

Abstract
Depression results from changes in the central nervous system (CNS) that may result from immunological abnormalities. The immune system affects the CNS through cytokines, which regulate brain activities and emotions. Cytokines affect two biological systems that are most associated with the pathophysiology of depression: The hypothalamic-pituitary-adrenal axis and the catecholamine/sympathetic nervous system. Neuroinflammation and cytokines affect the brain signal patterns involved in the psychopathology of depression and the mechanisms of antidepressants, and they are associated with neurogenesis and neural plasticity. These observations suggest that neuroinflammation and cytokines might cause and/or maintain depression, and that they might be useful in the diagnosis and prognosis of depression. This psychoneuroimmunologic perspective might compensate for some of the limitations of the monoamine theory by suggesting that depression is a result of a failure to adapt to stress and that inflammatory responses and cytokines are involved in this process. In this review, the interactions of cytokines with the CNS, neuroendocrine system, neurotransmitters, neurodegeneration/neurogenesis, and antidepressants are discussed. The roles of cytokines in the etiology and psychopathology of depression are examined. The use of cytokine inhibitors or anti-inflammatory drugs in depression treatment is explored. Finally, the significance and limitations of the cytokine hypothesis are discussed.
Keywords: Depression, Neurogenesis, Antidepressant, Cytokine, Neuroinflammation, Psychoneuroimmunology, Neuroendocrine, Neurotransmitter
Core tip: We investigated the etiology and the pathogenesis of depression regarding the cytokine network. It was concluded that depression may be caused by neuroinflammation and cytokine imbalances, which are closely connected with the central nerve system, hypothalamic-pituitary-adrenal axis, neurotransmitter, autonomic nerve system, neural plasticity, and antidepressants.
INTRODUCTION
Severe psychological and/or physical stress can result in homeostatic imbalances and abnormal immune responses. Several hypotheses have proposed that immunologic imbalances affect the central nervous system (CNS) and result in psychopathology. Depression is a disease that is associated with changes in the CNS that might be caused by immunological abnormalities. Recent clinical and experimental studies have confirmed that internal and external stress significantly affects the expression of depressive symptoms and their persistence in vulnerable individuals with immunological abnormalities[1]. Moreover, cytokines affect the activity of the two biological systems that are most associated with the pathophysiology of depression: The hypothalamic-pituitary-adrenal (HPA) axis and the catecholamine/sympathetic nervous system[2].
The CNS affects the immune system through the autonomic nervous system and the neuroendocrine system. Reciprocally, the immune system affects the CNS through cytokines secreted by immune cells that regulate brain activities and emotions[3]. Thus, the immune system can be regarded as a sensory organ that recognizes internal or external stress. Stress can trigger overall changes in the immune system, neurotransmitters, neuroendocrine system, and CNS, and their interactions contribute to the expression, continuation, and termination of depressive symptoms.
This psychoneuroimmunologic perspective suggests that depression is mediated by inflammatory responses and cytokines, and that the disease results from a failure to adapt to stress. This view might compensate for some of the limitations of the monoamine theory, which is an important psychopathologic model of depression.
In this review, the interactions of cytokines with the CNS, neuroendocrine system, neurotransmitters, neurogenesis, and antidepressants are investigated. The roles of cytokines in the etiology and psychopathology of depression are examined. In addition, the use of cytokine inhibitors and anti-inflammatory drugs in the treatment of depression are explored, and the significance and limitations of the cytokine hypothesis are discussed.
CYTOKINE SYSTEM
Peripheral and central cytokines and neuroimmune circuits
Cytokines mediate signaling among immune cells. They activate or inhibit other immune cells, which results in a complicated circuit. Cytokines act on cell membrane receptors like neurotransmitters or on intracellular receptors like hormones to transmit information to cells. They are mainly secreted from monocytes (or macrophages) or lymphocytes as well as from brain cells, such as neurons, endothelial cells, astrocytes, and microglia. Cytokines are divided into various types, including interleukins (ILs), chemokines, tumor necrosis factors (TNFs), interferons (IFNs), and transforming growth factors (TGFs).
Proinflammatory cytokines include IL-1, IL-2, IL-6, IFN-γ, and TNF-α. Anti-inflammatory cytokines include IL-4, IL-10, IL-11, IL-13, and TGF-β[4]. The proinflammatory cytokines activate cyclo-oxygenase-2 (COX-2), increase the levels of prostaglandin E2 (PGE2), activate inflammatory cells, and induce inflammatory reactions. They interact with each other to maintain balance. For example, IL-10 reduces TNF production, and the IL-1 receptor antagonist (IL-1ra) antagonizes the IL-1 receptor. In chronic inflammation, the proinflammatory cytokines are increased and the anti-inflammatory cytokines are decreased, which results in the onset of various diseases[5].
The production of the peripheral cytokines that are secreted from monocytes or macrophages is determined by the level of immune activity. In pathologic states, such as acute or chronic inflammation or tissue damage, immune function and macrophages are activated to increase the levels of proinflammatory cytokines. Cortisol secreted from the adrenal cortex as a result of HPA-axis activation is most important in peripheral cytokine production. When cortisol levels are low, the production of proinflammatory cytokines increases, while their production is inhibited when cortisol levels are high[6]. Neurotransmitters regulate peripheral cytokines through cortisol levels. For example, acetylcholine (Ach), dopamine (DA), and noradrenaline (NA) promote the secretion of corticotropin-releasing hormone (CRH) in the hypothalamus, and serotonin (5-HT) inhibits the secretion of CRH in the hypothalamus and adrenocorticotropic hormone (ACTH) in the pituitary[7]. In addition, the autonomic nervous system regulates peripheral cytokine production. Parasympathetic nerves directly affect the immune system, while sympathetic nerves affect the immune system through NA secretion from the peripheral sympathetic ganglia. The vagus nucleus, which is located in the pons, inhibits immune functions and cytokine production through the secretion of Ach from the vagus nerve[8] (Figure 1).
Figure 1.
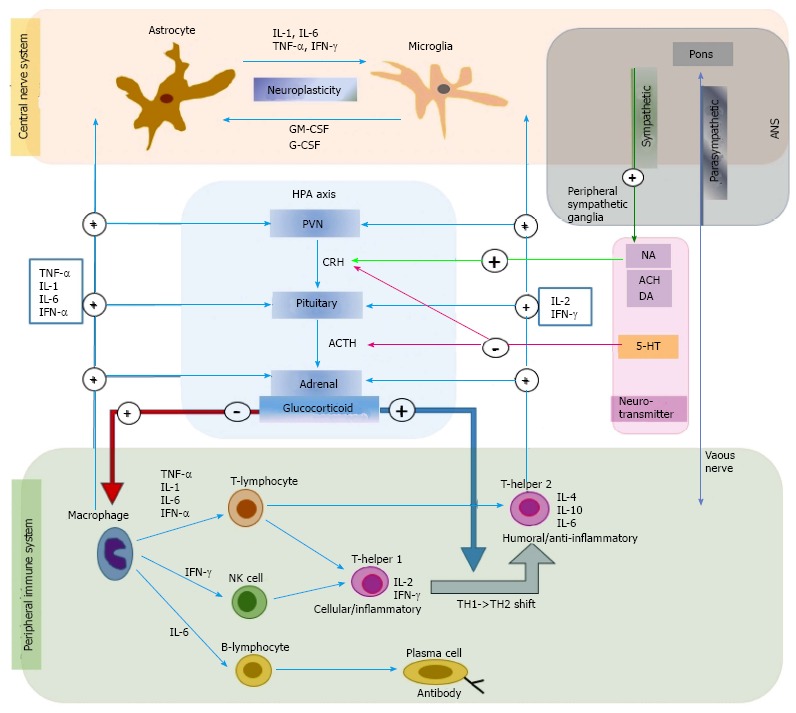
The role of cytokine network in depression in connection with immune system, hypothalamic-pituitary-adrenal axis, neurotransmitter, and autonomic nerve system. The figure shows communication between peripheral and central cytokine system. Early innate proinflammatory cytokines released by macrophage (TNF-α, IL-1, IL-6 and INF-α), and late acquired T cell cytokines (IL-2 and INF-γ) stimulate glucocorticoid secretion by acting at all three levels of the HPA axis. Glucocorticoids are negatively feedback on the peripheral immune system to suppress the production of proinflammatory cytokines. Glucocorticoids also play an important role in causing a shift from cellular (T-helper 1) to humoral (T-helper 2) immune responses. The central cytokines are usually secreted from the astrocyte or microglia. Central cytokines (IL-1, IL-6, TNF-α, and IFN-γ) are considered to be involved in neuroplasticity in brain. The neurotransmitters (NA, ACH, and 5-HT) regulate the peripheral cytokines by changing the cortisol concentration level. The Ach, DA, and NA promote the secretion of the CRH in hypothalamus, and 5-HT inhibits the secretion of the CRH in hypothalamus and the ACTH in pituitary. The ANS also regulates the peripheral cytokine production. The parasympathetic nerve directly reaches the immune system while the sympathetic nerve affects the immune system through the NA secretion from the peripheral sympathetic ganglia. 5-HT: Serotonin; ACH: Acetylcholine; ACTH: Adrenocorticotropic hormone; ANS: Autonomic nerve system; CRH: Corticotropin-releasing factor; DA: Dopamine; HPA: Hypothalamic-pituitary-adrenal; NA: Noradrenalin; PVN: Paraventricular nucleus of the hypothalamus; TH: Helper T cell; IL: Interleukins; TNF: Tumor necrosis factor; IFN: Interferons.
Because peripheral cytokines are hydrophilic and have large molecular weights, they are unable to pass through the blood-brain barrier (BBB) in their normal state. However, they can pass through the BBB in pathological states that involve increased BBB permeability. Moreover, cytokines are also able to affect the CNS through mediators, such as nitric oxide or prostaglandins released in response to cytokines[9,10]. IL-1 receptors are densely distributed in glial cells near arterioles or the plexus choroideus[11]. This suggests that the IL-1 receptors in the CNS and IL-1 in the peripheral blood actively communicate with each other. Additional channels through which peripheral cytokines transmit immune signals to the CNS include passive diffusion through the circumventricular organs (brain regions that do not have a BBB), active transport to the CNS, and nerve conduction pathways through the vagus nerve[6].
Central cytokines are usually secreted from astrocytes or microglia, but neurons can secrete them in certain conditions[12]. Central cytokines are produced in a number of brain regions, including the circumventricular region, hypothalamus, hippocampus, cerebellum, forebrain, basal ganglia, and brain stem nuclei[13]. IL-1, which is secreted from the brain, is found in the hypothalamus and hippocampus[14]. The roles of central cytokines in the brain are not fully understood. However, the proinflammatory cytokines IL-1, IL-6, TNF-α, and IFN-γ have been implicated in neuronal development, neuroplasticity, synaptogenesis, and tissue repair[15]. Proinflammatory cytokines promote neuronal necrosis after traumatic brain injuries[16].
Cytokine receptors are located in the immune system and various tissues, including the peripheral nervous system and CNS. For example, IL-1, IL-2, IL-6, and TNF-α receptors are densely distributed in the hippocampus and hypothalamus[17]. IL-1 has two receptor types: Type I and type II. The nuclear transcription factor nuclear factor kappa B (NFκB) is activated and intracellular signals can be transmitted through the type I receptor. A role of cytokines in specific mental functions and/or mental diseases has been suggested because of the locations of their receptors in the CNS and not because of the specific functions of the cytokines. The important CNS structures that are affected by central cytokines include the locus coeruleus, hippocampus, prefrontal cortex, and hypothalamus. These CNS structures are associated with the biological processes that underlie psychological changes[18].
THE CYTOKINE HYPOTHESIS OF DEPRESSION
Stress-cytokine-inflammation-depression
According to the cytokine hypothesis (Figure 2), internal or external stress induces cytokine imbalances that play important roles in the expression and continuity of depressive symptoms in vulnerable individuals[19]. A number of major research findings support the cytokine hypothesis.
Figure 2.
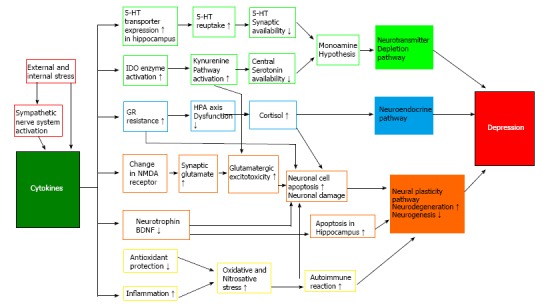
Schematic representation of neuroinflammatory pathways in the pathogenesis of depression. Cytokine production is initially activated by stress and sympathetic nerve system activation. In turn, cytokines have an important role by acting via neurotransmitter depletion pathway, neuroendocrine pathway, and neural plasticity pathway. There are multiple interactions between these pathways suggesting existence of a complex model for pathogenesis of depression. 5-HT: Serotonin; BDNF: Brain derived neurotrophic factor; GR: Glucocorticoid receptor; HPA: Hypothalamic-pituitary-adrenal; IDO: Indoleamine-2,3-dioxygenase; NMDA: N-methyl-D-aspartate.
First, the injection of cytokines into animals and humans induces depression-like symptoms. Depression occurs frequently in patients with hepatitis C undergoing INF treatment. Of note in one study, 23% of patients during INF treatment satisfied the diagnostic criteria for major depressive disorders; in 74% of them depression occurred within 2 mo after the start of INF treatment[20]. The levels of IL-6 and TNF-α, which increase after IFN-α administration, are significantly associated with the severity of depression[21]. Polymorphisms in the 5-hydroxytryptamine (5-HT) transporter and IL-6 genes contribute to the fatigue and depressive symptoms that are observed after IFN-α administration[22].
Second, increases in the levels of proinflammatory cytokines, such as IL-1, IL-6, IL-12, TNF-α, prostaglandin E2 (PGE2), and negative immunoregulatory cytokines have been observed in patients with depression[23,24].
Third, cytokines trigger activity in the HPA axis and the catecholamine/sympathetic nervous system, two biological systems that are closely associated with the pathophysiology of depression[2]. Cytokines stimulate corticotrophin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH), and activate the HPA axis[25]. In addition, cytokines activate indoleamine-2,3-dioxygenase (IDO), which catalyzes the metabolism of the 5-HT precursor tryptophan to kynurenine, and inhibits 5-HT synthesis in the brain[26]. The proinflammatory cytokine, NA, and DA promote CRF secretion, activate the sympathetic nerve system, and promote immune reactions. During this process, the temperature of the CNS increases and sickness behaviors may be induced[27]. Sickness behaviors refer to behavioral changes that are observed during an infection period. These include feelings of helplessness, depressive mood, anxiety, hypersomnia, loss of appetite, and inattention. Based on findings that patients with depression exhibit increased levels of proinflammatory cytokines in the plasma[23,24], decreased levels of anti-inflammatory cytokines[28], and increased levels of PGE2 in the cerebrospinal fluid[29], depression is considered a sickness behavior.
Fourth, antidepressants improve depressive symptoms by inhibiting cytokine secretion from immune cells or by acting as an antagonist of cytokine receptors. Antidepressants inhibit proinflammatory cytokine secretion from monocytes or macrophages, act as chemotaxis inhibitors, and increase the production of anti-inflammatory cytokines[30]. An in vitro study reported anti-inflammatory reactions with therapeutic doses of antidepressants that involved the inhibition of IFN-γ and increased IL-10[31]. In addition, antidepressants significantly inhibit the lipopolysaccharide-induced production of IL-1β, IL-6, and TNF-α, as well as the secretion of IL-2 and IFN-γ in T cells[32].
In summary, neuroinflammation and cytokines, which affect patterns of brain signal transmission, are important in the psychopathology of depression and mechanism of antidepressants. Furthermore, they are associated with neurogenesis and neural plasticity in the brain. Thus, neuroinflammation and cytokines appear to cause or continue depression and might be useful for determining the diagnosis and prognosis of depression. Epidemiological studies support the view that increased levels of IL-6, IL-1ra, and C-reactive protein (CRP) can be harnessed to predict the occurrence of depression[33]. A recent meta-analysis demonstrated that the markers of inflammation with relatively consistent increases in patients with depression are IL-6, TNF-α, TNF-β1, IFN, and CRP[34].
ARE CYTOKINES A CAUSE OF DEPRESSION?
Cytokine, HPA-axis activation, and glucocorticoid receptor resistance
HPA-axis activation is one of the most important biological findings in depression research. The activation results in increased cortisol concentrations in the plasma, urine, and cerebrospinal fluid, and exaggerated cortisol responses against ACTH[35]. HPA-axis activation has been suggested to result from excessive secretion of CRF, which triggers depressive mood, loss of appetite, and sleep disturbance[36]. These suggestions are supported by findings of increased CRF levels in the cerebrospinal fluid, increased levels of CRF mRNA in the paraventricular nucleus of the hypothalamus, blunted ACTH responses in CRH tests due to the down-regulation of CRF receptors in the pituitary gland in patients with depression, and also by the down-regulation of CRF receptors in the frontal cortex to compensate for CRF oversecretion in patients who committed suicide[35,36]. In patients with depression, CRF oversecretion can appear due to dysfunction of the negative feedback of glucocorticoids. Accordingly, cortisol is not inhibited in dexamethasone suppression tests in these patients. This might be due to deterioration in glucocorticoid receptor (GR) sensitivity. Glucocorticoid resistance results in absence of inhibition in the dexamethasone suppression test and CRF oversecretion.
Cytokines can cause HPA-axis activation, increased CRF, and glucocorticoid resistance. Proinflammatory cytokines, such as IL-1β and IL-6, stimulate CRH secretion from the paraventricular nucleus of the hypothalamus, activate the HPA axis, and promote ACTH and glucocorticoid secretion[37] (Figure 1). In early depression research, the cortisol oversecretion from HPA-axis activation was thought to inhibit immune function[32]. However, more recently, immune cells are thought to be unaffected by cortisol in chronic stress and depression because of the inhibition of GR function in immune cells[38]. This may be a result of the oversecretion of proinflammatory cytokines from increased cell-mediated immunity[38]. In support of this, IL-1 inhibits the translocation of the GR from the cytoplasm into the cell nucleus and GR-mediated gene transcription[39]. These findings suggest that cytokines directly affect GR function and induce glucocorticoid resistance. Moreover, antidepressants like desipramine stimulate GR translocation from the cytoplasm to the nucleus and increase GR-mediated gene transcription, which eventually promotes the feedback inhibition that is mediated by glucocorticoids in the HPA axis[40].
Theoretically, the glucocorticoid increase that is induced by HPA-axis activation and the cytokine increase that results from immune activation are not likely to occur at the same time in depression, but an inverse correlation should be observed because the synthetic glucocorticoids that are used to treat inflammatory diseases inhibit the release of proinflammatory cytokines and their synthesis, which results in anti-inflammatory effects[41]. However, no inverse correlations between the concentrations of plasma glucocorticoids and cytokines have been observed in patients with depression. Because the negative inhibitory mechanism of cortisol that prevents increased levels of CRF is impaired in patients with depression, the negative inhibitory mechanism of cytokine secretion in immune cells against increased levels of cortisol is also impaired in these patients. In other words, depression is characterized as a dysfunction of the cortisol feedback inhibitory mechanism to the GR, which is the mechanism for inhibiting CRF oversecretion, and to immune cell receptors, which is the mechanism for inhibiting cytokine oversecretion. These impairments may be directly related to the etiology of depression.
Cytokines and central neurotransmission
Stress simultaneously activates the HPA axis and the sympathoadrenal system (sympathetic nervous system and adrenal medulla). The most important stress response is activation of the noradrenergic (NA) neurons, which show stress responses through pathways from the locus coeruleus to the cortex, hippocampus, and cerebellum, and from the nucleus tractus solitarius to the hypothalamus. Dopaminergic (DA) neurons display stress responses through the nigrostriatal, mesolimbic, and mesocortical pathways. The mesocortical system, which connects the prefrontal cortex and cingulate, is the most important. The stress responses of the serotonergic (5-HT) system result in significant increases in tryptophan in all brain regions. These increases are not localized to the specific brain areas where 5-HT neurons are present. During stress responses, changes in the metabolism and secretion of neurotransmitters, such as Ach and γ-aminobutyric acid (GABA), are observed. Neuropeptides of the peptidergic system, which include CRF, are also involved in the stress response.
According to the monoamine depletion hypothesis, depression develops as a result of decreased availability of monoamine neurotransmitters, especially 5-HT and NA, in the synapse. Proinflammatory cytokines significantly affect the peripheral and central 5-HT systems. Peripheral injections of IL-1β and TNF-α increase the extracellular levels of 5-hydroxyindoleacetic acid (5-HIAA) in the nucleus raphe dorsalis, and central injection (intracerebroventricular application) of IL-1β, IFN-γ, and TNF-α stimulates the 5-HT transmission in the nucleus raphe dorsalis[42]. Peripheral injections of IL-1 increase NA turnover in the hypothalamus and hippocampus, 5-HT turnover in the hippocampus and prefrontal cortex, and DA turnover in the prefrontal cortex[43]. In an in vitro study, IL-1β increased the activity of the 5-HT transporter[44], which has a critical role in 5-HT transmission because it facilitates 5-HT reuptake. If the 5-HT transporter is activated in central 5-HT neurons, the amount of 5-HT removed from the synapses increases, which results in a deterioration of 5-HT-mediated functions. In addition, IL-1β receptors are expressed in 5-HT neurons and IL-1β is synthesized in neurons and glia[45]. IL-1 and IFN-γ increase the activity of IDO, which promotes the metabolism of tryptophan and decreases 5-HT synthesis in the brain[26]. IL-1β acts on 5-HT transporters to increase 5-HT reuptake in the synapse, and the decreased concentrations of serum tryptophan decrease the usefulness of the 5-HT system, which eventually induces depression (Figure 2).
The neurodegeneration hypothesis of depression: Cytokine-5-HT interaction
According to the monoamine hypothesis, patients with depression have a vulnerable 5-HT system. Their 5-HT turnover is increased and then becomes depleted, inducing 5-HT2 receptor up-regulation. The levels of tryptophan, a precursor of 5-HT, in the blood of patients with depression are decreased compared with those of healthy people[46]. Eating tryptophan-deficient foods worsens mood, while the administration of tryptophan improves depressive symptoms[47].
Depression is significantly associated with old age, chronic medical diseases (e.g., coronary heart diseases, diabetes, Parkinson’s disease, stroke, and cancer), and chronic stress. Nevertheless, not all elderly or chronically ill people experience depression. How can these individual differences be explained? One hypothesis that can explain these differences is the neurodegeneration hypothesis: Cytokine-5-HT interaction[47,48].
According to this hypothesis, acute psychological stress triggers tryptophan defects and mood swings. To correct the 5-HT imbalance, 5-HT synthesis and receptor expression are modified. This is the first stage of coping with psychological stress. If the psychological stress is chronic, the levels of proinflammatory cytokines increase. The levels of proinflammatory cytokines also increase in cases of physical stress or chronic diseases. These increases in proinflammatory cytokines trigger an increase in the levels of anti-inflammatory cytokines as a compensatory mechanism in order to maintain balance. This is the second stage. If the balance is not maintained and the levels of proinflammatory cytokines increase excessively, animals show sickness behaviors, while humans show depressive symptoms. The increased levels of proinflammatory cytokines activate the IDO enzyme and accelerate the metabolism of tryptophan to kynurenine. The level of 5-HT in the brain decreases, which further aggravates the symptoms of depression in individuals vulnerable to depression. Through the complicated tryptophan metabolism process, the neurodegenerative quinolinate and the neuroprotective kynurenate are formed in the brain. This is the third stage, which is important for maintaining balance between neurodegeneration and neuroprotection (Figure 2).
Minor neurodegeneration can occur in all people. However, in the elderly population or in individuals with severe stress or chronic diseases, the balance between the proinflammatory cytokines and anti-inflammatory cytokines is lost, and the process of metabolizing tryptophan to kynurenine is accelerated, which lowers the concentration of 5-HT in the brain. If the balance between neurodegeneration and neuroprotection is also lost, neurodegeneration begins. Neurodegeneration in brain regions including the hippocampus and frontal lobe results in cognitive and memory impairments. As a result, the neurodegeneration process inhibits all of the brain strategies that might cope with the stress, which induces depression or treatment-resistant depression. The neurodegeneration process is further deteriorated due to neurotoxicity from cortisol oversecretion from stress-induced HPA-axis activation[49].
The neurodegeneration hypothesis of depression can explain the development of depression in the elderly and chronically ill. In addition, it suggests methods for coping with various stresses in various stages according to the stress intensity and period. In a recent study[49], patients with depression showed a significantly higher tryptophan breakdown index and lower kynurenic acid concentration level compared with those in the normal population. These findings imply that patients with depression exhibit decreased levels of neuroprotection markers, which supports the neurodegeneration hypothesis.
Cytokines, microglia, and neurogenesis
Cytokines have been reported to promote neuronal differentiation and remodeling in the brain. Accordingly, their roles in neurodegenerative diseases are of interest. The brains of patients with chronic depression show increased cell apoptosis with decreased volumes of the hippocampus, prefrontal cortex, and amygdala and increased ventricular volume. The chances for developing dementia increase accordingly in these patients, and chronic inflammatory responses are thought to be involved in this process[50]. Proinflammatory cytokines reduce neuroplasticity by increasing the levels of quinolinic acid, which is a strong agonist of the N-methyl-D-aspartate (NMDA) receptor[51] (Figure 2).
Stress induces inflammatory responses through cytokine secretion. Cytokines are secreted from peripheral immune cells and central immune cells. Chronic stress activates brain microglia, which secrete cytokines and in turn affect neurogenesis. Neurogenesis is either inhibited or stimulated according to the level of microglia activation[52]. This means that various microglia perform various functions, such as stimulating or inhibiting neurons[52]. Inflammation and cytokines usually directly inhibit neurogenesis. Proinflammatory cytokines, such as TNF-α and INF-α, inhibit neurogenesis through IL-1 regulation[53]. The decline of neurogenesis is prevented by inhibiting IL-1β activity[54], confirming the important role of cytokines in inhibiting neurogenesis in the brain. In contrast, the administration of drugs that inhibit inflammation recovered or increased neurogenesis[55]. In summary, chronic stress promotes cytokine secretion in the peripheral blood and brain microglia, and cytokines affect neurogenesis.
CYTOKINES AND ANTIDEPRESSANTS
The mechanisms of antidepressants relative to cytokines
Stress induces proinflammatory cytokine oversecretion, which results in depressive symptoms. Antidepressants may inhibit the production and function of peripheral and brain cytokines. As mentioned above, the use of antidepressants decreases the levels of proinflammatory cytokines and increases the levels of anti-inflammatory cytokines[30]. These findings imply that antidepressants inhibit cytokine secretion in immune cells and/or antagonize cytokine receptors to improve the depressive symptoms.
How do antidepressants regulate cytokine secretion and improve depressive symptoms? Several hypotheses have been suggested[19,56-58]. First, the changes in peripheral and central cytokines after antidepressant treatment might be secondary results of the neurotransmitter changes that are induced by antidepressants. Stress-induced increases in IL-6 levels are inhibited by pretreatment with propranolol, a β-adrenoceptor antagonist, which suggests that the IL-6 increases could be mediated by the sympathetic nervous system and increased adrenalin in the adrenal medulla. Immune cells have neurotransmitter receptors, and antidepressants act on these receptors to regulate immune cell activity. T lymphocytes express 5-HT receptors (5-HT1A and 5-HT2A/2C) and high-affinity 5-HT transporters. Macrophages have a 5-HT uptake system that is similar to the system in platelets. Antidepressants can have negative immunoregulatory effects by causing deficiencies in intracellular 5-HT storage, increases in extracellular 5-HT, and blocking 5-HT2A/2C receptors; second, antidepressants restore the cytokine-induced GR resistance. In addition, they restore the inhibition of the negative feedback of the HPA axis and normalize HPA axis function; third, antidepressants inhibit the nitric oxide and PGE2 production that is increased by the cytokines; fourth, antidepressants inhibit IDO activity; fifth, antidepressants directly act on macrophages and lymphocytes to stimulate the production of anti-inflammatory cytokines.
In meta-analyses of 22 types of antidepressants, treatments with antidepressants, especially selective serotonin reuptake inhibitors (SSRIs), result in decreased levels of IL-1β and IL-6[58]. Inflammation increases the activities of the microglial cells and induce astroglial loss, which consequently induces glutamate release and an upregulation of NMDA receptors[59]. The anti-inflammatory effects of riluzole and ketamine, which are glutamatergic modulators, are being studied. Riluzole and ketamine prevent neurotoxicity and relieve inflammation by inhibiting glutamate secretion and modulating NMDA receptors[60].
Promising cytokine-related antidepressants
If cytokines are associated with the pathophysiology of depression, then receptor antagonists that can regulate inflammatory cytokines, anti-cytokine antibodies, and anti-inflammatory cytokines might improve depressive symptoms. Although the therapeutic usefulness of cytokine inhibitors in depression treatment has not been fully investigated, the possibility has been suggested by the results of experimental studies.
The long-term administration of antidepressants in mice results in significant increases in the mRNA levels of IL-1ra in the hypothalamus, hippocampus, frontal lobe, and diencephalon. The learned helplessness that is induced by inescapable shocks is inhibited in mice that were pretreated with IL-1ra[61]. These results suggest that stress-induced IL-1 secretion is a primary cause of the behavioral disturbances shown in the learned helplessness model of depression. CRF receptor antagonists could prevent learned helplessness[62] because the behavioral changes induced by IL-1 occur through central CRF secretion. The cytokine antagonists with a broad action range, such as IL-4 and IL-10, might be more effective than cytokine antagonists, such as IL-1ra, which inhibit specific cytokines, in the treatment of depression. In one study, seven severely depressed patients given low-dose lipopolysaccharide showed improved symptoms the next day, when the levels of anti-inflammatory cytokines were expected to peak[63]. These mood changes were transient, and the patients’ previous conditions returned after several days. TNF-α has most recently drawn attention as a treatment that can change the course of bipolar disorder (disease-modifying treatment)[64]. Current evidence suggests that TNF-α regulates apoptotic cascades that may be associated with neuronal and glial loss in bipolar disorder. TNF-α antagonists, such as adalimumab, etanercept, and infliximab, have been used as therapeutic agents for rheumatic diseases, and are currently being used in clinical trials to treat the depressive episodes of patients with bipolar disorder[64].
The antidepressant effects of anti-inflammatory drugs
If inflammatory responses contribute to the pathogenesis of depression, anti-inflammatory agents are expected to be effective for treating depression. The use of celecoxib, a COX-2 inhibitor, to augment SSRI treatment resulted in better treatment effects compared with the use of the SSRI alone[65]. Celecoxib augmentation therapy can accelerate the treatment responses in depressive episodes of patients with bipolar disorder[66]. In addition, acetylsalicylic acid (aspirin) improves the effects of SSRIs[67]. However, the combination of SSRIs and non-steroidal anti-inflammatory drugs inhibits the effects of the antidepressant. The effects of citalopram, which regulates TNF-α and IFN-γ in mouse frontal lobe, are inhibited by ibuprofen[68]. These laboratory results have been confirmed in clinical trials; the combined use of citalopram and anti-inflammatory drugs resulted in more treatment failures than the use of citalopram alone. These results were not found in subsequent studies[69]. These discrepancies suggest that inflammatory drug reactions vary according to depression subtype.
Eicosapentaenoic acid and docosahexaenoic acid, which are omega-3 fatty acids, can be used to treat rheumatoid arthritis, psoriasis, asthma, and inflammatory bowel diseases because they reduce proinflammatory cytokines. In addition, they can be used as a supplemental agent of antidepressants[70]. Angiotensin receptor blockers, which are hypertension agents, are thought to have anti-inflammatory effects in the CNS. When their mechanism is fully understood, they can be used in depression treatment.
LIMITATIONS
Limitations of the cytokine hypothesis of depression
Many clinical studies have suggested that the neuroimmune activation that results from the production of proinflammatory cytokines is significantly involved in the etiology and pathogenesis of depression. However, the cytokine hypothesis of depression is still controversial due to the following limitations.
First, the cytokine hypothesis states that increased levels of proinflammatory cytokines cause secondary changes, such as neurotransmitter depletion, HPA-axis activation, and the expression of depressive symptoms. Nevertheless, it has not been clarified if the increased levels of proinflammatory cytokines are the cause of depression or are a concomitant phenomenon that maintains the depressive symptoms regardless of the depression etiology. When IFN-α was used in cancer patients as an immunotherapy, the patients developed depression. When the immunotherapy was discontinued or antidepressants were administered, the depressive symptoms improved[20,21]. Accordingly, the proinflammatory cytokines probably function as a causative factor in patients with depression from medical diseases or immunotherapies. However, it is still unclear how the proinflammatory cytokines function as a causative factor in patients with depression that is caused by etiologies other than diseases or immunotherapies[71].
Second, the effects of antidepressant treatments are not always associated with decreased levels of proinflammatory cytokines[72,73]. The effects of antidepressant treatment may not be caused by decreased cytokine release or synthesis but rather by disturbance of the actions of peripheral-released cytokines on the CNS regardless of the concentration of released cytokines. Thus, antidepressants may not directly inhibit immune activation but, rather, indirectly regulate immune functions.
Third, previous studies have shown that the increased levels of cytokines in depression are within a low range compared with those in systemic infection and inflammation. In acute infection, the huge amounts of cytokines are produced act on the brain functions, which often results in the progression of depression. However, in most clinical conditions, such as chronic infection and inflammation, only low amounts of cytokines circulate. Thus, there is a question whether and how low amounts of peripheral cytokines act on the brain and develop depression, even under baseline conditions. The difference in brain function affected by low and high levels of cytokines is another issue. Low levels of peripheral cytokines have a similar effect on sleep-awake behaviors compared with a high dose of peripheral cytokines[74]. On the other hand, low levels of cytokines promote non-REM sleep, but this stage of sleep is suppressed by high level of cytokines[75]. It is still unclear, if the doses have similar effects on depression[74]. Because insufficient research for depression and brain functions in various levels of cytokines, the answer to these questions depend on further experimental studies.
It is unclear whether depression is caused by increased neuroinflammation or vice versa because depression diagnoses are made by examining patients’ histories of perceptible symptoms and the intrinsic heterogeneity and various environmental factors of the patients are not controlled. We may consider various aspects of the cytokine hypothesis of depression. These include genetic aspects, role of early life stress and trauma, information on modulators of cytokine activity in depression (diet, obesity, gut health, physical condition, sleep deprivation, vitamin D deficiency), medical illness, differences of cytokine activities between animal models and human, inflammatory markers in suicide, and the influence of treatments like antidepressant drugs, psychotherapy, and electroconvulsive therapy.
CONCLUSION
Depression is considered a syndrome that includes diverse symptoms and a mental disorder with various causes. No single mechanism that explains every aspect of depression exists. Some depressive symptoms appear in association with the cell proteins that are produced by the complicated intracellular signal transmission of neurotransmitters, such as 5-HT and NE. However, in some cases, childhood stress sustains CRF hyperactivity and increases stress responses in adulthood, which then results in the oversecretion of cerebral CRF and eventually leads to depression[75,76]. In other cases, increases in the levels of cytokines from immune system activation activate the HPA axis and increase neurotransmitter turnover, thus leading to depression. These findings suggest that either various etiologies can be observed at the same time in a single patient with depression or that a specific etiology can be dominant.
From a psychoneuroimmunological point of view, the immune, endocrine, and neurotransmission systems closely interact with each other, and inflammation acts as an allostatic load to disconnect them. Depression can be caused by these functional impairments. The sickness behaviors that are observed under inflammatory conditions are similar to depressive symptoms, and some cytokine treatments lead to depression. These results confirm the association between inflammation and depression. Cytokines including IL-1, IL-2, IL-6, IFN-γ, and TNF-α, and hormones like CRF and glucocorticoid have been suggested as inflammation markers. Inflammatory responses that are thought to affect the synthesis and transmission of neurotransmitters, glucocorticoid resistance, and neurodegeneration/neurogenesis contribute to the onset of depression and inhibit recovery. Although no definitive markers of inflammation have been established, they could at least be used in vulnerable patients with depression. Future studies on the mechanisms of neuroinflammation are expected to help overcome the limitations of the monoamine theory and contribute to finding new solutions for the diagnosis and treatment of depression.
Footnotes
Conflict-of-interest statement: No potential conflicts of interest. No financial support.
Manuscript source: Invited manuscript
Specialty type: Psychiatry
Country of origin: South Korea
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B, B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: May 9, 2016
First decision: June 13, 2016
Article in press: August 29, 2016
P- Reviewer: Chakrabarti S, Gabriel A, Gazdag G, Tsai SJ S- Editor: Qiu S L- Editor: A E- Editor: Lu YJ
References
- 1.Maes M. The cytokine hypothesis of depression: inflammation, oxidative & amp; nitrosative stress (IO& amp; NS) and leaky gut as new targets for adjunctive treatments in depression. Neuro Endocrinol Lett. 2008;29:287–291. [PubMed] [Google Scholar]
- 2.Leonard BE. HPA and immune axes in stress: involvement of the serotonergic system. Neuroimmunomodulation. 2006;13:268–276. doi: 10.1159/000104854. [DOI] [PubMed] [Google Scholar]
- 3.Szelényi J. Cytokines and the central nervous system. Brain Res Bull. 2001;54:329–338. doi: 10.1016/s0361-9230(01)00428-2. [DOI] [PubMed] [Google Scholar]
- 4.Kronfol Z, Remick DG. Cytokines and the brain: implications for clinical psychiatry. Am J Psychiatry. 2000;157:683–694. doi: 10.1176/appi.ajp.157.5.683. [DOI] [PubMed] [Google Scholar]
- 5.Felger JC, Miller AH. Neurotherapeutic implications of brain-immune interactions. Neuropsychopharmacology. 2014;39:242–243. doi: 10.1038/npp.2013.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watkins LR, Nguyen KT, Lee JE, Maier SF. Dynamic regulation of proinflammatory cytokines. Adv Exp Med Biol. 1999;461:153–178. doi: 10.1007/978-0-585-37970-8_10. [DOI] [PubMed] [Google Scholar]
- 7.Calogero AE, Gallucci WT, Chrousos GP, Gold PW. Catecholamine effects upon rat hypothalamic corticotropin-releasing hormone secretion in vitro. J Clin Invest. 1988;82:839–846. doi: 10.1172/JCI113687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 9.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gądek-Michalska A, Tadeusz J, Rachwalska P, Bugajski J. Cytokines, prostaglandins and nitric oxide in the regulation of stress-response systems. Pharmacol Rep. 2013;65:1655–1662. doi: 10.1016/s1734-1140(13)71527-5. [DOI] [PubMed] [Google Scholar]
- 11.Wong ML, Bongiorno PB, Gold PW, Licinio J. Localization of interleukin-1 beta converting enzyme mRNA in rat brain vasculature: evidence that the genes encoding the interleukin-1 system are constitutively expressed in brain blood vessels. Pathophysiological implications. Neuroimmunomodulation. 1995;2:141–148. doi: 10.1159/000096884. [DOI] [PubMed] [Google Scholar]
- 12.Freidin M, Bennett MV, Kessler JA. Cultured sympathetic neurons synthesize and release the cytokine interleukin 1 beta. Proc Natl Acad Sci USA. 1992;89:10440–10443. doi: 10.1073/pnas.89.21.10440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maier SF, Watkins LR. Cytokines for psychologists: implications of bidirectional immune-to-brain communication for understanding behavior, mood, and cognition. Psychol Rev. 1998;105:83–107. doi: 10.1037/0033-295x.105.1.83. [DOI] [PubMed] [Google Scholar]
- 14.Goshen I, Yirmiya R. Interleukin-1 (IL-1): a central regulator of stress responses. Front Neuroendocrinol. 2009;30:30–45. doi: 10.1016/j.yfrne.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Khairova RA, Machado-Vieira R, Du J, Manji HK. A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int J Neuropsychopharmacol. 2009;12:561–578. doi: 10.1017/S1461145709009924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothwell NJ, Luheshi G, Toulmond S. Cytokines and their receptors in the central nervous system: physiology, pharmacology, and pathology. Pharmacol Ther. 1996;69:85–95. doi: 10.1016/0163-7258(95)02033-0. [DOI] [PubMed] [Google Scholar]
- 18.Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35:2–11. doi: 10.1080/07853890310004075. [DOI] [PubMed] [Google Scholar]
- 19.Schiepers OJ, Wichers MC, Maes M. Cytokines and major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:201–217. doi: 10.1016/j.pnpbp.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Horikawa N, Yamazaki T, Izumi N, Uchihara M. Incidence and clinical course of major depression in patients with chronic hepatitis type C undergoing interferon-alpha therapy: a prospective study. Gen Hosp Psychiatry. 2003;25:34–38. doi: 10.1016/s0163-8343(02)00239-6. [DOI] [PubMed] [Google Scholar]
- 21.Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB, Miller AH. Neurobehavioral effects of interferon-alpha in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology. 2002;26:643–652. doi: 10.1016/S0893-133X(01)00407-9. [DOI] [PubMed] [Google Scholar]
- 22.Bull SJ, Huezo-Diaz P, Binder EB, Cubells JF, Ranjith G, Maddock C, Miyazaki C, Alexander N, Hotopf M, Cleare AJ, et al. Functional polymorphisms in the interleukin-6 and serotonin transporter genes, and depression and fatigue induced by interferon-alpha and ribavirin treatment. Mol Psychiatry. 2009;14:1095–1104. doi: 10.1038/mp.2008.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myint AM, Leonard BE, Steinbusch HW, Kim YK. Th1, Th2, and Th3 cytokine alterations in major depression. J Affect Disord. 2005;88:167–173. doi: 10.1016/j.jad.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 24.Lee KM, Kim YK. The role of IL-12 and TGF-beta1 in the pathophysiology of major depressive disorder. Int Immunopharmacol. 2006;6:1298–1304. doi: 10.1016/j.intimp.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 25.Besedovsky HO, del Rey A. The cytokine-HPA axis feed-back circuit. Z Rheumatol. 2000;59 Suppl 2:II/26–II/30. doi: 10.1007/s003930070014. [DOI] [PubMed] [Google Scholar]
- 26.Raison CL, Borisov AS, Majer M, Drake DF, Pagnoni G, Woolwine BJ, Vogt GJ, Massung B, Miller AH. Activation of central nervous system inflammatory pathways by interferon-alpha: relationship to monoamines and depression. Biol Psychiatry. 2009;65:296–303. doi: 10.1016/j.biopsych.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silverman MN, Pearce BD, Biron CA, Miller AH. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005;18:41–78. doi: 10.1089/vim.2005.18.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kubera M, Maes M, Holan V, Basta-Kaim A, Roman A, Shani J. Prolonged desipramine treatment increases the production of interleukin-10, an anti-inflammatory cytokine, in C57BL/6 mice subjected to the chronic mild stress model of depression. J Affect Disord. 2001;63:171–178. doi: 10.1016/s0165-0327(00)00182-8. [DOI] [PubMed] [Google Scholar]
- 29.Song C, Lin A, Bonaccorso S, Heide C, Verkerk R, Kenis G, Bosmans E, Scharpe S, Whelan A, Cosyns P, et al. The inflammatory response system and the availability of plasma tryptophan in patients with primary sleep disorders and major depression. J Affect Disord. 1998;49:211–219. doi: 10.1016/s0165-0327(98)00025-1. [DOI] [PubMed] [Google Scholar]
- 30.Vogelzangs N, Duivis HE, Beekman AT, Kluft C, Neuteboom J, Hoogendijk W, Smit JH, de Jonge P, Penninx BW. Association of depressive disorders, depression characteristics and antidepressant medication with inflammation. Transl Psychiatry. 2012;2:e79. doi: 10.1038/tp.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki E, Shintani F, Kanba S, Asai M, Nakaki T. Induction of interleukin-1 beta and interleukin-1 receptor antagonist mRNA by chronic treatment with various psychotropics in widespread area of rat brain. Neurosci Lett. 1996;215:201–204. doi: 10.1016/0304-3940(96)12985-2. [DOI] [PubMed] [Google Scholar]
- 32.Nabriski D, Saperstein A, Brand H, Jain R, Zwickler D, Hutchinson B, Rosenzweig S, Hollander CS, Audhya T. Role of corticotropin-releasing factor in immunosuppression. Trans Assoc Am Physicians. 1991;104:238–247. [PubMed] [Google Scholar]
- 33.Glover AT, Shaw SM, Williams SG, Fildes JE. Can inflammation be an independent predictor of depression? Brain Behav Immun. 2010;24:173; author reply 174–175. doi: 10.1016/j.bbi.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 34.Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctôt KL. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–457. doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 35.Pariante CM, Lightman SL. The HPA axis in major depression: classical theories and new developments. Trends Neurosci. 2008;31:464–468. doi: 10.1016/j.tins.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Nemeroff CB. The corticotropin-releasing factor (CRF) hypothesis of depression: new findings and new directions. Mol Psychiatry. 1996;1:336–342. [PubMed] [Google Scholar]
- 37.Dunn AJ. Cytokine activation of the HPA axis. Ann N Y Acad Sci. 2000;917:608–617. doi: 10.1111/j.1749-6632.2000.tb05426.x. [DOI] [PubMed] [Google Scholar]
- 38.Miller AH, Pariante CM, Pearce BD. Effects of cytokines on glucocorticoid receptor expression and function. Glucocorticoid resistance and relevance to depression. Adv Exp Med Biol. 1999;461:107–116. doi: 10.1007/978-0-585-37970-8_7. [DOI] [PubMed] [Google Scholar]
- 39.Pariante CM, Pearce BD, Pisell TL, Sanchez CI, Po C, Su C, Miller AH. The proinflammatory cytokine, interleukin-1alpha, reduces glucocorticoid receptor translocation and function. Endocrinology. 1999;140:4359–4366. doi: 10.1210/endo.140.9.6986. [DOI] [PubMed] [Google Scholar]
- 40.Pariante CM, Pearce BD, Pisell TL, Owens MJ, Miller AH. Steroid-independent translocation of the glucocorticoid receptor by the antidepressant desipramine. Mol Pharmacol. 1997;52:571–581. doi: 10.1124/mol.52.4.571. [DOI] [PubMed] [Google Scholar]
- 41.Almawi WY, Beyhum HN, Rahme AA, Rieder MJ. Regulation of cytokine and cytokine receptor expression by glucocorticoids. J Leukoc Biol. 1996;60:563–572. doi: 10.1002/jlb.60.5.563. [DOI] [PubMed] [Google Scholar]
- 42.Clement HW, Buschmann J, Rex S, Grote C, Opper C, Gemsa D, Wesemann W. Effects of interferon-gamma, interleukin-1 beta, and tumor necrosis factor-alpha on the serotonin metabolism in the nucleus raphe dorsalis of the rat. J Neural Transm (Vienna) 1997;104:981–991. doi: 10.1007/BF01273312. [DOI] [PubMed] [Google Scholar]
- 43.Zalcman S, Green-Johnson JM, Murray L, Nance DM, Dyck D, Anisman H, Greenberg AH. Cytokine-specific central monoamine alterations induced by interleukin-1, -2 and -6. Brain Res. 1994;643:40–49. doi: 10.1016/0006-8993(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 44.Ramamoorthy S, Ramamoorthy JD, Prasad PD, Bhat GK, Mahesh VB, Leibach FH, Ganapathy V. Regulation of the human serotonin transporter by interleukin-1 beta. Biochem Biophys Res Commun. 1995;216:560–567. doi: 10.1006/bbrc.1995.2659. [DOI] [PubMed] [Google Scholar]
- 45.Cunningham ET, De Souza EB. Interleukin 1 receptors in the brain and endocrine tissues. Immunol Today. 1993;14:171–176. doi: 10.1016/0167-5699(93)90281-o. [DOI] [PubMed] [Google Scholar]
- 46.Feder A, Skipper J, Blair JR, Buchholz K, Mathew SJ, Schwarz M, Doucette JT, Alonso A, Collins KA, Neumeister A, et al. Tryptophan depletion and emotional processing in healthy volunteers at high risk for depression. Biol Psychiatry. 2011;69:804–807. doi: 10.1016/j.biopsych.2010.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miura H, Ozaki N, Sawada M, Isobe K, Ohta T, Nagatsu T. A link between stress and depression: shifts in the balance between the kynurenine and serotonin pathways of tryptophan metabolism and the etiology and pathophysiology of depression. Stress. 2008;11:198–209. doi: 10.1080/10253890701754068. [DOI] [PubMed] [Google Scholar]
- 48.Myint AM, Kim YK. Network beyond IDO in psychiatric disorders: revisiting neurodegeneration hypothesis. Prog Neuropsychopharmacol Biol Psychiatry. 2014;48:304–313. doi: 10.1016/j.pnpbp.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 49.Myint AM, Kim YK, Verkerk R, Scharpé S, Steinbusch H, Leonard B. Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord. 2007;98:143–151. doi: 10.1016/j.jad.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 50.Leonard BE, Myint A. Inflammation and depression: is there a causal connection with dementia? Neurotox Res. 2006;10:149–160. doi: 10.1007/BF03033243. [DOI] [PubMed] [Google Scholar]
- 51.Ting KK, Brew BJ, Guillemin GJ. Effect of quinolinic acid on human astrocytes morphology and functions: implications in Alzheimer’s disease. J Neuroinflammation. 2009;6:36. doi: 10.1186/1742-2094-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ekdahl CT, Kokaia Z, Lindvall O. Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience. 2009;158:1021–1029. doi: 10.1016/j.neuroscience.2008.06.052. [DOI] [PubMed] [Google Scholar]
- 53.Kaneko N, Kudo K, Mabuchi T, Takemoto K, Fujimaki K, Wati H, Iguchi H, Tezuka H, Kanba S. Suppression of cell proliferation by interferon-alpha through interleukin-1 production in adult rat dentate gyrus. Neuropsychopharmacology. 2006;31:2619–2626. doi: 10.1038/sj.npp.1301137. [DOI] [PubMed] [Google Scholar]
- 54.Koo JW, Duman RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci USA. 2008;105:751–756. doi: 10.1073/pnas.0708092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- 56.Leonard BE. The concept of depression as a dysfunction of the immune system. Curr Immunol Rev. 2010;6:205–212. doi: 10.2174/157339510791823835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Janssen DG, Caniato RN, Verster JC, Baune BT. A psychoneuroimmunological review on cytokines involved in antidepressant treatment response. Hum Psychopharmacol. 2010;25:201–215. doi: 10.1002/hup.1103. [DOI] [PubMed] [Google Scholar]
- 58.Hannestad J, DellaGioia N, Bloch M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology. 2011;36:2452–2459. doi: 10.1038/npp.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McNally L, Bhagwagar Z, Hannestad J. Inflammation, glutamate, and glia in depression: a literature review. CNS Spectr. 2008;13:501–510. doi: 10.1017/s1092852900016734. [DOI] [PubMed] [Google Scholar]
- 60.Zarate C, Machado-Vieira R, Henter I, Ibrahim L, Diazgranados N, Salvadore G. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry. 2010;18:293–303. doi: 10.3109/10673229.2010.511059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maier SF, Watkins LR. Intracerebroventricular interleukin-1 receptor antagonist blocks the enhancement of fear conditioning and interference with escape produced by inescapable shock. Brain Res. 1995;695:279–282. doi: 10.1016/0006-8993(95)00930-o. [DOI] [PubMed] [Google Scholar]
- 62.Mansbach RS, Brooks EN, Chen YL. Antidepressant-like effects of CP-154,526, a selective CRF1 receptor antagonist. Eur J Pharmacol. 1997;323:21–26. doi: 10.1016/s0014-2999(97)00025-3. [DOI] [PubMed] [Google Scholar]
- 63.Bauer J, Hohagen F, Gimmel E, Bruns F, Lis S, Krieger S, Ambach W, Guthmann A, Grunze H, Fritsch-Montero R. Induction of cytokine synthesis and fever suppresses REM sleep and improves mood in patients with major depression. Biol Psychiatry. 1995;38:611–621. doi: 10.1016/0006-3223(95)00374-x. [DOI] [PubMed] [Google Scholar]
- 64.Brietzke E, Kapczinski F. TNF-alpha as a molecular target in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1355–1361. doi: 10.1016/j.pnpbp.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 65.Nery FG, Monkul ES, Hatch JP, Fonseca M, Zunta-Soares GB, Frey BN, Bowden CL, Soares JC. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Hum Psychopharmacol. 2008;23:87–94. doi: 10.1002/hup.912. [DOI] [PubMed] [Google Scholar]
- 66.Wang Y, Yang F, Liu YF, Gao F, Jiang W. Acetylsalicylic acid as an augmentation agent in fluoxetine treatment resistant depressive rats. Neurosci Lett. 2011;499:74–79. doi: 10.1016/j.neulet.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 67.Warner-Schmidt JL, Vanover KE, Chen EY, Marshall JJ, Greengard P. Antidepressant effects of selective serotonin reuptake inhibitors (SSRIs) are attenuated by antiinflammatory drugs in mice and humans. Proc Natl Acad Sci USA. 2011;108:9262–9267. doi: 10.1073/pnas.1104836108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uher R, Carver S, Power RA, Mors O, Maier W, Rietschel M, Hauser J, Dernovsek MZ, Henigsberg N, Souery D, et al. Non-steroidal anti-inflammatory drugs and efficacy of antidepressants in major depressive disorder. Psychol Med. 2012;42:2027–2035. doi: 10.1017/S0033291712000190. [DOI] [PubMed] [Google Scholar]
- 69.Lin PY, Mischoulon D, Freeman MP, Matsuoka Y, Hibbeln J, Belmaker RH, Su KP. Are omega-3 fatty acids antidepressants or just mood-improving agents? The effect depends upon diagnosis, supplement preparation, and severity of depression. Mol Psychiatry. 2012;17:1161–1163. doi: 10.1038/mp.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davies NM, Kehoe PG, Ben-Shlomo Y, Martin RM. Associations of anti-hypertensive treatments with Alzheimer’s disease, vascular dementia, and other dementias. J Alzheimers Dis. 2011;26:699–708. doi: 10.3233/JAD-2011-110347. [DOI] [PubMed] [Google Scholar]
- 71.de Beaurepaire R. Questions raised by the cytokine hypothesis of depression. Brain Behav Immun. 2002;16:610–617. doi: 10.1016/s0889-1591(02)00005-3. [DOI] [PubMed] [Google Scholar]
- 72.Weizman R, Laor N, Podliszewski E, Notti I, Djaldetti M, Bessler H. Cytokine production in major depressed patients before and after clomipramine treatment. Biol Psychiatry. 1994;35:42–47. doi: 10.1016/0006-3223(94)91166-5. [DOI] [PubMed] [Google Scholar]
- 73.Maes M. Evidence for an immune response in major depression: a review and hypothesis. Prog Neuropsychopharmacol Biol Psychiatry. 1995;19:11–38. doi: 10.1016/0278-5846(94)00101-m. [DOI] [PubMed] [Google Scholar]
- 74.Pollmächer T, Haack M, Schuld A, Reichenberg A, Yirmiya R. Low levels of circulating inflammatory cytokines--do they affect human brain functions? Brain Behav Immun. 2002;16:525–532. doi: 10.1016/s0889-1591(02)00004-1. [DOI] [PubMed] [Google Scholar]
- 75.Heim C, Newport DJ, Bonsall R, Miller AH, Nemeroff CB. Altered pituitary-adrenal axis responses to provocative challenge tests in adult survivors of childhood abuse. Am J Psychiatry. 2001;158:575–581. doi: 10.1176/appi.ajp.158.4.575. [DOI] [PubMed] [Google Scholar]
- 76.Carpenter LL, Tyrka AR, McDougle CJ, Malison RT, Owens MJ, Nemeroff CB, Price LH. Cerebrospinal fluid corticotropin-releasing factor and perceived early-life stress in depressed patients and healthy control subjects. Neuropsychopharmacology. 2004;29:777–784. doi: 10.1038/sj.npp.1300375. [DOI] [PubMed] [Google Scholar]