that the influence of hexose phosphorylation diminishes during fruit development. Plant Physiol. 133, 84–99. ( 10.1104/pp.103.023572) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Birkemeyer C, Kolasa A, Kopka J. 2003. Comprehensive chemical derivatization for gas chromatography–mass spectrometry-based multi-targeted profiling of the major phytohormones. J. Chromatogr. A 993, 89–102. ( 10.1016/S0021-9673(03)00356-X) [DOI] [PubMed] [Google Scholar]
- 132.Engelberth J, Schmelz EA, Alborn HT, Cardoza YJ, Huang J, Tumlinson JH. 2003. Simultaneous quantification of jasmonic acid and salicylic acid in plants by vapor phase extraction and gas chromatography–chemical ionization–mass spectrometry. Anal. Biochem. 312, 242–250. ( 10.1016/S0003-2697(02)00466-9) [DOI] [PubMed] [Google Scholar]
- 133.Schmelz EA, Engelberth J, Alborn HT, O'Donnell P, Sammons M, Toshima H, Tumlinson JH. 2003. Simultaneous analysis of phytohormones, phytotoxins, and volatile organic compounds in plants. Proc. Natl Acad. Sci. USA 100, 10 552–10 557. ( 10.1073/pnas.1633615100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rawlinson C, Kamphuis LG, Gummer JPA, Singh KB, Trengove RD. 2015. A rapid method for profiling of volatile and semi-volatile phytohormones using methyl chloroformate derivatisation and GC-MS. Metabolomics 11, 1922–1933. ( 10.1007/s11306-015-0837-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Patterson JH, Newbigin E, Tester M, Bacic A, Roessner U. 2009. Metabolic responses to salt stress of barley (Hordeum vulgare L.) cultivars, Sahara and Clipper, which differ in salinity tolerance. J. Exp. Bot. 6, 4089–4103. ( 10.1093/jxb/erp243) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lugan R, Niogret MF, Leport L, Guégan JP, Larher FR, Savouré A, Kopka J, Bouchereau A. 2010. Metabolome and water homeostasis analysis of Thellungiella salsuginea suggests that dehydration tolerance is a key response to osmotic stress in this halophyte. Plant J. 64, 215–229. ( 10.1111/j.1365-313X.2010.04323.x) [DOI] [PubMed] [Google Scholar]
- 137.Bowne JB, Erwin TA, Juttner J, Schnurbusch T, Langridge P, Bacic A, Roessner U. 2012. Drought responses of leaf tissues from wheat cultivars of differing drought tolerance at the metabolite level. Mol. Plant 5, 418–429. ( 10.1093/mp/ssr114) [DOI] [PubMed] [Google Scholar]
- 138.Witt S, Galicia L, Lisec J, Cairns J, Tiessen A, Araus JL, Palacios-Rojas N, Fernie AR. 2012. Metabolic and phenotypic responses of greenhouse-grown maize hybrids to experimentally controlled drought stress. Mol. Plant 5, 401–417. ( 10.1093/mp/ssr102) [DOI] [PubMed] [Google Scholar]
- 139.Sulpice R, et al. 2013. Impact of the carbon and nitrogen supply on relationships and connectivity between metabolism and biomass in a broad panel of Arabidopsis accessions. Plant Physiol. 162, 347–363. ( 10.1104/pp.112.210104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Florian A, Timm S, Nikoloski Z, Tohge T, Bauwe H, Araújo WL, Fernie AR. 2014. Analysis of metabolic alterations in Arabidopsis following changes in the carbon dioxide and oxygen partial pressures. J. Integr. Plant Biol. 56, 941–959. ( 10.1111/jipb.12237) [DOI] [PubMed] [Google Scholar]
- 141.Obata T, Witt S, Lisec J, Palacios-Rojas N, Florez-Sarasa I, Yousfi S, Araus JL, Cairns JE, Fernie AR. 2015. Metabolite profiles of maize leaves in drought, heat and combined stress field trials reveal the relationship between metabolism and grain yield. Plant Physiol. 169, 2665–2683. ( 10.1104/pp.15.01164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Richter JA, Erban A, Kopka J, Zörb C. 2015. Metabolic contribution to salt stress in two maize hybrids with contrasting resistance. Plant Sci. 233, 107–115. ( 10.1016/j.plantsci.2015.01.006) [DOI] [PubMed] [Google Scholar]
- 143.António C, Päpke C, Rocha M, Diab H, Limami AM, Obata T, Fernie AR, van Dongen JT. 2016. Regulation of primary metabolism in response to low oxygen availability as revealed by carbon and nitrogen isotope redistribution. Plant Physiol. 170, 43–56. ( 10.1104/pp.15.00266) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hu C, et al. 2016. Identification of conserved and diverse metabolic shifts during rice grain development. Sci. Rep. 6, 1–12. ( 10.1038/srep20942) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Müller A, Duchting P, Weiler EW. 2002. A multiplex GCMS/MS technique for the sensitive and quantitative single-run analysis of acidic phytohormones and related compounds, and its application to Arabidopsis thaliana. Planta 216, 44–56. ( 10.1007/s00425-002-0866-6) [DOI] [PubMed] [Google Scholar]
- 146.Schmelz EA, Engelberth J, Tumlinson JH, Block A, Alborn HT. 2004. The use of vapor phase extraction in metabolic profiling of phytohormones and other metabolites. Plant J. 39, 790–808. ( 10.1111/j.1365-313X.2004.02168.x) [DOI] [PubMed] [Google Scholar]
- 147.Vallarino JG, Osorio S. 2016. Simultaneous determination of plant hormones by GC-TOF-MS. In Plant signal transduction: methods and protocols. In Plant signal transduction: methods and protocols (eds JR Botella, MA Botella). Methods in Molecular Biology, vol. 1363, pp. 229–237. Totowa, NJ: Humana Press. () [DOI] [PubMed] [Google Scholar]
- 148.Villas-Bôas SG, Delicado DG, Åkesson M, Nielsen J. 2003. Simultaneous analysis of amino and nonamino organic acids as methyl chloroformate derivatives using gas chromatography–mass spectrometry. Anal. Biochem. 322, 134–138. ( 10.1016/j.ab.2003.07.018) [DOI] [PubMed] [Google Scholar]