

Abstract
Aims
Invaginations of the cellular membrane called t-tubules are essential for maintaining efficient excitation–contraction coupling in ventricular cardiomyocytes. Disruption of t-tubule structure during heart failure has been linked to dyssynchronous, slowed Ca2+ release and reduced power of the heartbeat. The underlying mechanism is, however, unknown. We presently investigated whether elevated ventricular wall stress triggers remodelling of t-tubule structure and function.
Methods and results
MRI and blood pressure measurements were employed to examine regional wall stress across the left ventricle of sham-operated and failing, post-infarction rat hearts. In failing hearts, elevated left ventricular diastolic pressure and ventricular dilation resulted in markedly increased wall stress, particularly in the thin-walled region proximal to the infarct. High wall stress in this proximal zone was associated with reduced expression of the dyadic anchor junctophilin-2 and disrupted cardiomyocyte t-tubular structure. Indeed, local wall stress measurements predicted t-tubule density across sham and failing hearts. Elevated wall stress and disrupted cardiomyocyte structure in the proximal zone were also associated with desynchronized Ca2+ release in cardiomyocytes and markedly reduced local contractility in vivo. A causative role of wall stress in promoting t-tubule remodelling was established by applying stretch to papillary muscles ex vivo under culture conditions. Loads comparable to wall stress levels observed in vivo in the proximal zone reduced expression of junctophilin-2 and promoted t-tubule loss.
Conclusion
Elevated wall stress reduces junctophilin-2 expression and disrupts t-tubule integrity, Ca2+ release, and contractile function. These findings provide new insight into the role of wall stress in promoting heart failure progression.
Keywords: Left ventricular wall stress, Heart failure, T-tubules, Ca2+ homeostasis, Junctophilin-2
1. Introduction
In cardiac myocytes, contraction is triggered by a transient elevation of cytosolic [Ca2+] during the action potential. This increase in Ca2+ levels is initiated by the opening of L-type Ca2+ channels, which triggers additional Ca2+ release from ryanodine receptors in the sarcoplasmic reticulum (SR). This process of Ca2+-induced Ca2+ release occurs at functional units called dyads, where invaginations of the cellular membrane, called t-tubules, place Ca2+ channels in close apposition to the ryanodine receptors.1 A dense t-tubule network in ventricular cardiomyocytes ensures efficient and synchronous triggering of Ca2+ release across the cell.2
During diseases such as heart failure (HF) and atrial fibrillation, t-tubules are lost and disorganized.3,4 Such dyadic disruption reduces the efficiency5,6 and synchrony of Ca2+-induced Ca2+ release across the cell,7–10 and has been linked to slowed contraction.11,12 The majority of studies that have examined t-tubule alterations in pathological conditions have been descriptive in nature, and given little mechanistic insight into the signals that lead to their remodelling. However, a link to altered mechanical load has been proposed.13 Specifically, the restoration of t-tubule organization in failing hearts has been observed following resynchronization therapy,14 treatment with β-blockers,15 and unloading of the heart by heterotopic abdominal heart transplantation.16 We presently hypothesized that elevated ventricular wall stress triggers t-tubule degradation during HF. We observed that wall stress varied across post-infarction failing rat hearts, with the highest levels observed proximal to the infarct. Elevated wall stress was associated with reduced levels of junctophilin-2, a critical dyadic anchor,17 disruption of cardiomyocyte t-tubule organization and Ca2+ homeostasis, and impaired local in vivo contractile function. Ex vivo studies on loaded papillary muscles confirmed a direct role of wall stress in the regulation of junctophilin-2 expression and t-tubular structure.
2. Methods
Refer to the Supplementary material online, Methods.
2.1 Rat model of HF following myocardial infarction
All animal protocols were performed in accordance with the Norwegian Animal Welfare Act and NIH Guidelines (NIH publication No. 85-23, revised 2011) and were approved by the Ethics Committee of the University of Oslo. Myocardial infarction was induced by coronary artery ligation in anaesthetized male Wistar rats (mask ventilated with a mixture of 2.5% isoflurane, 97.5% oxygen) to enable HF progression over the following 6 weeks. Sham-operated rats (Sham) served as controls. Cardiac geometry and function were examined by MRI. Peak systolic and end-diastolic blood pressures were measured in the left ventricle via the right common carotid artery.
2.2 Magnetic resonance imaging
Anaesthesia was induced in a chamber with a mixture of 4.0–5.0% isoflurane and O2, and maintained by a mixture of 1.0–2.0% isoflurane and O2. Animals were freely breathing during MRI experiments, and respiration rate, heart rate, and body temperature were monitored continuously. Heart and respiration rates were kept stable by minor adjustments of anaesthesia level. Animal temperature was maintained by thermostat-regulated air. MRI acquisitions were triggered by the R-peak of the ECG signal and gated for respiration.
Velocity-encoded MRI data were acquired using a phase-contrast black-blood gradient echo sequence. 3D myocardial motion was measured in three short-axis slices: basal, mid-ventricular, and apical. Data acquisition time was 45–60 min, depending on heart and respiration rate.
In each slice of the velocity-encoded MRI data sets, the myocardium was manually segmented and automatically divided into 32 circumferential sectors. In each sector, myocardial curvature, wall thickness, and circumferential strain were calculated as previously described in detail.18,19
2.3 Cardiomyocyte imaging
Cardiomyocytes were isolated as previously described20 and plated on laminin-coated coverslips, mounted in a perfusion chamber. T-tubules were visualized with di-8-ANEPPS using an LSM 710 confocal scanning microscope (Zeiss GmbH, Jena, Germany). T-tubule organization was assessed for density and the prominence of transverse and longitudinal components, as described in the Supplementary material online, Methods and Figure S1.
For measurements of Ca2+ transients, cells were loaded with fluo-4 AM and superfused with a 37°C HEPES Tyrode solution. Cardiomyocytes were scanned every 1.5 ms by an LSM 7 Live scanning system (Zeiss), with a 1024 pixel line drawn along the longitudinal axis of the cell. Dyssynchrony of Ca2+ release was quantified as previously described.7
2.4 Western blotting
Primary antibodies for immunoblotting were Junctophilin-2 (1:1000) (Santa Cruz Biotechnology Inc., Texas, USA, sc-51313) and GAPDH (1:1000) (Santa Cruz, sc-20357). Secondary antibodies were anti-rabbit or anti-mouse IgG HRP-linked whole antibody (GE Healthcare, Oslo, Norway).
2.5 Papillary muscle experiments
Anterior and posteromedial papillary muscles were harvested from anaesthetized 11-week-old Wistar rats and mounted with sutures in a myobath system (Myobath II Multi-Channel Tissue Bath System, World Precision Instruments). Muscles were superfused with Tyrode's buffer solution and electrically stimulated at 0.5 Hz, while force was recorded during isometric contraction. Pre-load (diastolic wall stress) was set by stretching muscles. Diastolic, systolic, and developed force were sampled for 1 min every hour. Following 48 h of force recording, muscles were frozen and cut into transverse 20 µm sections, and t-tubules were labelled with Caveolin-3 antibody (Abcam, ab2912).
2.6 Statistics
Statistical difference was tested with t-tests or when applicable, ANOVA tests. Linear regression analyses were performed, and Pearson's Product Moment test was utilized to test for significant relationships using SigmaPlot (SyStat Software Inc., San Jose, CA, USA). All correlations were based on data points from individual cells and hearts. P < 0.05 was considered statistically significant. All data are presented as mean ± SEM.
3. Results
3.1 Regional variation in left ventricular wall stress across post-infarction failing rat hearts
In vivo left ventricular geometry and function were assessed by cine phase-contrast MRI (Figure 1A) and blood pressure measurements (Figure 1B). In comparison with Sham, rats that had developed HF following myocardial infarction exhibited a marked reduction in left ventricular ejection fraction (25.5 ± 2.9% in HF vs. 67.1 ± 3.4% in Sham, P < 0.05) and elevated diastolic pressure (P < 0.05, Figure 1B). Transmural infarctions were observed to cover 42.0 ± 1.9% of the left ventricle in failing hearts, and remaining viable myocardium was subdivided into equal sized regions, termed the proximal, medial, and distal zones (Figure 1A). Regional wall stress was estimated by first calculating local surface tension 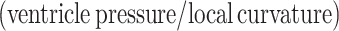 during the cardiac cycle. In comparison with Sham, the elevation of diastolic blood pressure in HF and ventricular dilation (reduced curvature; Figure 1C; Supplementary material online, FigureS2E) resulted in high surface tension across failing hearts (Figure 1D; see Supplementary material online, FigureS2F and Video SV1). Wall stress
during the cardiac cycle. In comparison with Sham, the elevation of diastolic blood pressure in HF and ventricular dilation (reduced curvature; Figure 1C; Supplementary material online, FigureS2E) resulted in high surface tension across failing hearts (Figure 1D; see Supplementary material online, FigureS2F and Video SV1). Wall stress 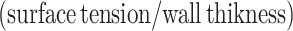 was also elevated in HF but was particularly high in the proximal zone (Figure 1F; see Supplementary material online, Video SV2) due to local thinning of the ventricular wall (Figure 1E; see Supplementary material online, FigureS2G). This regional disparity in wall stress measurements in HF was apparent during both the diastolic and systolic phases of the cardiac cycle, but elevation from Sham values was most pronounced during end-diastole (Figure 1G). Curvature, surface tension, and wall stress were similar across Sham hearts, as measured in regions equivalent in position to the proximal, medial, distal, and infarct zones of failing hearts (see Supplementary material online, Figure S2).
was also elevated in HF but was particularly high in the proximal zone (Figure 1F; see Supplementary material online, Video SV2) due to local thinning of the ventricular wall (Figure 1E; see Supplementary material online, FigureS2G). This regional disparity in wall stress measurements in HF was apparent during both the diastolic and systolic phases of the cardiac cycle, but elevation from Sham values was most pronounced during end-diastole (Figure 1G). Curvature, surface tension, and wall stress were similar across Sham hearts, as measured in regions equivalent in position to the proximal, medial, distal, and infarct zones of failing hearts (see Supplementary material online, Figure S2).
Figure 1.

Graded elevation of wall stress across the post-infarction, failing heart. MRI recordings showed left ventricular dilation in post-infarction rats with heart failure (HF) compared with sham-operated controls (A), and haemodynamic measurements indicated elevation of diastolic blood pressure (B). Division of failing hearts into equally sized zones proximal, medial, and distal to the infarct revealed reduction in curvature across the failing heart relative to Sham (C), and increased surface tension throughout the cardiac cycle (D). Due to a thinner ventricular wall in the proximal zone (E), wall stress was markedly elevated in this region (F). Wall stress declined with increasing distance from the infarct but was elevated in all regions relative to Sham, particularly during diastole (G). avo, aortic valve opening; avc, aortic valve closing; mvo, mitral valve opening; mvc, mitral valve closing. (nSham hearts= 3; nHF hearts= 4; *P < 0.05 vs. Sham).
3.2 T-tubule structure and Ca2+ homeostasis are disrupted in ventricular regions with elevated wall stress
The relationship between regional left ventricular wall stress and cardiomyocyte structure/function was investigated by isolating cardiomyocytes from the proximal, medial, and distal zones of HF and equivalent regions in Sham. T-tubules were imaged by di-8 ANEPPS staining and confocal scanning microscopy (Figure 2, left panels). In Sham hearts, somewhat higher t-tubule density was observed in the proximal equivalent zone compared with other zones (0.21 ± 0.01 vs. 0.18 ± 0.01 in both distal and medial equivalent zones, P < 0.05), although an organized, striated pattern of t-tubule staining was observed across these hearts. This organized t-tubule pattern was markedly disrupted in the proximal zone of failing hearts, as t-tubule density was reduced. In contrast, t-tubule density was maintained in the medial and distal zones in HF. Further analyses revealed that transversely oriented tubules were lost in all zones, while in the distal and medial zones, overall t-tubule density was maintained due to increased amounts of longitudinal elements (Figure 2, right panels).
Figure 2.

T-tubules are most markedly disrupted proximal to the infarction. di-8-ANEPPS stains were employed to examine t-tubule density and organization. In comparison with sham-operated controls, overall t-tubule density was only reduced in the proximal zone of failing hearts. However, transverse elements were observed to be lost in all regions, but compensated by growth of longitudinal tubules in the medial and distal zones. (nSham cells = 183 from 3 hearts, nHF cells= 246 from 4 hearts; *P < 0.05 vs. Sham).
The net effect of these complex changes in t-tubule pattern was examined by calculating the distances from all points across the cell to the nearest t-tubule or surface membrane. Distance maps (Figure 3A; see Supplementary material online) showed a gradient across the failing heart, as distances to the nearest membrane were markedly increased in the proximal zone but not significantly different from Sham values in the distal zone. Importantly, wall stress measurements significantly predicted both t-tubule density and distance-to-nearest membrane (see Supplementary material online, Figure S3A and S3B, respectively, P < 0.05). Elevated wall stress in the proximal zone of failing hearts was also associated with loss of junctophilin-2 (Figure 3B), supporting recent reports that this dyadic anchor is necessary for maintaining t-tubule density.21–23
Figure 3.

T-tubule disruption is associated with reduced junctophilin-2 expression. Images of di-8-ANEPPS-stained cardiomyocytes were used to create ‘distance maps’ (A, left panel), indicating distance from all points in the cytosol to the nearest t-tubule or surface sarcolemma. Average distance was increased with greater proximity to the infarction (right panel). (nSham cells = 183 from 3 hearts, nHF cells= 246 from four hearts). (B) Junctophilin-2 (JP2) protein abundance was assessed by western blotting and normalized to GAPDH. Mean JP2 levels were significantly down-regulated in the proximal zone only. (nhearts= 6 in Sham and HF). (*P < 0.05 vs. Sham).
Ca2+ homeostasis was examined across Sham and failing hearts. In Sham hearts, Ca2+ release was somewhat less synchronous in the distal equivalent zone than other zones (1.57 ± 0.13 vs. 1.20 ± 0.05 in medial equivalent, P < 0.05, 1.32 ± 0.09 in proximal equivalent, P = NS). In agreement with an observed gradient disruption of t-tubule structure in HF, desynchronized Ca2+ release was observed proximal but not distal to the infarct in comparison with Sham (Figure 4A and B). Dyssynchronous Ca2+ release in the proximal zone resulted in a marked slowing of the rise time of the Ca2+ transient (Figure 4C). Ca2+ release dyssynchrony across Sham and failing hearts tended to be correlated with diastolic wall stress (see Supplementary material online, Figure S4A, P = 0.10) and was significantly predicted by junctophilin-2 expression (see Supplementary material online, Figure S4B, P < 0.05).
Figure 4.

Ca2+ transients are progressively desynchronized with greater proximity to the infarct. As predicted by distance maps in Figure 3, confocal line-scan images (A) showed progressively more dyssynchronous and slowed Ca2+ transients approaching the infarct. Mean measurements of dyssynchrony index and time to 50% rise of the Ca2+ transient are shown in (B and C), respectively (nSham cells = 82 from three hearts, nHF cells= 83 from four hearts) (*P < 0.05 vs. Sham).
The observed alterations in cardiomyocyte structure and function in the proximal zone could theoretically result from release of signalling molecules from the infarct or inadequate tissue perfusion. To investigate this possibility, we examined t-tubule structure and Ca2+ transients in non-failing, infarcted hearts (mean infarct size = 14.3 ± 3.7%) which exhibited significantly lower wall stress than in HF. Normal t-tubular structure was observed in the proximal zone of these hearts (see Supplementary material online, Figure S5), indicating that the mere presence of a nearby infarct does not directly disrupt t-tubules.
3.3 Elevation of wall stress disrupts cardiomyocyte structure in cultured papillary muscles
The above findings showed marked correlation between ventricular wall stress and cardiomyocyte structure/function. These correlations do not establish causation. Therefore, to investigate whether elevated wall stress directly disrupts t-tubular structure, we developed an in situ model where rat papillary muscles were kept in culture conditions under different levels of simulated wall stress (see Methods and Figure 5A). One group of muscles was stretched to estimate the diastolic wall stress observed during end-diastole in the proximal zone of failing hearts (≈ 15–20 kN/m2). A second group was exposed to modest diastolic stress (≈ 4 kN/m2). Tension development was well sustained in both groups during 48 h of isometric stimulation at 0.5 Hz (Figure 5A; P = NS vs. 1 h). Muscles exposed to low wall stress maintained an organized t-tubule network during the incubation period, while muscles exposed to high wall stress revealed extensive t-tubule loss (Figure 5B and C), which was due to degradation of both transverse and longitudinal elements (Figure 5D and E). In congruence with decreased t-tubule density, juntophilin-2 gene expression was reduced in muscles exposed to high wall stress (Figure 5F). Importantly, acute exposure of muscles to high wall stress conditions did not disrupt t-tubule organization (see Supplementary material online, Figure S6), indicating that observed t-tubule loss during the 48 h treatment period was a regulated process. These data support a direct role of elevated wall stress in triggering junctophilin-2 down-regulation and t-tubule disruption.
Figure 5.

High wall stress induces t-tubule disruption and loss in vitro. Papillary muscles were isolated, mounted in a Myobath system, and maintained under culture conditions for 48 h during 0.5 Hz stimulation. Muscles were exposed to modest pre-load (diastolic stress ≈4 kN/m2) or elevated diastolic stress approximating that observed during end-diastole in the proximal zone (15–20 kN/m2). (A) Representative recordings of diastolic and systolic stress for the two treatment groups during the protocol. Visualization of t-tubules by caveolin-3 immunostaining after the 48 h incubation period revealed well-maintained t-tubular structure in the low-wall stress group, but marked t-tubule disruption following high wall stress (B). Overall t-tubule density was reduced by exposure to high wall stress (C) and included loss of both transverse and longitudinal elements [D and E, low wall stress: n = 67 cells from five muscles (4 hearts), high wall stress: n = 64 cells from five muscles (three hearts)]. T-tubule loss was associated with a marked decrease in relative junctophilin-2 mRNA expression in the high-wall stress group (F, nmuscles = 4, 3 in low, high wall stress groups, respectively). LWS, low wall stress; HWS, high wall stress (*P < 0.05 vs. low wall stress).
3.4 In vivo consequences of wall stress-induced cardiomyocyte remodelling
The observed wall stress-induced disruption of t-tubule structure and Ca2+ homeostasis in the proximal zone would suggest that local contractile function would be depressed in this region of the failing heart. To test this prediction, we assessed strain (local contraction) across the left ventricle. Figure 6A depicts regional peak circumferential strain in a representative Sham and HF heart, while strain during the cardiac cycle is illustrated in Figure 6B. Average strain recordings showed relatively uniform strain and rate of strain development across the left ventricle in sham-operated hearts (Figure 6D and E; see Supplementary material online, FigureS7A). In failing hearts, no contraction was observed in the infarcted zone, and very little strain was observed in the proximal zone, indicating that these regions contracted nearly isometrically. Peak strain and strain rate increased with distance from the infarction towards the distal zone, where contraction was comparable to Sham values (Figure 6B, D, and E). We additionally examined contractility across the heart with stress–strain loops (Figure 6C; see Supplementary material online, FigureS7B) and defined a ‘contractility index’ calculated as regional end-systolic strain/wall stress.9–11 These calculations indicated markedly reduced contractility with greater proximity to the infarction in failing hearts (Figure 6F). Importantly, local contractility was correlated with levels of junctophilin-2 (P < 0.01), t-tubule density (P < 0.01), and the synchrony of Ca2+ release (P = 0.01) observed in isolated myocytes across sham and HF hearts, linking wall stress-induced cardiomyocyte remodelling to in vivo dysfunction.
Figure 6.

Local in vivo contractility is reduced in regions with disrupted cardiomyocyte structure and Ca2+ handling. (A) Representative measurements of MRI-determined circumferential strain show graded contraction across the failing heart. Mean strain and peak strain rate were markedly reduced in the proximal zone but near-normal in the distal zone (B, D and E). (C) Wall stress–strain loops were employed to estimate a ‘contractility index’, calculated as end-systolic strain/stress. In comparison with Sham, the contractility index was significantly reduced in regions of failing hearts near the infarction (F, nSham hearts= 3, nHF hearts= 4). avo, aortic valve opening; avc, aortic valve closing; mvo, mitral valve opening; mvc, mitral valve closing. (*P < 0.05 vs. Sham).
4. Discussion
In the present study, we observed a gradient of ventricular wall stress across failing rat hearts, with highest values close to the infarct (proximal zone) and lower levels in distal regions. This pattern coincided with regional variability in the disruption of cardiomyocyte structure and function, as elevated wall stress was associated with decreased junctophilin-2 expression, t-tubule density, Ca2+ release synchrony, and local in vivo contractility. Verifying a causative link between wall stress and disruption of cardiomyocyte structure/function, exposure of papillary muscles to high wall stress approximating in vivo conditions in the proximal zone triggered decreased junctophilin-2 expression and marked t-tubule remodelling. These data provide novel insight into the mechanistic link between elevation of ventricular wall stress and disrupted contraction during HF.
While the pathways controlling t-tubule structure are unknown, an unspecified involvement of ventricular workload has been suggested, since increased workload is associated with t-tubule disruption.13,24,25 Conversely, reducing the workload of failing hearts, either pharmacologically,15,26 by implanting assist devices14 or by heterotopic transplantation16 reverses t-tubule remodelling and relieves HF progression. The present results support the paradigm of a workload-regulated t-tubule network and specifically point to the importance of the physical stress placed on the ventricular wall. Of note, we did not observe significant correlation between wall stress and various measures of cardiomyocyte t-tubule structure and Ca2+ release across failing hearts (see Supplementary material online, Figures S3 and S4). However, significant correlations were observed when measures across Sham hearts were also included (see Supplementary material online, Figure S3 and S4), and reproducing elevated wall stress conditions by ex vivo stretch markedly disrupted t-tubule structure (Figure 5). In apparent agreement with our findings, dyssynchronous HF following left bundle-branch block was observed to promote t-tubule disruption in late-activated regions, which experience abnormal stretch/wall stress as contraction occurs in early activated regions.14 Although experimental measurements of wall stress and t-tubule integrity have not been previously paired, it is important to note that many studies have reported t-tubule disruption in conditions of eccentric, dilated hypertrophy,6–8,10,22 which is associated with elevated diastolic wall stress.27 Our present data indicate that eccentric hypertrophy following myocardial infarction may be particularly prone to t-tubule disruption, as the myocardial wall is thinned (and wall stress further elevated) near the scar. Importantly, our data from non-failing infarcted hearts indicate that the presence of a nearby infarct alone does not promote t-tubule disruption in the absence of sufficiently elevated wall stress.
The mechanosensors and signalling pathways that trigger wall stress-induced t-tubule disruption are unclear. One candidate player is titin and its associated ligands, which play an important role in cardiomyocyte mechanotransduction by regulating the interaction between wall stress, the extracellular matrix, and the sarcomere.28 Titin-cap (T-cap) may be critically involved in such transduction, as T-cap down-regulation during HF has been linked to t-tubule disruption.29 Furthermore, T-cap knockout leads to progressive loss of t-tubules and accelerates progression of HF following aortic banding.30 Membrane-mediated mechanosensation by stretch-activated channels, integrins, proteoglycans, and angiotensin II type I receptors has also been reported, and shown to activate a variety of pathways, including MAPK, AKT, and calcineurin-NFAT.31,32 Particularly intriguing are recent data indicating that the t-tubules themselves can transduce these signals during stretch and/or contraction,33,34 raising the possibility that stress sensing within t-tubules could regulate their own structure. It is critical that the upstream regulators of junctophilin-2 are identified, as previous work22,35 and our present findings show that even relatively small reductions in junctophilin-2 expression are associated with significant t-tubule alterations. Of note, the NFAT pathway may be involved, since its downstream effector mir-24 has been shown to reduce junctophilin-2 levels and promote t-tubule loss.36 Conversely, mir-24 suppression is reported to protect against t-tubule disruption and HF progression.37
Since cardiomyocytes can sense cell shortening (strain), it might be argued that t-tubular degradation in the proximal zone of failing hearts is signalled by reduced strain, rather than elevated wall stress in this region. However, our in vitro experiments showed that t-tubule structure was well maintained during isometric stimulation when wall stress was normal. Therefore, while reduced contractility may signal other changes in structure such as cell thickening during concentric hypertrophy,38 it does not appear that altered cell shortening signals t-tubule remodelling. Rather, our data suggest that the reduced strain and contractility observed in the proximal zone is likely a consequence of remodelling of t-tubules and Ca2+ homeostasis. A significant correlation was observed between cellular structure/function (junctophilin-2 level, t-tubular structure, Ca2+ release synchrony) and regional in vivo contractility. While in vivo hypocontractility proximal to the infarcted zone has been reported to occur despite normal perfusion,39,40 it is important to point out that tethering of the proximal zone to the immobile infarct likely additionally contributes to the contractile deficit.40
In agreement with other studies (reviewed in Ref. 4), we observed an increased fraction of longitudinally oriented tubules in failing cells. Unlike loss of transverse elements, we have previously shown that growth of these longitudinal elements is compensatory, as they facilitate greater trans-sarcolemmal Ca2+ cycling.41 We presently found no association between wall stress and longitudinal t-tubule density; rather, we saw the highest fraction of these tubules in the medial and distal zones (Figure 3B). This finding suggests that transverse and longitudinal tubules are regulated by different pathways.
In conclusion, the current study provides critical new insight into the link between mechanical stress and remodelling of cardiomyocyte structure and function during HF. We show that wall stress regulates junctophilin-2 protein level, t-tubule integrity, cellular Ca2+ release characteristics, and contractile function in rat hearts. These findings shed new light on the beneficial effects of unloading the heart for HF patients.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This project has received funding from the European Union's Horizon 2020 research and innovation programme (Consolidator grant, WEL) under grant agreement No 647714. Additional support was provided by European Union Project No. FP7-HEALTH-2010.2.4.2-4 (‘MEDIA-Metabolic Road to Diastolic Heart Failure’), The South-Eastern Norway Regional Health Authority, Anders Jahre's Fund for the Promotion of Science, The Norwegian Institute of Public Health, Oslo University Hospital Ullevål, and the University of Oslo. Funding to pay the Open Access publication charges for this article was provided by the European Union's Horizon 2020 research and innovation programme (Consolidator grant, WEL) under grant agreement No 647714.
Acknowledgements
The authors thank the Section of Comparative Medicine, Oslo University Hospital Ullevål (Oslo, Norway) for animal care. We thank Dr Peter V. Skov for providing the myobath system and Dr Ståle Nygård for statistical assistance.
Conflict of interest: none declared.
References
- 1.Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys J 1999;77:1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo A, Zhang C, Wei S, Chen B, Song LS. Emerging mechanisms of T-tubule remodelling in heart failure. Cardiovasc Res 2013;98:204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louch WE, Sejersted OM, Swift F. There goes the neighborhood: pathological alterations in T-tubule morphology and consequences for cardiomyocyte Ca2+ handling. J Biomed Biotechnol 2010;2010:503906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roe AT, Frisk M, Louch WE. Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr Pharm Des 2014;21:431–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation–contraction coupling in experimental cardiac hypertrophy and heart failure. Science 1997;276:800–806. [DOI] [PubMed] [Google Scholar]
- 6.Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, Claus P, Dymarkowski S, Maes F, Bogaert J, Rademaker F, D'Hooge J, Sipido K. Remodeling of T–tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res 2008;102:338–346. [DOI] [PubMed] [Google Scholar]
- 7.Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I, Sejersted OM. T–tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol 2006;574:519–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci USA 2006;103:4305–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, Mubagwa K, Sipido K. Reduced synchrony of Ca2+ release with loss of T–tubules – a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res 2004;62:63–73. [DOI] [PubMed] [Google Scholar]
- 10.Oyehaug L, Loose KO, Jolle GF, Roe AT, Sjaastad I, Christensen G, Sejersted OM, Louch WE. Synchrony of cardiomyocyte Ca2+ release is controlled by t–tubule organization, SR Ca2+ content, and ryanodine receptor Ca2+ sensitivity. Biophys J 2013;104:1685–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mork HK, Sjaastad I, Sejersted OM, Louch WE. Slowing of cardiomyocyte Ca2+ release and contraction during heart failure progression in postinfarction mice. Am J Physiol Heart Circ Physiol 2009;296:H1069–H1079. [DOI] [PubMed] [Google Scholar]
- 12.Bokenes J, Aronsen JM, Birkeland JA, Henriksen UL, Louch WE, Sjaastad I, Sejersted OM. Slow contractions characterize failing rat hearts. Basic Res Cardiol 2008;103:328–344. [DOI] [PubMed] [Google Scholar]
- 13.Ibrahim M, Terracciano CM. Reversibility of T–tubule remodelling in heart failure: mechanical load as a dynamic regulator of the T–tubules. Cardiovasc Res 2013;98:225–232. [DOI] [PubMed] [Google Scholar]
- 14.Sachse FB, Torres NS, Savio–Galimberti E, Aiba T, Kass DA, Tomaselli GF, Bridge JH. Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy. Circ Res 2012;110:588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen B, Li Y, Jiang S, Xie YP, Guo A, Kutschke W, Zimmerman K, Weiss RM, Miller FJ, Anderson ME, Song LS. Beta–adrenergic receptor antagonists ameliorate myocyte T–tubule remodeling following myocardial infarction. FASEB J 2012;26:2531–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ibrahim M, Navaratnarajah M, Siedlecka U, Rao C, Dias P, Moshkov AV, Gorelik J, Yacoub MH, Terracciano CM. Mechanical unloading reverses transverse tubule remodelling and normalizes local Ca2+–induced Ca2+ release in a rodent model of heart failure. Eur J Heart Fail 2012;14:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell 2000;6:11–22. [DOI] [PubMed] [Google Scholar]
- 18.Espe EK, Aronsen JM, Skardal K, Schneider JE, Zhang L, Sjaastad I. Novel insight into the detailed myocardial motion and deformation of the rodent heart using high–resolution phase contrast cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2013;15:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espe EK, Aronsen JM, Eriksen GS, Zhang L, Smiseth OA, Edvardsen T, Sjaastad I, Eriksen M. Assessment of regional myocardial work in rats. Circ Cardiovasc Imaging 2015;8:e002695. [DOI] [PubMed] [Google Scholar]
- 20.Frisk M, Koivumaki JT, Norseng PA, Maleckar MM, Sejersted OM, Louch WE. Variable t–tubule organization and Ca2+ homeostasis across the atria. Am J Physiol Heart Circ Physiol 2014;307:H609–H620. [DOI] [PubMed] [Google Scholar]
- 21.van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, De Almeida AC, Skapura DG, Rudy Y, Burns AR, Ackerman MJ, Wehrens XH. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation 2011;123:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS. T–tubule remodeling during transition from hypertrophy to heart failure. Circ Res 2010;107:520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landstrom AP, Kellen CA, Dixit SS, van Oort RJ, Garbino A, Weisleder N, Ma J, Wehrens XH, Ackerman MJ. Junctophilin–2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium–handling. Circ Heart Fail 2011;4:214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ibrahim M, Kukadia P, Siedlecka U, Cartledge JE, Navaratnarajah M, Tokar S, Van Doorn C, Tsang VT, Gorelik J, Yacoub MH, Terracciano CM. Cardiomyocyte Ca2+ handling and structure is regulated by degree and duration of mechanical load variation. J Cell Mol Med 2012;16:2910–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aistrup GL, Gupta DK, Kelly JE, O'Toole MJ, Nahhas A, Chirayil N, Misener S, Beussink L, Singh N, Ng J, Reddy M, Mongkolrattanothai T, El–Bizri N, Rajarnani S, Shryock JC, Belardinelli L, Shah SJ, Wasserstrom JA. Inhibition of the late sodium current slows t–tubule disruption during the progression of hypertensive heart disease in the rat. Am J Physiol Heart Circ Physiol 2013;305:H1068–H1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie YP, Chen B, Sanders P, Guo A, Li Y, Zimmerman K, Wang LC, Weiss RM, Grumbach IM, Anderson ME, Song LS. Sildenafil prevents and reverses transverse–tubule remodeling and Ca2+ handling dysfunction in right ventricle failure induced by pulmonary artery hypertension. Hypertension 2012;59:355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest 1975;56:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linke WA. Sense and stretchability: the role of titin and titin–associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res 2008;77:637–648. [DOI] [PubMed] [Google Scholar]
- 29.Lyon AR, Nikolaev VO, Miragoli M, Sikkel MB, Paur H, Benard L, Hulot JS, Kohlbrenner E, Hajjar RJ, Peters NS, Korchev YE, Macleod KT, Harding SE, Gorelik J. Plasticity of surface structures and beta(2)–adrenergic receptor localization in failing ventricular cardiomyocytes during recovery from heart failure. Circ Heart Fail 2012;5:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ibrahim M, Siedlecka U, Buyandelger B, Harada M, Rao C, Moshkov A, Bhargava A, Schneider M, Yacoub MH, Gorelik J, Knoll R, Terracciano CM. A critical role for Telethonin in regulating t–tubule structure and function in the mammalian heart. Hum Mol Genet 2013;22:372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dostal DE, Feng H, Nizamutdinov D, Golden HB, Afroze SH, Dostal JD, Jacob JC, Foster DM, Tong C, Glaser S, Gerilechaogetu F. Mechanosensing and regulation of cardiac function. J Clin Exp Cardiolog 2014;5:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lammerding J, Kamm RD, Lee RT. Mechanotransduction in cardiac myocytes. Ann N Y Acad Sci 2004;1015:53–70. [DOI] [PubMed] [Google Scholar]
- 33.McNary TG, Bridge JH, Sachse FB. Strain transfer in ventricular cardiomyocytes to their transverse tubular system revealed by scanning confocal microscopy. Biophys J 2011;100:L53–L55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dyachenko V, Husse B, Rueckschloss U, Isenberg G. Mechanical deformation of ventricular myocytes modulates both TRPC6 and Kir2.3 channels. Cell Calcium 2009;45:38–54. [DOI] [PubMed] [Google Scholar]
- 35.Zhang HB, Li RC, Xu M, Xu SM, Lai YS, Wu HD, Xie XJ, Gao W, Ye H, Zhang YY, Meng X, Wang SQ. Ultrastructural uncoupling between T–tubules and sarcoplasmic reticulum in human heart failure. Cardiovasc Res 2013;98:269–276. [DOI] [PubMed] [Google Scholar]
- 36.Xu M, Wu HD, Li RC, Zhang HB, Wang M, Tao J, Feng XH, Guo YB, Li SF, Lai ST, Zhou P, Li LL, Yang HQ, Luo GZ, Bai Y, Xi JJ, Gao W, Han QD, Zhang YY, Wang XJ, Meng X, Wang SQ. Mir–24 regulates junctophilin–2 expression in cardiomyocytes. Circ Res 2012;111:837–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li RC, Tao J, Guo YB, Wu HD, Liu RF, Bai Y, Lv ZZ, Luo GZ, Li LL, Wang M, Yang HQ, Gao W, Han QD, Zhang YY, Wang XJ, Xu M, Wang SQ. In vivo suppression of microRNA–24 prevents the transition toward decompensated hypertrophy in aortic–constricted mice. Circ Res 2013;112:601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guterl KA, Haggart CR, Janssen PM, Holmes JW. Isometric contraction induces rapid myocyte remodeling in cultured rat right ventricular papillary muscles. Am J Physiol Heart Circ Physiol 2007;293:H3707–H3712. [DOI] [PubMed] [Google Scholar]
- 39.Mazhari R, Omens JH, Covell JW, McCulloch AD. Structural basis of regional dysfunction in acutely ischemic myocardium. Cardiovasc Res 2000;47:284–293. [DOI] [PubMed] [Google Scholar]
- 40.Ashikaga H, Mickelsen SR, Ennis DB, Rodriguez I, Kellman P, Wen H, McVeigh ER. Electromechanical analysis of infarct border zone in chronic myocardial infarction. Am J Physiol Heart Circ Physiol 2005;289:H1099–H1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swift F, Franzini-Armstrong C, Oyehaug L, Enger UH, Andersson KB, Christensen G, Sejersted OM, Louch WE. Extreme sarcoplasmic reticulum volume loss and compensatory T–tubule remodeling after Serca2 knockout. Proc Natl Acad Sci USA 2012;109:3997–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]