

Abstract
Although qualitative strategies based on direct injection mass spectrometry (DIMS) have recently emerged as an alternative for the rapid classification of food samples, the potential of these approaches in quantitative tasks has scarcely been addressed to date. In this paper, the applicability of different multivariate regression procedures to data collected by DIMS from simulated mixtures has been evaluated. The most relevant factors affecting quantitation, such as random noise, the number of calibration samples, type of validation, mixture complexity and similarity of mass spectra, were also considered and comprehensively discussed. Based on the conclusions drawn from simulated data, and as an example of application, experimental mass spectral fingerprints collected by direct thermal desorption coupled to mass spectrometry were used for the quantitation of major volatiles in Thymus zygis subsp. zygis chemotypes. The results obtained, validated with the direct thermal desorption coupled to gas chromatography–mass spectrometry method here used as a reference, show the potential of DIMS approaches for the fast and precise quantitative profiling of volatiles in foods.
This article is part of the themed issue ‘Quantitative mass spectrometry’.
Keywords: direct injection mass spectrometry, mass spectral fingerprints, quantitation, multivariate regression, volatiles, Thymus zygis subsp. zygis
1. Introduction
The analysis of food volatile composition is a topic of high relevance in the food science and technology field, as several properties such as food aroma greatly determine the acceptance of foods by consumers. Therefore, authentication and quality control of many food samples are often based on the analysis of their volatile composition.
Thymus zygis is a widespread endemic plant in the Iberian Peninsula extensively used for culinary purposes and in popular medicine. Three subspecies of T. zygis have been recognized by Morales Valverde [1], and four chemotypes with a different volatile profile have also been reported for these species [2,3]. To select the most appropriate chemotype for collection or cultivation, the rapid characterization of T. zygis subsp. zygis based on its content of volatile secondary metabolites would be of great interest.
The coupling of gas chromatography to mass spectrometry (GC-MS) has become the technique of choice for the characterization of the volatile composition of food samples, as it allows in a single analysis the separation, identification and quantitation of the complex mixtures of volatiles usually present in foods. However, interest in the analytical platforms that allow the analysis time (mainly associated with the chromatographic separation) to be reduced has promoted the development of non-separative approaches based on direct injection mass spectrometry (DIMS) for the high-throughput and cost-effective characterization of food samples.
A number of mass spectral fingerprint approaches based on the so-called MS noses or mass sensors have been described in the food field [4–6]. Direct sampling by static or dynamic headspace techniques [7–9] and solid-phase microextraction (SPME) [7,10,11] are the most common sample introduction systems, whereas electron impact (EI) and quadrupole are the preferred ionization method and mass analyser, respectively.
DIMS strategies have mostly dealt with the discrimination of food samples for authentication tasks [7,9,10,12–15] or the prediction of different food quality parameters from mass spectral fingerprints [8,13]. However, the number of contributions addressing quantitative analysis is much more limited, papers usually addressing the quantitation of a limited number of target compounds [7,13,16]. Strategies for compensating for the effect of temporal instability of mass spectral fingerprints, which are critical to achieve precise quantitation with MS sensors, have also been reviewed by Pérez Pavón et al. [4]. However, other factors noticeably affecting quantitative results such as data processing have not been comprehensively evaluated yet.
The multi-channel nature of the mass spectrometer signal, associated with a high number of selective mass/charge (m/z) ratios for every compound determined, makes it possible to mathematically model the variation in the mass spectral fingerprint versus the concentration of the analyte under study by applying multivariate regression procedures. Moreover, the mass spectrum is a type of multivariate signal known for being virtually discrete and additive. This allows the estimation of the concentration of analyte i (ci) in a mixture after analysis of the global spectrum (total intensities for all recorded m/z ratios) by multivariate regression procedures [17].
Different supervised and unsupervised statistical methods have been reported for multivariate regression of DIMS data: multiple linear regression (MLR), principal component regression (PCR), partial least-squares regression (PLSR), support vector machine (SVM), neural networks (NN), etc. [4,6,17]. Among them, PCR and PLSR have become the most popular as they are easy-to-use approaches available from different statistical packages and whose mathematical basis is well known [18–20]. However, the comparison of the results obtained after data processing with more than one of these methods is scarcely addressed in the literature [6,10], although it is advisable for every intended application.
In view of the above, this paper aims to evaluate the applicability of different multivariate regression procedures to quantitative DIMS data obtained from simulated mixtures. Moreover, in an attempt to comprehensively assess the most relevant factors affecting quantitative response, the effect of random noise, the number of calibration samples, type of validation, mixture complexity and similarity of mass spectra were also studied. As an example of application, the conclusions drawn from simulated data were applied to the fast quantitation by direct thermal desorption (DTD)-MS of major volatiles representing each of the T. zygis subsp. zygis chemotypes. Results of this approach were validated with those obtained by conventional DTD-GC-MS analysis.
2. Experimental set-up
(a). Samples and standard mixtures
Thymus zygis subsp. zygis (hereafter abbreviated as thyme) plants were collected at the flowering stage in two sampling areas in the north of Madrid province (Spain) under botanical surveillance. An amount of 200–300 mg of thyme leaves was used to obtain representative samples from 53 individual plants. Leaves were air-dried at room temperature before analysis.
Analytical standards of benzene, tetrahydrofuran and n-alkanes (from n-decane to n-hexadecane) were purchased from Merck Co. (Darmstadt, Germany). Different mixtures were prepared from these standards: Mnoise, consisting of equal volumes of benzene, tetrahydrofuran and n-nonane, was used for setting the noise level of simulated data; Malkanes, consisting of equal volumes of n-alkanes (from n-C10 to n-C16), was used to evaluate the dispersion of T. zygis subsp. zygis chemotypes.
(b). Simulation of mass spectral fingerprints for volatile mixtures
The mass spectra of the different volatile mixtures evaluated (hereafter referred to as mass spectral fingerprints) were simulated as the linear combination of the mass spectra for every individual mixture component. In a general case, for a multi-component mixture, the global mass spectral fingerprint consisting of multiple m/z data can be expressed as
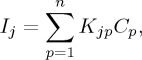 |
2.1 |
where Ij is the total intensity of the jth m/z, Kjp is the intensity of the jth m/z for the p component, Cp is the concentration of the pth component and n is the total number of components in the mixture.
Writing this expression using matrix notation
| 2.2 |
where I is a single column intensity matrix, corresponding to the global mass spectral fingerprint of the mixture considered; K is a column-wise matrix, where each column vector corresponds to the mass spectrum of a pure component; and C is a single column concentration matrix, where the concentrations of the mixture components are placed as a column vector.
To maximize the number of simulated data available for this study and to avoid instrumental artefacts, simulated mass spectral fingerprints were used rather than experimental mixture spectra. Thus, global mass spectral fingerprints (I) for different mixtures of a varied number of components with different functionality were simulated using the individual mass spectra (K) obtained from the NIST/EPA/NIH Mass Spectral Library 2.0 for benzene (Bz), tetrahydrofuran (THF), n-nonane (C9), n-decane (C10), 1,4-dioxane (Diox), n-butyl acetate (ButAc), 2-heptanone (Hept), 1-pentanenitrile (Pent), 1-nitropropane (Nitro) and 1-octanol (Octa), according to equation (2.2).
(c). Experimental data
(i). Gas chromatography–mass spectrometry analysis
Analyses were carried out on a GC 8000 gas chromatograph (Fisons, Milan, Italy) coupled to an MD 800 mass detector (Fisons, Manchester, UK), provided with two sample introduction systems: (i) a split/splitless injector and (ii) a module for DTD (ATD 400; Perkin-Elmer, Norwalk, CT, USA). Mass spectra were recorded in EI mode at 70 eV, scanning the 35–350 m/z range. Interface and source temperatures were 250°C and 200°C, respectively. Separation was carried out on a poly(100% methylsiloxane) CPSIL-5CB column (25 m × 0.25 mm i.d., 0.25 µm film thickness) (Agilent J&W, Santa Clara, CA, USA) temperature programmed from 70°C (10 min) to 180°C (at 4°C min−1) and then to 250°C (at 10°C min−1) for 15 min. Helium at 2.1 ml min−1 was used as the carrier gas.
For DTD-GC-MS analyses, 2 mg (accurately weighed) of dry thyme leaves (or 1 µl of Malkanes mixture on silanized glass wool) was introduced into a polytetrafluoroethylene tube (52 × 4 mm i.d.) which was then placed into a stainless steel desorption cartridge (89 × 4.5 mm i.d. × 6.5 mm o.d.). Volatile compounds were desorbed at 180°C for 15 min under a 45 ml min−1 helium (99.995% purity) flow and then cryofocused on a Tenax TA trap at −30°C. This trap was then heated to 320°C at 40°C s−1, remaining at the maximum temperature for 4 min. The desorbed volatiles were transferred to the GC-MS system through a fused silica line heated to 225°C. Inlet and outlet split flows were set at 90 and 55 ml min−1, respectively. Each plant was analysed in triplicate.
For experiments related to the evaluation of noise level, 1 µl of Mnoise mixture was injected (n = 10) using a split ratio of 100 : 1 and separated on the CPSIL-5CB column under isocratic conditions (50°C).
MassLab v. 1.4 software (Finnigan, Manchester, UK) was used for data acquisition and for exporting mass spectra to other programs. Qualitative analysis was based on the comparison of experimental spectra with those of the Wiley mass spectral library [21] and was further confirmed by using linear retention indices and published data [22]. Semi-quantitative data (percentage of total volatile composition) were directly calculated from total ion current peak areas, assuming no differences in response factor for all volatiles quantified.
(ii). Direct thermal desorption–mass spectrometry analysis
For DIMS determinations, the chromatographic column of the DTD-GC-MS system described above was replaced by a short fused silica tube (30 cm length × 0.32 mm i.d.). A second silica tube, acting as an interface between the first tube and the mass spectrometer, was used to transfer the molecules into the mass detector and as a restrictor to maintain the required vacuum level. Optimization of the dimensions of this interface was done using the Mnoise mixture. Oven temperature and carrier gas flow rate were set at 230°C and 6.0 ml min−1, respectively. Other experimental conditions were as previously described in §2c(i). Each plant was analysed in triplicate.
(d). Multivariate regression procedures
The most used regression procedures, as well as those with a more solid mathematical and statistical base, were evaluated. The selected methods were MLR, stepwise multiple linear regression (SMLR), ridge regression (RR), PCR and PLSR. Several steps were followed in the application of these regression models:
First, a calibration model was built using a series of samples (calibration sample set) for which the concentration of every mixture component was known. Equation (2.3) shows the type of predictive equations obtained,
| 2.3 |
where Ci is the concentration of the compound i, (m/z)j is the intensity of the different ions of its spectrum and a, b, c, d,… are empirical adjustable parameters.
The matrix where the mass spectra of the calibration samples are placed as column vectors is known as the calibration matrix, and its quality is critical to develop a good mathematical model. In this work, calibration matrices were built according to Brereton [23], that is, 25 calibration mixtures split into five concentration levels and five replicates per level.
Once the model was developed, its efficiency was checked, usually against another set of samples (validation sample set), which were placed as column vectors in the validation matrix. The validation sample set should be as large as possible and representative of the expected variability. In the present work, 250 validation samples were used unless specified.
For quality control of the results obtained by the different regression procedures studied, the graphical representation of the residuals and the value of the root mean square error of prediction (RMSEP) calculated according to equation (2.4) were used,
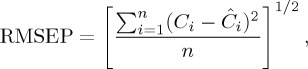 |
2.4 |
where Ci is the actual concentration of a compound in sample i, 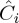 is its estimated concentration and n is the number of samples. Furthermore, because the present work will deal with high-collinearity data matrices, correlation matrices were also calculated.
is its estimated concentration and n is the number of samples. Furthermore, because the present work will deal with high-collinearity data matrices, correlation matrices were also calculated.
The number of independent variables selected in the SMLR method was determined by the forward procedure and using as the entry criterion a value of 4 for the F-Snedecor statistic. The number of components selected for the PCR and PLSR methods was obtained on the basis of the minimum RMESP value.
Statgraphics® Centurion XV (Statistical Graphics Corporation), Unscrambler® 10.1 (CAMO ASA) and Microsoft® Excel 2010 (Microsoft Co.) were the software packages used for data handling and for application of the different regression procedures.
3. Results and discussion
(a). Evaluation of multivariate regression procedures on simulated data
Before carrying out the quantitation of the major volatiles in thyme samples, further described in §3b(i), it was necessary to establish the robustness of the regression procedures under study against those factors that could have a relevant effect on quantitative results. To that aim, a comprehensive study of the simulated data was performed.
First, a calibration set of 25 mixtures, with concentration ranging between 0 and 100 mg ml−1, was built according to Brereton [23] using the mass spectra of Bz, C9 and THF taken from the NIST/EPA/NIH database. Using the same procedure, 250 simulated mixtures were also obtained to be used as the external validation set.
To provide simulated data with a realistic level of random noise, evaluation of this parameter was carried out on the experimental mass spectral fingerprint for the Mnoise mixture analysed as described in §2c(i). Except for m/z ions below 1% of relative intensity, whose relative standard deviation (RSD, %) was noticeably higher, the RSD (%) values for m/z ions of experimental spectra were 5% on average. Therefore, a 5% level of random noise was added to simulated spectra, and m/z ions below 1% were discarded before applying the selected regression procedures.
Results obtained using calibration and validation sets corrected for the level of noise are shown in table 1. As can be seen, RR is the method that presented the smallest RMSEP (from 0.88 to 1.67) for all the compounds studied, these values being up to three times smaller than those of the regression procedure with the highest error. Regarding the performance of the other methods, MLR and SMLR provided better results than data compression methods (PCR and PLSR). The graphical representation of the residuals obtained for each regression model showed the expected heteroscedastic nature of the data, but did not reveal any anomaly. However, the analysis of variance showed the dramatic effect that the collinearity has for the correct estimation of the regression parameters (electronic supplementary material, table S1). Very high errors were obtained for the parameter estimators in MLR and SMLR, being in many cases even greater than the absolute value of the corresponding estimator. Thus, although MLR and SMLR produced lower RMSEP values, the high uncertainty of their regression coefficients makes these regression procedures not recommendable for the quantitation of mixtures represented by data matrices with high collinearity. In other words, the degree of correlation between mass spectra should be known for the reliable application of MLR or SMLR.
Table 1.
Results obtained by applying the regression procedures under study to simulated mixtures with three components and 5% random noise (average for n = 250 replicates).
| compound |
||||
|---|---|---|---|---|
| regression | parameter | Bz | C9 | THF |
| MLR | RMSEP | 1.95 | 1.22 | 2.02 |
| SMLR | RMSEP | 1.73 | 1.27 | 2.08 |
| selected m/z | 38, 50, 74, 78 | 55, 57, 70, 85, 98, 128 | 40, 72 | |
| RR | RMSEP | 0.88 | 1.03 | 1.67 |
| θa | 0.029 | 0.030 | 0.028 | |
| PCR | RMSEP | 2.45 | 2.09 | 3.58 |
| selected PCsb | 1 | 2 | 2 | |
| PLSR | RMSEP | 2.45 | 2.08 | 3.55 |
| selected PCsb | 1 | 2 | 2 | |
aθ, ridge factor [24].
bSelected PCs, number of principal components for the minimum value of RMSEP.
In addition, several other factors that may affect the performance of the regression models under study were also evaluated, as follows.
(i). Size of the calibration set
To study the effect of the number of calibration samples used for quantitation, the three following possibilities were considered: (i) calibration matrix 1, designed to include a total of five samples, one for each concentration level; (ii) calibration matrix 2, with 25 samples, five samples per level; and (iii) calibration matrix 3, with a total of 50 samples, 10 per concentration level. As an example, table 2 lists the results obtained by using these three matrices in the quantitation of benzene.
Table 2.
Results obtained in the quantitation of benzene using calibration sets with a different number of samples (average for n = 250 replicates).
| calibration samples |
||||
|---|---|---|---|---|
| regression | parameter | 5 | 25 | 50 |
| MLR | RMSEP | n.c.a | 1.947 | 1.038 |
| SMLR | RMSEP | 2.377 | 1.731 | 1.240 |
| selected m/z | 63, 78 | 38, 50, 74, 78 | 37, 38, 50, 52, 74, 78 | |
| RR | RMSEP | n.c. | 0.880 | 0.842 |
| θb | n.c. | 0.0286 | 0.0330 | |
| PCR | RMSEP | 2.676 | 2.449 | 2.416 |
| selected PCsc | 1 | 1 | 1 | |
| PLS | RMSEP | 2.676 | 2.448 | 2.415 |
| selected PCsc | 1 | 1 | 1 | |
an.c., not computable; the number of calibration samples is lower than the number of independent variables.
bθ, ridge factor [24].
cSelected PCs, number of principal components for the minimum value of RMSEP.
Irrespective of the compound and model considered, the RMSEP value always diminished when increasing the number of samples used in the calibration set. This decrease was very noticeable for SMLR and MLR regressions, whereas it was less important for the compression methods (PCR and PLSR) between five and 25 samples, and hardly noticeable for 25–50 samples. With regards to RR, it showed a very good behaviour with almost no differences when using 25 or 50 samples. However, RR presented the same disadvantage as MLR, since it cannot be applied when there are fewer samples than the independent m/z variables. Therefore, it seems evident that, when the number of samples available for calibration is small, PCR and PLSR are the recommended procedures.
(ii). Validation method
In previous sections, a very high number of test samples (250) was used to draw conclusions about statistical significance. Nevertheless, in analytical laboratories, it is not always feasible to use the time and economic resources to carry out so many analyses. For that reason, the usual practice is either to work with a much smaller set of validation samples or to apply cross-validation. Bearing this in mind, RMSEP values obtained for subsets of validation samples including 25, 10 and five samples and that obtained in the cross-validation procedure were compared with RMSEP data for the set of reference (250) samples. The results shown in figure 1 indicate that, irrespective of the regression method considered, the difference between the RMSEP value for the reference set and that obtained for external validation diminishes with the size of the validation set. The use of at least 25 validation samples was considered advisable, and where this was not feasible, the cross-validation procedure provided similar results.
Figure 1.
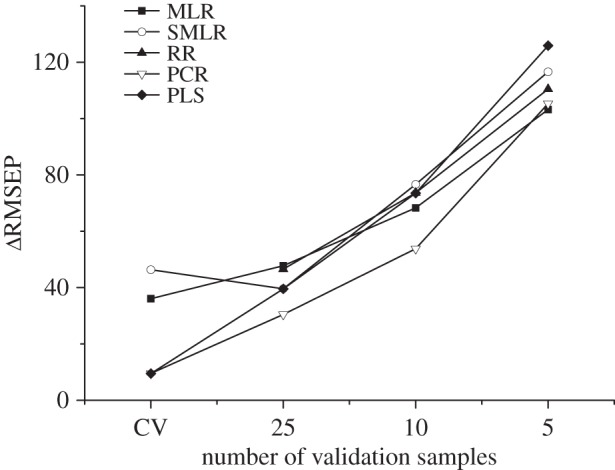
Differences between the reference value of RMSEP (250 samples) and those obtained by the external validation set (25, 10 and 5 samples) and cross-validation (CV).
(iii). Number of compounds and similarity of mass spectra
The results described in previous sections were obtained on mixtures with three components. However, a regression model applicable to real samples should show good behaviour when the number of mixture components is much higher. Therefore, in this subsection, the different regression methods were tested on mixtures of 10 components: Bz, THF, C9, C10, Diox, ButAc, Hept, Pent, Nitro and Octa. The concentrations of the calibration mixtures were obtained according to Brereton [23], and the concentrations of the validation set (25 samples) were randomly generated between 0 and 100 mg l−1. In general, results obtained on mixtures with 10 components (table 3) were of the same order of magnitude as those for three-compound mixtures. Nevertheless, some particularities should be mentioned. SMLR provided acceptable RMSEP values in all cases, but the selection of variables frequently led to the choice of a single representative m/z. Therefore, SMLR must be used with caution, since in these cases the advantages provided by the use of a multivariate regression technique are lost. Data compression methods gave rise to models with a high complexity, due to the presence of many principal components, although the errors obtained were not much higher. The case of the quantitation of 1,4-dioxane by PCR should also be highlighted, since a higher error was obtained than in the remaining methods, even when a distinctive m/z ratio was available (m/z 88). After a more detailed study of the case, the reason seemed to be the scarce number of m/z ratios available to carry out the quantitation (m/z 43, 44, 45, 57, 58, 88). Consequently, principal component analysis (PCA) is not able to extract all the necessary information for the development of a regression model good enough for this compound. Although this conclusion cannot be extrapolated to other compounds showing a similar behaviour (e.g. Pent and Nitro), it can be concluded that the presence of distinctive m/z ratios is not always enough to achieve a good quantitation by PCR.
Table 3.
Results obtained by applying the regression procedures under study to mixtures with 10 components and 5% random noise (average for n = 25 replicates).
| component |
||||||
|---|---|---|---|---|---|---|
| regression | parameter | THF | C9 | Bz | C10 | Diox |
| MLR | RMSEP | 1.269 | 3.484 | 6.666 | 3.156 | 3.245 |
| SMLR | RMSEP | 1.224 | 3.115 | 7.500 | 2.753 | 3.160 |
| selected m/z | 37, 50, 51, 74, 76, 77, 78 | 128 | 42, 43, 71, 72 | 142 | 45 | |
| RR | RMSEP | 1.027 | 3.343 | 6.638 | 3.030 | 3.125 |
| θa | 0.0383 | 0.0304 | 0.0277 | 0.0331 | 0.0207 | |
| PCR | RMSEP | 2.480 | 4.240 | 7.342 | 5.273 | 10.432 |
| selected PCsb | 1 | 13 | 6 | 5 | 4 | |
| PLSR | RMSEP | 2.474 | 3.640 | 7.335 | 4.918 | 7.338 |
| selected PCsb | 1 | 13 | 5 | 13 | 5 | |
| ButAc | Hept | Pent | Nitro | Octa | ||
|---|---|---|---|---|---|---|
| MLR | RMSEP | 1.873 | 2.225 | 3.006 | 3.055 | 1.787 |
| SMLR | RMSEP | 1.873 | 2.225 | 2.993 | 2.972 | 1.787 |
| selected m/z | 39, 41, 43, 44, 55, 56, 57, 61, 73 | 59, 114 | 54 | 46 | 43, 56, 68, 82, 83 | |
| RR | RMSEP | 1.983 | 2.293 | 3.166 | 3.189 | 1.545 |
| θa | 0.0297 | 0.0358 | 0.0345 | 0.0336 | 0.0378 | |
| PCR | RMSEP | 1.949 | 2.519 | 3.004 | 1.898 | 2.362 |
| selected PCsb | 7 | 10 | 6 | 6 | 7 | |
| PLSR | RMSEP | 3.352 | 2.478 | 3.024 | 5.951 | 3.338 |
| selected PCsb | 6 | 9 | 5 | 7 | 4 |
aθ, ridge factor [24].
bSelected PCs, number of principal components for the minimum value of RMSEP.
When comparing the behaviour of those compounds present in both mixtures (3 and 10 component mixtures), benzene and C9 showed higher values of RMSEP when they were quantified in the 10-component mixture. Nevertheless, the errors for THF were similar and in some cases even higher for the three-compound mixture. Therefore, direct quantitation seems to be feasible even when using complex volatile mixtures.
Another factor to be considered is the similarity (degree of correlation) between mass spectra, which is more probable when a higher number of compounds are included in the mixture. In the present case, the pairs C9/C10 and Pent/Nitro showed the highest correlation, with r = 0.983 and 0.845, respectively (electronic supplementary material, table S2). As the regression method is not able to distinguish the contribution of every individual compound, it is difficult to achieve good quantitation (low RMSEP values) for these pairs. The joint measurement of both compounds can be considered in these cases as an alternative to provide better quantitation results (as experimentally evidenced in §3b(ii)).
(b). Analysis of the volatile composition of Thymus zygis subsp. zygis
The study carried out on simulated data showed that each of the regression procedures evaluated has its own characteristics, but all of them can provide good quantitation results depending on the specific dataset. Furthermore, some important issues such as the appropriate size of the calibration set, the more suitable validation method or the effect of sample complexity and spectral similarity on the final results were also established. Thus, the next step was the application of the regression methods under study to the quantitation of volatiles in real thyme samples. In addition to the wide number of applications this aromatic plant has in different fields, this study was carried out on T. zygis subsp. zygis because aspects not previously addressed in DIMS approaches such as the differentiation of plant chemotypes could also be evaluated from the analysis of this Lamiaceae plant. Results gathered in the DTD-GC-MS analysis of thyme samples (§3b(i)) were considered as a reference for the further study of the application of regression methods to mass spectral fingerprints collected by DTD-MS (§3b(ii)).
(i). Direct thermal desorption–gas chromatography–mass spectrometry analysis
In an attempt to identify chemotypes in the 53 thyme samples under study, quantitative results (relative data) obtained in the DTD-GC-MS analysis (n = 3) of major volatiles were subjected to PCA. In agreement with previous studies [1,25], four chemotypes were distinguished (figure 2a). The number of samples clustered in each of these chemotypes, together with their characteristic compounds and relative concentrations, is summarized in table 4. As can be seen, the first chemotype is dominated by geraniol (22%) and geranyl acetate (42%), the second one by linalool (60%), in the third one the main compounds are the aromatics thymol (49%) and carvacrol (17%), and in the fourth chemotype α-terpineol (19%) and α-terpinyl acetate (55%) are the major components. These clusters will hereafter be referred as geraniol, linalool, aromatics and α-terpineol chemotypes. A representative chromatogram of each of these chemotypes is shown in the electronic supplementary material, figure S1.
Figure 2.

Chemotypes identified in Thymus zygis subsp. zygis using (a) relative areas from DTD-GC-MS analysis and (b) DTD-MS fingerprints.
Table 4.
Chemotypes and main components identified in Thymus zygis subsp. zygis samples (average for n = 3 replicates).
| area (%) |
||||
|---|---|---|---|---|
| compound | tR (min) | minimum | maximum | average |
| chemotype geraniol | ||||
| trans-sabinene hydrate | 14.3 | 0.00 | 8.11 | 1.01 |
| linalool | 16.0 | 0.00 | 7.55 | 2.41 |
| geraniol | 23.3 | 15.72 | 27.55 | 22.30 |
| α-terpinyl acetate | 26.9 | 0.00 | 15.59 | 2.04 |
| geranyl acetate | 28.2 | 23.07 | 76.38 | 42.08 |
| chemotype linalool | ||||
| γ-terpinene | 13.7 | 0.00 | 3.26 | 0.17 |
| cis-linalool oxide | 14.5 | 0.41 | 5.08 | 2.96 |
| trans-linalool oxide | 15.3 | 0.48 | 4.76 | 3.06 |
| linalool | 16.1 | 43.95 | 78.61 | 59.69 |
| borneol | 19.2 | 0.00 | 8.16 | 1.89 |
| monoterpenediol (isomer I) | 20.4 | 2.30 | 10.38 | 6.51 |
| linalyl acetate | 23.1 | 0.00 | 20.25 | 7.69 |
| monoterpenediol (isomer II) | 24.0 | 3.16 | 14.61 | 8.32 |
| thymol | 24.7 | 0.00 | 2.03 | 0.25 |
| geranyl acetate | 28.0 | 0.00 | 3.12 | 0.17 |
| chemotype aromatics | ||||
| p-cymene | 11.9 | 4.50 | 26.80 | 12.41 |
| γ-terpinene | 13.8 | 0.18 | 16.51 | 6.94 |
| linalool | 16.0 | 0.00 | 3.61 | 1.63 |
| borneol | 19.2 | 0.00 | 6.14 | 0.70 |
| thymoquinone | 23.0 | 0.00 | 14.73 | 1.25 |
| thymol | 25.1 | 0.00 | 87.14 | 49.24 |
| carvacrol | 25.2 | 1.03 | 69.83 | 16.94 |
| C10H14O2a | 30.3 | 0.28 | 11.78 | 5.13 |
| C10H14O2b | 34.0 | 0.00 | 4.13 | 1.10 |
| chemotype α-terpineol | ||||
| myrcene | 9.90 | 0.33 | 5.53 | 3.59 |
| linalool | 16.1 | 0.00 | 29.10 | 5.45 |
| borneol | 19.2 | 0.00 | 5.10 | 0.72 |
| α-terpineol | 20.5 | 5.56 | 34.14 | 18.84 |
| α-terpinyl acetate | 26.9 | 24.35 | 91.10 | 55.07 |
| carvone acetate | 34.7 | 0.00 | 3.44 | 1.43 |
| C12H20O3c | 36.7 | 0.00 | 3.98 | 1.28 |
am/z ratios and abundances: 77(16), 79(11), 91(9), 105(41), 133(24), 151(100), 166(38).
bm/z ratios and abundances: 77(14), 79(12), 95(21), 123(17), 151(100), 166(42).
cm/z ratios and abundances: 43(100), 71(47), 93(27), 108(42), 109(37), 126(30), 137(17), 152(13), 170(2).
As shown in table 4, the quantitative data (relative areas) showed a great variability for samples of the same chemotype. To know whether such variability was due to instrumental errors or to differences in metabolic pathways giving rise to these volatile secondary metabolites, a study to evaluate data reproducibility was carried out. To this aim, the n-alkane mixture (Malkanes, §2a) covering the elution range of thyme volatiles was first analysed (n = 10) by DTD-GC-MS. Similarly, 10 samples of the same T. zygis subsp. zygis plant (linalool chemotype) were further analysed. The relative areas obtained and their corresponding standard deviations are listed in the electronic supplementary material, table S3. Whereas the average RSD (%) for n-alkanes was 2.3%, dispersion data for thyme volatiles were almost an order of magnitude higher, except for thymol due to its high per cent area. It can be concluded therefore that for these types of samples the main contribution to variability seems to arise from biological sources.
(ii). Direct thermal desorption–mass spectrometry analysis
As previously mentioned, the DTD-MS analysis of thyme samples required the analytical system to be redesigned for its use without a chromatographic column. Particular emphasis was paid to the dimensioning of the interface leading the desorbed volatiles into the mass spectrometer. A silica tube of 80 cm length × 0.1 mm i.d. was selected as a trade-off to provide good sensitivity and good vacuum value.
After optimization of the experimental set-up, the 53 thyme samples were analysed in triplicate by DTD-MS. As no chromatographic column was now used, all compounds eluted together in a single and almost Gaussian peak at a retention time of approximately 0.5 min (electronic supplementary material, figure S2). The mass spectra obtained during the elution of this peak were averaged, and the resulting spectrum normalized to the sum of all detected ions. This type of normalization was previously applied to chromatographic areas of all recorded peaks, and was chosen to minimize the effects of data closure [26].
Direct injection mass spectra for the thyme samples under study were subjected to PCA, and the resulting score plot is shown in figure 2b. As can be seen, the four chemotypes previously identified by DTD-GC-MS analysis were also observed from DTD-MS data. These results highlight the usefulness of mass spectral fingerprints for the representative characterization of the volatile composition of T. zygis subsp. zygis samples and for the intended classification of these samples into different chemotypes.
After checking the good quality of the DTD-MS data, the selected regression procedures were applied to the mass spectral fingerprints obtained for samples of each chemotype. A comprehensive summary of the results obtained is shown in the electronic supplementary material, tables S4–S7. As can be seen, the behaviour of the different regression methods with experimental data for thyme samples was very similar to that previously described for simulated data, or even better. In general, the performance of all regression methods with DTD-MS data was good, giving rise to very low RMSEP values. Moreover, errors obtained for every specific compound and chemotype were very similar irrespective of the regression procedure considered. RR was the only method tending to outperform the remaining methods, as it usually gave rise to the lowest RMSEP values. This could be explained by the high collinearity between mass spectra shown for the DTD-MS data and the good performance of the RR method for ‘ill-conditioned’ data.
Finally, and to get a simple overview of the reliability of the DTD-MS results, values obtained by DTD-MS and DTD-GC-MS were compared. As an example, figure 3 shows the differences observed in the quantitation of the two main components for each chemotype. Considering the best regression method for each compound, differences between 2% and 10% were obtained in most cases. However, carvacrol showed an anomalous behaviour, with an error as high as 50% for the best regression method (RR). As previously stated for simulated data, the high degree of similarity between the mass spectral fingerprints of the isomers thymol and carvacrol (r = 0.992) could justify these results. In agreement with this, a decrease down to values of approximately 7% was obtained when the joint quantitation of thymol–carvacrol was considered by using the different regression models under study.
Figure 3.
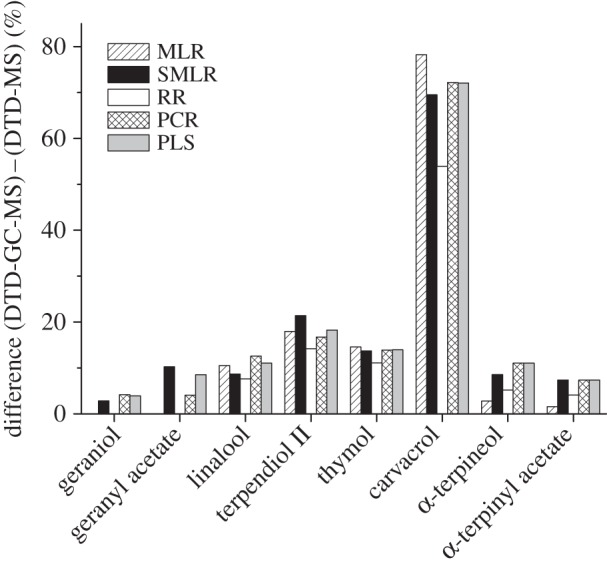
Comparison between DTD-GC-MS and DTD-MS quantitation.
4. Conclusion
The present work has been developed to establish the real possibilities and limitations of quantitative profiling of volatiles in food samples by DIMS in combination with multivariate regression methods. To this aim, quantitative global responses (mass spectral fingerprints) from simulated volatile mixtures, rather than data for specific standards or samples (which is the common approach in the literature), were processed under very diverse experimental conditions and subjected to different multivariate regression methods. Although less well known than other multivariate regression procedures here evaluated, RR was shown to be a robust method for the precise quantitation of simulated data with high collinearity. MLR and SMLR also provided low RMSEP values, but they should be applied with caution, because their reliability is inversely correlated with data collinearity. As far as PCR and PLSR are concerned, they are the methods of choice when the calibration set includes a low number of samples.
On the other hand, application of the different regression methods under study to experimental mass spectral fingerprints collected by DTD-MS has been shown to be useful for the quantitation of major volatiles in T. zygis subsp. zygis chemotypes. In this sense, it is worth noting that the advantages in terms of no sample pretreatment provided by the use of DTD as the sample introduction system are in good agreement with the development of the fast analytical methodologies for volatile quantitation here addressed. The results obtained by DTD-MS, validated using as reference a DTD-GC-MS method, show the potential of DIMS approaches as high-throughput and cost-effective methodologies for the precise quantitative profiling of volatiles in foods.
Supplementary Material
Supplementary Material
Data accessibility
The datasets supporting this article are available in the electronic supplementary material.
Authors' contributions
All authors contributed equally to this work.
Competing interests
We have no competing interests.
Funding
This work has been funded by Comunidad de Madrid (Spain) and European funding from the FEDER program (S2013/ABI-3028 AVANSECAL-CM). A.C.S. thanks MINECO for a Ramón y Cajal contract and Fundación Ramón Areces for financial support.
References
- 1.Morales Valverde R. 1986. Taxonomía de los géneros Thymus (excluida la sección Serpillum) y Thimbra en la Península Ibérica. Madrid, Spain: RJB (CSIC). [Google Scholar]
- 2.Rota MC, Herrera A, Martínez RM, Sotomayor JA, Jordán MJ. 2008. Antimicrobial activity and chemical composition of Thymus vulgaris, Thymus zygis and Thymus hyemalis essential oils. Food Control 19, 681–687. ( 10.1016/j.foodcont.2007.07.007) [DOI] [Google Scholar]
- 3.Saez F. 1995. Essential oil variability of Thymus zygis growing wild in southeastern Spain. Phytochemistry 40, 819–825. ( 10.1016/0031-9422(95)00347-a) [DOI] [Google Scholar]
- 4.Pérez Pavón JL, del Nogal Sánchez M, Pinto CG, Fernández Laespada ME, Cordero BM, Peña AG. 2006. Strategies for qualitative and quantitative analyses with mass spectrometry-based electronic noses. Trends Anal. Chem. 25, 257–266. ( 10.1016/j.trac.2005.09.003) [DOI] [Google Scholar]
- 5.Cevoli C, Cerretani L, Gori A, Caboni MF, Gallina Toschi T, Fabbri A. 2011. Classification of Pecorino cheeses using electronic nose combined with artificial neural network and comparison with GC-MS analysis of volatile compounds. Food Chem. 129, 1315–1319. ( 10.1016/j.foodchem.2011.05.126) [DOI] [PubMed] [Google Scholar]
- 6.Peris M, Escuder-Gilabert L. 2013. On-line monitoring of food fermentation processes using electronic noses and electronic tongues: a review. Anal. Chim. Acta 804, 29–36. ( 10.1016/j.aca.2013.09.048) [DOI] [PubMed] [Google Scholar]
- 7.Ampuero S, Bogdanov S, Bosset JO. 2004. Classification of unifloral honeys with an MS-based electronic nose using different sampling modes: SHS, SPME and INDEX. Eur. Food Res. Technol. 218, 198–207. ( 10.1007/s00217-003-0834-9) [DOI] [Google Scholar]
- 8.Gamboa-Santos J, Soria AC, Perez-Mateos M, Carrasco JA, Montilla A, Villamiel M. 2013. Vitamin C content and sensorial properties of dehydrated carrots blanched conventionally or by ultrasound. Food Chem. 136, 782–788. ( 10.1016/j.foodchem.2012.07.122) [DOI] [PubMed] [Google Scholar]
- 9.Pena F, Cardenas S, Gallego M, Valcarcel M. 2002. Characterization of olive oil classes using a ChemSensor and pattern recognition techniques. J. Am. Oil Chem. Soc. 79, 1103–1108. ( 10.1007/s11746-002-0611-6) [DOI] [Google Scholar]
- 10.Cheng P, Fan W, Xu Y. 2013. Quality grade discrimination of Chinese strong aroma type liquors using mass spectrometry and multivariate analysis. Food Res. Int. 54, 1753–1760. ( 10.1016/j.foodres.2013.09.002) [DOI] [Google Scholar]
- 11.Marsili RT. 2001. SPME-MS-MVA as a rapid technique for assessing oxidation off-flavors in foods. In Headspace analysis of foods and flavors: theory and practice (eds Rouseff RL, Cadwallader KR), pp. 89–100. New York, NY: Springer. [DOI] [PubMed] [Google Scholar]
- 12.Dirinck I, Van Leuven I, Dirinck P. 2006. ChemSensor classification of red wines. In Flavour science: recent advances and trends (eds Bredie WLP, Petersen MA), pp. 521–524. Amsterdam, The Netherlands: Elsevier. [Google Scholar]
- 13.Berna AZ, Trowell S, Clifford D, Cynkar W, Cozzolino D. 2009. Geographical origin of Sauvignon Blanc wines predicted by mass spectrometry and metal oxide based electronic nose. Anal. Chim. Acta 648, 146–152. ( 10.1016/j.aca.2009.06.056) [DOI] [PubMed] [Google Scholar]
- 14.Cerrato Oliveros C, Boggia R, Casale M, Armanino C, Forina M. 2005. Optimisation of a new headspace mass spectrometry instrument. J. Chromatogr. A 1076, 7–15. ( 10.1016/j.chroma.2005.04.020) [DOI] [PubMed] [Google Scholar]
- 15.Goodner KL, Rouseff RL. 2001. Using an ion-trap MS sensor to differentiate and identify individual components in grapefruit juice headspace volatiles. J. Agric. Food Chem. 49, 250–253. ( 10.1021/jf000867g) [DOI] [PubMed] [Google Scholar]
- 16.Marti MP, Boque R, Riu M, Busto O, Guasch J. 2003. Fast screening method for determining 2,4,6-trichloroanisole in wines using a headspace-mass spectrometry (HS-MS) system and multivariate calibration. Anal. Bioanal. Chem. 376, 497–501. ( 10.1007/s00216-003-1940-z) [DOI] [PubMed] [Google Scholar]
- 17.Scott SM, James D, Ali Z. 2006. Data analysis for electronic nose systems. Microchim. Acta 156, 183–207. ( 10.1007/s00604-006-0623-9) [DOI] [Google Scholar]
- 18.Geladi P, Kowalski BR. 1986. Partial least-squares regression: a tutorial. Anal. Chim. Acta 185, 1–17. ( 10.1016/0003-2670(86)80028-9) [DOI] [Google Scholar]
- 19.Miller JN, Miller JC. 2005. Statistics and chemometrics for analytical chemistry. Harlow, UK: Pearson. [Google Scholar]
- 20.Brereton RG. 2003. Chemometrics: data analysis for the laboratory and chemical plant. In Chemometrics (ed. Brereton RG.), pp. 1–489. Chichester, UK: Wiley. [Google Scholar]
- 21.Davies NW. 1990. Gas chromatographic retention indices of monoterpenes and sesquiterpenes on methyl silicon and Carbowax 20M phases. J. Chromatogr. A 503, 1–24. ( 10.1016/S0021-9673(01)81487-4) [DOI] [Google Scholar]
- 22.McLafferty FW, Stauffer DB. 1989. Wiley/NBS registry of mass spectral data. New York, NY: Wiley. [Google Scholar]
- 23.Brereton RG. 1997. Multilevel multifactor designs for multivariate calibration. Analyst 122, 1521–1529. ( 10.1039/a703654j) [DOI] [Google Scholar]
- 24.Hoerl AE, Kennard RW. 1970. Ridge regression-biased estimation for nonorthogonal problems. Technometrics 12, 55–67. ( 10.1080/00401706.1970.10488634) [DOI] [Google Scholar]
- 25.Velasco Negueruela A, Pérez-Alonso MJ. 1984. Aceites esenciales de tomillos ibéricos. III. Contribución al estudio de quimiotipos en el grupo Thymus zygis L. Anal. Bromatol. 36, 301–308. [Google Scholar]
- 26.Johansson E, Wold S, Sjodin K. 1984. Minimizing effects of closure on analytical data. Anal. Chem. 56, 1685–1688. ( 10.1021/ac00273a034) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting this article are available in the electronic supplementary material.