

Summary
The development of dengue antivirals and vaccine has been hampered by the incomplete understanding of molecular mechanisms of dengue virus (DENV) infection and pathology, partly due to the limited suitable cell culture or animal models that can capture the comprehensive cellular changes induced by DENV. In this study, we differentiated human pluripotent stem cells (hPSCs) into hepatocytes, one of the target cells of DENV, to investigate various aspects of DENV-hepatocyte interaction. hPSC-derived hepatocyte-like cells (HLCs) supported persistent and productive DENV infection. The activation of interferon pathways by DENV protected bystander cells from infection and protected the infected cells from massive apoptosis. Furthermore, DENV infection activated the NF-κB pathway, which led to production of proinflammatory cytokines and downregulated many liver-specific genes such as albumin and coagulation factor V. Our study demonstrates the utility of hPSC-derived hepatocytes as an in vitro model for DENV infection and reveals important aspects of DENV-host interactions.
Graphical Abstract
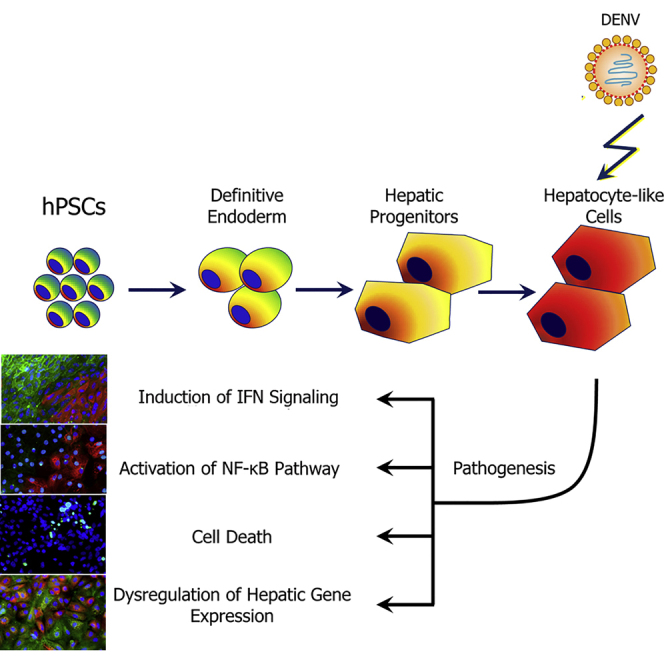
Highlights
-
•
Hepatocyte-like cells (HLCs) derived from hPSCs support productive DENV infection
-
•
DENV infection of HLCs leads to cellular responses that are relevant to pathology
-
•
HLCs represent a new model system to study dengue virus infection of liver
Infection/disease models modeling the comprehensive cellular changes induced by DENV infection are limited. Here, Tang and colleagues use pluripotent stem cells to produce a renewable cell model that captures in vitro phenotypes relevant for dengue pathogenesis. Combining targeted differentiation and viral infection represents a new model to study the interface of host genetics and dengue infection and pathogenesis.
Introduction
Dengue is emerging as one of the most widespread vector-borne infectious diseases in humans. A recent large-scale mapping of global dengue distribution estimates 390 million dengue infections per year worldwide (Bhatt et al., 2013). The pathogenic agent dengue virus (DENV) is a member of the Flaviviridae, a family of positive-strand RNA viruses that include many additional important human pathogens such as West Nile virus and hepatitis C virus (HCV). Infections of humans by DENV cause dengue fever and the more severe dengue diseases, dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS). DHF is characterized by increased vascular permeability and hemorrhagic manifestations, and may progress to the life-threatening DSS. It is proposed that proinflammatory cytokines and chemokines as a result of DENV infection form a “cytokine storm” and lead to increased vascular permeability.
DENV infection in human affects a variety of organ functions, and viral nucleic acids and proteins have been detected in many tissues (Acosta et al., 2014). As such, many different infection systems can be used to model different aspects of DENV infection, although each has its limitations. Laboratory animals by and large sustain only low levels of infection and do not recapitulate natural disease progression (Rothman, 2011). Primary human cells, including monocytes, macrophages, hepatocytes, and endothelial cells, represent the most physiologically relevant model in vitro. However, these cells are limited in supply and often exhibit significant donor-to-donor variability.
The monocyte-lineage cells are considered a major target of DENV in vivo (Blackley et al., 2007, Wu et al., 2000, Pham et al., 2012, Jessie et al., 2004), but DENV also infects liver in vivo (Halstead, 2007, Povoa et al., 2014, Balsitis et al., 2009). The clinical manifestations of DENV infection of liver range from asymptomatic mild liver function abnormality in most cases to acute liver failure. In vitro, DENV has been demonstrated to infect hepatoma cell lines (Marianneau et al., 1997, Cabrera-Hernandez et al., 2007, Hilgard and Stockert, 2000, Heaton and Randall, 2010) as well as primary human hepatocytes (PHHs) (Suksanpaisan et al., 2007), but these cells are either genetically abnormal or not universally accessible. The stem cell-derived, differentiated human hepatocyte-like cells (HLCs/DHHs/iHeps) have been shown to support productive infection by HCV (Wu et al., 2012, Schwartz et al., 2012, Roelandt et al., 2012) and hepatitis B virus (HBV) (Shlomai et al., 2014). In the current study, we apply the HLC model to study the interactions between DENV and hepatic cells. We demonstrate that pluripotent stem cells are refractory to DENV infection but that DENV can efficiently and productively infect the HLCs. The permissiveness transition correlates with the induction of putative DENV receptors. The profound changes caused by DENV infection of HLCs include stimulation of innate immunity, apoptosis, and cytokine and chemokine induction, all of which recapitulate important aspects of dengue-host interaction. In addition, we found that DENV infection leads to a partial dysregulation of hepatic functions, such as the downregulation of essential components of the complement and coagulation systems, which might be relevant to DENV pathology. The HLCs thus represent a valuable stem cell-based model for investigations into DENV-hepatic cell interactions.
Results
Differentiation of Human Pluripotent Stem Cells into Hepatocyte-like Cells
A chemically defined differentiation protocol was used to derive human HLCs from human pluripotent stem cells (hPSCs) in vitro (Si-Tayeb et al., 2010, Touboul et al., 2010), as shown in Figure 1A. The undifferentiated human embryonic stem cells (hESCs) expressed pluripotent marker genes, including octamer-binding transcription factor 4 (OCT4), NANOG, sex determining region Y-box 2 (SOX2), and stage-specific embryonic antigen 4 (SSEA4) (Figure 1B). Upon differentiation to definitive endoderm, the endodermic markers sex determining region Y-box 17 (SOX17) and chemokine (C-X-C motif) receptor type 4 (CXCR4) started to express (Figure 1C). Hepatic specification resulted in a hepatic progenitor population that expressed α-fetoprotein (AFP), cytokeratin 19 (CK19), and hepatocyte nuclear factor 4α (HNF4A) (Figure 1D). These cells were further differentiated toward the hepatic lineage and started to adopt distinct hepatocyte morphology, including defined and canaliculated borders and a polygonal appearance (Figure S1A). Many more liver-specific genes such as albumin (ALB), α1-antitrypsin (A1AT), sodium/taurocholate cotransporting polypeptide (NTCP), and coagulation factor V (F5) were also expressed in the HLCs (Figure 1E). These cells also gained the ability to form lipid droplets (Figure 1E), a site of particle assembly for several viruses (Miyanari et al., 2007, Samsa et al., 2009). In the same way, human induced pluripotent stem cells (iPSCs) were differentiated along the hepatic lineage with each stage expressing the specific markers (Figure S1B). RNA-sequencing (RNA-seq) analysis showed that the expression levels of many liver-specific genes were upregulated in day-12 compared with day-5 cells (Figure 1F and Table S1), and thus were the core hepatic transcription factors (Figure S1C). Together, these data are consistent with efficient hepatic differentiation in vitro.
Figure 1.

Differentiation of Hepatocyte-like Cells from Human Pluripotent Stem Cells
(A) Schematic description of the four stages of hepatocyte differentiation.
(B) Undifferentiated hESCs. hESCs were positive for NANOG, SOX2, OCT4, and SSEA4 and maintained in the pluripotent state.
(C) Induction of definitive endoderm. The expression of specific marker genes SOX17 and CXCR4 were induced in hESC-derived definitive endoderm at day 3.
(D) Hepatic specification. At day 6 after differentiation, the differentiated hepatic progenitors were AFP and CK19 double-positive and homogenously expressed the hepatic gene HNF4A.
(E) Maturation of hepatic progenitors into hepatocyte-like cells. The HLCs underwent functional maturation from day 7. At day 17, the resulting cells expressed specific genes unique to hepatocytes, including AFP, ALB, A1AT, NTCP, and coagulation factor F5. Lipid droplets were formed in these cells.
(F) Induction of genes preferentially expressed in the liver during differentiation. Heatmap of RNA-seq expression values (log10(1 + FPKM)) of the 291 RefSeq genes preferentially expressed (Liu et al., 2008) in liver at day 5, day 10, and day 12 of differentiation.
The Hepatocyte-like Cells Express TAM3 and Support Productive Infection by Dengue Virus
Whether the HLCs are permissive for DENV infection has not been tested previously. We exposed the stem cells and their progeny cells during the course of the hepatic differentiation to dengue virus serotype 2 (DENV2) for 48 hr and then lysed the cells for detection of intracellular expression of dengue antigens. The signal for DENV NS3 and, to a lesser extent, capsid, became detectable at day 6 post differentiation, indicating successful infection of these cells (Figure 2A). The infection efficiency steadily increased in the subsequent days until day 11 when both the NS3 and the capsid signal reached saturation (Figure 2A). Immunostaining confirmed that the percentage of DENV NS3-positive cells increased from day 6 to day 10 (Figure 2B). At day 11, almost all HLCs can be infected by DENV at a high MOI. In contrast, hPSCs were refractory to DENV infection (Figure S2A). Furthermore, iPSC-derived HLCs were permissible to DENV infection (Figure S2B). The infection could be suppressed by known inhibitors of DENV infection such as interferon-α2a (IFN-α2a), chloroquine (CQ), and mycophenolic acid (MPA) (Figures 2C and 2D), indicating that the detected NS3 expression represents authentic viral infection and replication. Persistent replication of DENV in the infected HLCs was indicated by the continuous secretion of NS1 into the cell culture medium after repeated wash and medium change cycles (Figure 2E). Virus production from the infected HLCs was demonstrated by using supernatant from the infected cells to infect the mosquito cell line C6/36, the natural host cells in mosquito, and virus titration (Figures 2F and 2G). Analysis of the RNA-seq data revealed that the expression of a TAM family phosphatidylserine receptor, AXL receptor tyrosine kinase (AXL or TAM3), as well as its ligand protein S (PROS), were upregulated in HLCs compared with day-5 cells (Figure 2H), consistent with the report that these proteins play important roles in binding dengue virions and mediating entry into multiple cell types, including hepatocytes (Meertens et al., 2012). In addition, claudin-1 (CLDN1), which interacts with prM/M protein of DENV and has been implicated in DENV entry (Che et al., 2013), was also upregulated (Figure 2H).
Figure 2.

HLCs Supported Persistent and Productive Infection of DENV
(A) DENV2 infection of HLCs in a differentiation stage-dependent manner. The hESCs (day 0) and differentiated cells at various differentiation stages from day 1 to day 13 were inoculated with DENV2 (strain 16681) at an MOI of 2 for 48 hr, and cell lysates were collected for western blotting.
(B) Immunostaining analysis of DENV2-infected HLCs. hESCs (day 0), definitive endoderm (day 3), hepatic progenitors (day 6), and HLCs (day 10) were infected by DENV2 as described above. Cell were fixed and stained with anti-NS3 antibody. Scale bar, 50 μm.
(C) DENV2 infection was blocked in HLCs by inhibitors. HLCs at day 15 after differentiation were pretreated with IFN-α2a (1,000 U/mL), MPA (1 μM), CQ (5 μg/mL), or DMSO (0.1%, v/v) for 8 hr, then subjected to DENV2 infection as described above.
(D) qRT-PCR analysis of DENV RNA levels. Cells were treated as described above. The relative DENV RNA level in each sample was normalized to DMSO-treated samples.
(E) Persistent replication of DENV2 in HLCs. Day-15 HLCs were exposed to DENV2 for 6 hr before the cells were stringently washed. NS1 protein in the supernatant collected every 24 hr after infection was measured by ELISA assay.
(F) DENV2 particles released from HLCs efficiently infected C6/36 cells. HLCs at day 15 were infected with DENV2 at an MOI of 2 for 6 hr. The inoculum was then removed and the cells washed stringently with PBS five times before the fresh medium was added. The supernatants were collected every 24 hr from day 16 to day 20. After each change of the spent medium, cells were washed stringently as described above. Each medium supernatant was added to naive C6/36 cells and incubated for 24 hr. Cells were then lysed and subjected to western blotting analysis. DENV2 derived from C6/36 cells was used as a positive control.
(G) Virus titration. Virus titers were calculated in the above supernatants collected at 24, 48, and 72 hr post infection.
(H) Upregulation of DENV entry factors during the differentiation process. Total RNA from differentiated cells at the indicated days were extracted and subjected to real-time qRT-PCR to measure the cellular expressions of AXL, HAVCR1, PROS, and CLDN1. GAPDH was used as the reference and its expression was set as 1. All the genes were normalized to GAPDH to obtain their relative gene-expression ratio.
Three independent experiments were performed. Data are presented as mean ± SD. p Values were calculated by Student's t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figure S2.
Dengue Virus Infection of the HLCs Induces Innate Immune Response that Limits Virus Spread
To determine whether infection triggered IFN production and signaling in these cells, we performed RNA-seq on HLCs with or without DENV infection. A robust induction of antiviral proteins by DENV infection was detected, and many genes involved in the IFN pathway were remarkably upregulated (Figure S3A). Immunoblots to detect selected IFN-stimulated genes (ISGs) confirmed the strong induction of IFN-induced transmembrane protein 1 (IFITM1) and ubiquitin-specific peptidase 18 (USP18) in the HLCs upon infection (Figure 3A). Interestingly, the expression of IFITM1, but not that of USP18, was elevated in the undifferentiated cells to a comparable level with what could be induced by IFN (Figure 3A). We next determined whether IFN activation and signaling played a role in limiting DENV infection and spread in these cells. A Janus kinase inhibitor (JAKi) completely abolished the induction of IFITM1 by DENV infection and allowed robust expression of DENV NS3, even in the presence of exogenous IFN (Figure 3B). An inhibitor of TANK-binding kinase 1 (TBK1), a key kinase in IFN activation pathway, partially inhibited DENV-mediated IFITM1 induction, but only did so in the absence of exogenous IFN (Figure 3B). These results indicate that the complete IFN network is activated in these HLCs. Consistent with the hypothesis that IFN signaling is responsible for the observed self-limiting infection, treatment with JAKi resulted in rapid spreading of infection to 100% of the HLCs (Figures 3C and 3D). While IFN production can protect the uninfected cells in a paracrine fashion, it has been reported that DENV can inhibit IFN signaling via a variety of mechanisms in the infected cells. We indeed observed that despite the robust induction of ISGs in the uninfected cells, the expression of the IFITM family proteins was reduced in the infected cells (Figure S3B), supporting an efficient role of DENV in blocking IFN signaling. We further compared the HLCs with a hepatoma cell line, Huh 7.5, to evaluate the difference in DENV infection efficiency and immune response. As shown in Figure 3E, DENV infection did not spread to the whole cell population of the HLCs. In contrast, the virus infection rate and virus titer increased rapidly after DENV infected Huh 7.5 cells (Figures 3E–3G). Furthermore, no IFITM1 expression was induced in Huh 7.5 cells upon DENV infection (Figure 3H), suggesting a deficiency in innate immunity in this hepatoma cell line.
Figure 3.

Interferon Response Induced by DENV Infection in HLCs
(A) Upregulation of IFITM1 and USP18 upon DENV2 infection. The hESCs (day 0) and differentiated cells from day 1 to day 11 were inoculated with or without DENV2, and cell lysates were collected 48 hr post infection for western blotting. IFN-α2a-treated cells at day 12 were used as a positive control.
(B) Inhibition of IFN signaling pathway enhanced DENV2 infection. HLCs at day 15 were pretreated with DMSO (0.1%), JAK inhibitor (1 μM), or TBK1 inhibitor (2 μM) for 3 hr, followed by incubation with or without IFN-α2a (1,000 U/mL) for 8 hr. Cells were then infected with DENV2 at an MOI of 0.3 for 48 hr. Cell lysates were collected for western blotting.
(C) Inhibition of IFN signaling resulted in the spread of DENV2. Cells were treated as described above, and infected by DENV2 at an MOI of 0.01 for 72 hr. Cells were fixed and stained for immunostaining. Scale bar, 50 μm.
(D) Statistical analysis of the percentage of DENV infection. HLCs were pretreated with DMSO, JAKi, and IFN as described above, then infected by DENV2 at an MOI of 0.5 for 48 hr. The percentage of DENV infection in each sample was analyzed by immunostaining assay.
(E–G) Comparison of virus infection between HLCs and Huh 7.5 cells. Both HLCs and Huh 7.5 cells were infected by DENV2 at an MOI of 1 for 48 hr. Cell samples and the supernatants were collected every 24 hr for immunostaining analysis (E), infection rate calculation (F), and virus titration (G). Cells were washed stringently before medium change. Scale bar, 50 μm.
(H) No induction of IFITM1 in Huh 7.5 cells upon DENV2 infection. Cells were treated as described above. Cell samples were collected at 24 hr post infection for western blotting.
Three independent experiments were performed. Data are presented as mean ± SD. p Values were calculated by Student's t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figure S3.
Dengue Virus Infection Activates NF-κB Pathway and Triggers Proinflammatory Cytokine Expression
We observed that DENV infection led to significant accumulation of phosphorylated RELA/p65, the main activator of the nuclear factor κB (NF-κB) pathway, in the entire population regardless of the infection status of individual cells (Figures 4A and 4B). These suggested that the NF-κB pathway was activated not only by DENV itself, but in a paracrine fashion, such as tumor necrosis factor α (TNF) secretion. Furthermore, DENV infection of the HLCs also resulted in increased reactive oxygen species (ROS) production (Figure 4C), which, together with NF-κB activation, can lead to the expression of a large number of chemokines and proinflammatory cytokines, such as TNF, interleukin-6 (IL6), IL8, IL10, and IL12 (Figure 4D).
Figure 4.

DENV Infection Activated NF-κB Signaling Pathway
(A) Phosphorylated RELA/p65 entered the nuclei after DENV2 infection. HLCs were infected by DENV2 at an MOI of 2 for 48 hr and then subjected to staining. Scale bar, 50 μm.
(B) Statistical analysis of the ratio of phospho-NF-κB RELA/p65 positive cells in the whole population. Three independent experiments were performed. Data are presented as mean ± SD. p Values were calculated by Student's t test: ∗∗∗p < 0.001.
(C) DENV2 infection increased ROS production. HLCs were infected by DENV2 at an MOI of 2 for 48 hr during which time the cells were treated with or without PDTC as an antioxidant (10 μM). General oxidative stress indicator was used to detect ROS production. Scale bar, 50 μm.
(D) Transcriptional activation of cytokine and chemokine genes by DENV infection. Bar plot of log2(DENV2-treated/untreated) ratios of RNA-seq expression values for cytokines and chemokines targeted by NF-κB signaling (http://www.bu.edu/nf-kb/gene-resources/target-genes/). Bars are colored based on the significance of differential expression based on DESeq2. Note that some genes displayed apparently large differences in expression levels, but lack enough sequencing reads to support a significant change in expression, and these bars are colored black.
IFN Signaling Counteracts DENV-Induced Apoptosis of the HLCs
DENV infection of the HLCs led to cell death that showed characteristics of apoptosis, including morphological changes (Figure S4A), positivity in TUNEL assay (Figures 5A, S4B, and S4C), and the cleavage of caspase-3 (CASP3) and poly(ADP-ribose) polymerase (PARP), a hallmark event of apoptosis (Figure 5B). Several important pro-apoptotic genes, including TNF and Fas cell surface death receptor (FAS), were greatly upregulated by DENV infection (Figure S4D). Interestingly, cell death was dramatically exacerbated when the infected cells were also treated with JAKi. Under infection conditions of high MOI (MOI = 5 or greater), virtually all HLCs became infected within 24 hr, regardless of the presence or absence of JAKi. Without the inhibitor, cell deaths occurred in 25%–30% of cells but DENV-positive cells persisted up to 144 hr in culture, when the HLCs almost reached their normal lifespan in culture (Figure 5C, top panels). In the presence of JAKi, however, massive cell death occurred 72 hr after infection and no cells survived past 96 hr after infection (Figure 5C, bottom panels). JAKi alone in the absence of DENV infection did not cause cell death (Figure 5D). These data strongly suggest that DENV-infection-triggered apoptosis is counterbalanced by IFN signaling activation. Indeed, the cleavage of CASP3 and PARP became undetectable after 120 hr post infection, even though these cells were still productively infected with DENV (Figure 5E). Consistent with the hypothesis, DENV infection induced the rapid cell death of Huh 7.5 cells within 96 hr, compared with the HLCs (Figure 5F).
Figure 5.

Innate Immunity Protected HLCs from Massive Apoptosis Induced by DENV
(A) DENV2 infection led to apoptosis of HLCs. At 48 hr post DENV infection at an MOI of 2, HLCs were fixed for TUNEL assay. For the positive control, 0.5 μM doxorubicin, as an apoptosis inducer, was incubated with HLCs for 24 hr. Scale bar, 50 μm.
(B) DENV2 infection resulted in PARP and CASP3 cleavage. HLCs at day 15 were pretreated with JAKi (1 μM) or Z-Vad (50 μM) for 3 hr, followed by DENV2 infection as described above.
(C) Most of the DENV2-infected cells survived the infection. Day-15 HLCs were incubated with or without JAKi (1 μM) for 3 hr, then subjected to DENV2 infection at an MOI of 5 for 144 hr. Cell samples were collected at different time points after infection for immunostaining. Scale bar, 50 μm.
(D) The time course of apoptosis induced by DENV2. HLCs treated with or without IFN-α2a (1,000 U/mL) or JAKi (1 μM) were incubated with or without DENV2 at an MOI of 5 for 144 hr as described above. Cell samples were collected every 24 hr and subjected to MTT assay. Relative cell viability was calculated by comparing each sample with the mock.
(E) PARP and CASP3 cleavage during DENV2 infection. HLCs were infected by DENV2 at an MOI of 5 for 144 hr. Cell samples at different time points were collected for western blotting.
(F) Comparison of cell death between HLCs and Huh 7.5 cells after DENV2 infection. Both HLCs and Huh 7.5 cells were infected by DENV2 at an MOI of 5 for 96 hr. Cell samples were collected every 24 hr for MTT assay.
Three independent experiments were performed. Data are presented as mean ± SD. p Values were calculated by Student's t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figure S4.
Dengue Virus Infection Leads to Partial Loss of Hepatic Features In Vitro
We further investigated the effect of DENV infection on hepatic gene expression. Downregulation of E-cadherin and β-catenin was observed in the infected cells (Figures S5A and S5B), suggesting the loss of adherens junctions. To determine whether specific hepatic functions were affected, we analyzed genes that preferentially expressed in the liver from the RNA-seq dataset. In addition to a general reduction of many liver-specific genes in the infected cells (Figure 6A), we also observed a downregulation of several coagulation factors and a component of the complement system (Figure S5C). Immunostaining confirmed that ALB and F5 levels were reduced in the infected cells (Figure 6B), whereas those of other hepatic proteins such as cytokeratin 18 (CK18) and HNF4A were not affected (Figures S5D and S5E), indicating that the downregulation of ALB and F5 was not a result of non-specific inhibition of protein synthesis by DENV.
Figure 6.

DENV Infection Inhibited the Expression of ALB and F5
(A) Bar plot of log2(DENV2-treated/untreated) ratios of RNA-seq expression values for the 291 RefSeq genes preferentially expressed in liver (Liu et al., 2008). RNA-seq data were analyzed in the same way as described in Figure 4.
(B) Immunostaining of DENV2-infected HLCs. HLCs at day 15 were infected by DENV2 at an MOI of 2 for 72 hr. Cells were fixed for immunofluorescence analysis. Scale bar, 50 μm.
(C) ALB and F5 expression were downregulated in both the HLCs and PHHs after DENV2 infection. PHHs and HLCs were infected by DENV2 at an MOI of 5 for 72 hr and then subjected to western blotting.
(D) Other three serotypes of DENV reduced F5 and ALB expression in the HLCs and PHHs. HLCs and PHHs were infected by DENV type 1-Hawaii, DENV type 3-MK-594-87, DENV type 4-H241, and DENV type 4-D85-019 as described above. Cell lysates were collected for western blotting.
All Four Serotypes of DENV Can Downregulate ALB and F5 in Primary Human Hepatocytes
To determine whether the downregulation of ALB and F5 by DENV also occurred in PHHs, we performed parallel infections of HLCs and PHHs using high MOI. As shown in Figure 6C, substantial reduction of both ALB and F5 protein levels were seen in both infected HLCs and PHHs. We also investigated whether other serotypes of DENV can downregulate these liver proteins in the HLCs and PHHs. As shown in Figure 6D, infection by isolates representing all different serotypes efficiently downregulated ALB and F5 expression, suggesting that this is a conserved function of DENV.
Discussion
Human HLCs derived from pluripotent stem cells are permissive to infection by hepatotropic viruses, including HCV and HBV. Interestingly, the SEC14L2 gene, an essential host factor for supporting the replication of wild-type, unadapted HCV genomes, was induced during the hepatic differentiation (data not shown) but not expressed in the hepatoma cell line Huh 7.5 cells (Saeed et al., 2015). This difference may account for the ability of the HLCs to support infection by HCV patient serum (Wu et al., 2012, Saeed et al., 2015). Here we demonstrate that these cells also support productive infection and persistent replication of DENV. The timing of transition to viral permissiveness during the hepatic differentiation differs for the three viruses. For HCV, the transition required the induction of a liver-specific microRNA, miR-122 (Wu et al., 2012, Jopling et al., 2005), while HBV infection of the differentiated HLCs correlated with the induction of the HBV receptor, sodium taurocholate cotransporting peptide (NTCP) (Shlomai et al., 2014, Yan et al., 2012). Similar to the situation with HBV, transition to DENV permissiveness correlated with the induction of DENV entry factors, the TIM-TAM family of proteins and their ligand, for hepatic cells (Meertens et al., 2012). Elucidating gene-expression changes that are associated with the shift to virus susceptibility during differentiation represents a valuable approach with the potential of identifying cellular determinants that control viral infection. This approach is especially attractive in the case of DENV because of the extraordinarily broad tropism it exhibits in cell culture (Acosta et al., 2014). In fact, hPSCs and the early progenies are rare examples of cells that are refractory to DENV infection in vitro.
In the liver, hepatocytes are organized as polarized epithelial cell layers to separate sinusoidal blood from the secreted bile and perform hepatic functions, and specific cell-cell junctions are required for the maintenance of the polarized cell layers (Gissen and Arias, 2015). In some liver diseases, these junctions and the polarity of hepatocytes are affected. However, it is difficult to fully elucidate the pathophysiological features of hepatocytes using hepatoma cell lines because hepatocellular epithelial to mesenchymal transition and degeneration are common events in hepatocellular carcinoma progression, which result in the loss of epithelial features and some hepatic functions (van Zijl et al., 2009). In the case of DENV infection, it is has been reported that DENV can damage hepatocytes and disrupt liver functions, leading to elevations of serum bilirubin and transaminases, prolonged prothrombin time, and reduced serum albumin level (Huy et al., 2013, Roy et al., 2013, Samanta and Sharma, 2015). In our study, we utilized stem cell-derived HLCs to investigate the interactions between DENV and hepatocytes and observed downregulation of albumin, several coagulation factors, and a component of the complement pathway by DENV infection. These virus-induced changes in hepatic features may contribute to the effect of DENV on liver in vivo. The precise mechanism of the direct downregulation of these genes remains unclear at this time.
DENV infection activates an antiviral response in vitro and in vivo (Green et al., 2014). Our results showed that many reported anti-DENV ISGs, including IFITM1, IFTM3, IRF1, IRF7, and IFI6 (Schoggins et al., 2012, Brass et al., 2009), were highly increased in the DENV-treated HLCs, which probably is the main reason why DENV infection of hepatic cells is self-limiting. In vivo, the IFN response generated by the hepatic cells is likely complemented by similar responses from resident immune cells in the liver; in fact, IFN secreted from infected immune cells can also trigger IFN signaling in the hepatocytes and protect liver from massive infection. Together, these data may help explain the rareness of acute liver injuries in DENV infection.
DENV has also been shown to counteract IFN signaling in the infected cells, mainly through the expression of NS5 and NS4B (Munoz-Jordan et al., 2005, Ashour et al., 2009, Morrison et al., 2013). We indeed observed lessened ISG induction in the infected cells but not in the uninfected cells in the same virally exposed population. Consistent with these results, we found that although the infection of HLCs was normally self-limiting when a low MOI was used, virtually all cells could be infected if a high (>5) MOI was used in the initial inoculation, as no viral spread was needed in this case to infect all the cells and the IFN response was not able to clear the infection once it had been established. As reduced viral load generally correlates with lessened disease severity (Marques et al., 2015), therapeutic strategies aiming at reducing viral load early may better harness the benefit of host IFN response to thwart disease progression.
A “cytokine storm” triggered by DENV is thought to be the major event of DENV pathogenesis (Guzman and Harris, 2015). Several cytokines that have been proposed to be associated with plasma leakage, including TNF, IL-6, IL-8, IL-10, and IL-12 (Guzman and Harris, 2015), were dramatically upregulated in HLCs by DENV infection. Activation of an inflammatory response via the cellular NF-κB pathway by the secreted viral protein NS1 is one underlying mechanism of the cytokine activation (Modhiran et al., 2015, Beatty et al., 2015, Silva et al., 2011). We observed a remarkable upregulation of mRNA levels of TNF and CD137 (TNFRSF9), the major signal and receptor for the canonical NF-κB pathway. The expression of CD40 gene was also induced by DENV infection, but its ligand, CD40LG, was not expressed in these hepatic cells, suggesting that the non-canonical NF-κB pathway is not activated in the HLC system. Because the TNF gene can also be stimulated by type I IFN, its induction by DENV infection may represent the crosstalk before the cellular antiviral response and the inflammatory response at the tissue level. Furthermore, ROS induction by DENV infection can also influence the outcome of the NF-κB pathway activation (Morgan and Liu, 2011, Olagnier et al., 2014). Finally, direct activation of NF-κB pathway by DENV protease has also been reported. In this case, the DENV protease induces the degradation of NF-κB inhibitors IκBα and IκBβ via cleavage (Lin et al., 2014). Regardless of the mechanism of the initial activation, these signal molecules likely elicit an amplified response through both autocrine and paracrine pathways. The nuclear translocation of RELA/p65 was indeed detected in all the cells including the uninfected cells. Unlike the IFN signaling and ISG induction, we did not observe any evidence of DENV proteins interfering with NF-κB signaling in the infected cells. The unabated activation of NF-κB pathway and the release of cytokines from hepatocytes, either infected or exposed to soluble signals (NS1, cytokines) produced by infected blood cells, likely contribute to the inflammatory aspects of dengue pathology.
Induction of apoptosis of immune cells following DENV infection is a common feature in vivo. However, it has not been well documented whether DENV causes massive apoptosis of hepatocytes in the same way. We observed rapid cell death and CASP3 activation early in the infection time course. However, a population of the infected cells eventually survived and showed no signs of cleavage of CASP3 or PARP, but only when IFN signaling was not completely blocked. Together, these data point to an anti-apoptotic role of IFN signaling during DENV infection of the HLCs, such as the one reported for IFI6 in DENV-infected endothelial cells (Qi et al., 2015). In addition, viral protein-induced autophagy has been proposed as an additional mechanism for protecting the infected epithelial cells from apoptosis (McLean et al., 2011). In vivo, the infected hepatocytes may also be cleared by antibody-dependent cell-mediated cytotoxicity and T cell-mediated cytotoxicity through DENV antigen (such as NS1) displayed on the cell surface. Despite all of these factors, the strong capacity of regeneration of the liver likely contributes to the rareness of acute liver failure.
DENV, as one of the most widespread vector-borne viruses, infects millions of people each year worldwide. However, no clinically approved drugs are yet available against DENV. There is an urgent need to develop effective anti-DENV drugs for such treatment. In this study we tested IFN-α2a, chloroquine, and mycophenolic acid for their ability to inhibit DENV infection in our in vitro system. IFN, as a well-established antiviral cytokine, can block several RNA and DNA viruses at different stages from viral entry to release. Both chloroquine and mycophenolic acid have been shown to inhibit replication of DENV in the laboratory tests (Farias et al., 2013, Diamond et al., 2002). Consistent with previous studies, all of these inhibitors reduced DENV infection in HLCs at various levels, suggesting that our in vitro system may have potential utility for anti-DENV drug screening efforts.
In summary, we demonstrate that DENV can productively infect human HLCs derived from PSCs and produce in vitro phenotypes that reflect pathophysiologically relevant aspects of DENV-host interaction. These HLCs, which transit from refractory to permissive for DENV infection under defined growth conditions, are also useful tools for dissecting the molecular determinants of DENV susceptibility and the identification of new drug targets for this important human pathogen. Finally, coupling genome-editing technologies, patient-specific iPSCs, or both with the HLC-DENV system can enable research into the effect of host genetics on DENV infection and pathogenesis.
Experimental Procedures
Cells and Viruses
hESC line H9 (WA09) and iPSC line iPS DF19-9-11T.H were obtained from WiCell Research Institute and propagated on Matrigel (BD Biosciences)-coated plates in mTeSR1 medium (Stemcell Technologies). Freshly isolated PHHs (Life Technologies) were maintained in William's E media (Life Technologies) with Supplements CM4000 (Life Technologies) in collagen-coated plates according to the vendor's instructions. Aedes albopictus cell clone C6/36 cells (ATCC) were cultured in ATCC-formulated Eagle's minimum essential medium supplemented with 10% fetal bovine serum (FBS) (Life Technologies). Huh 7.5 cells were provided by Charles M. Rice and grown in DMEM (Life Technologies) with 10% FBS. Three serotypes of DENV, including DENV type 1-Hawaii, DENV type 3-MK-594-87, DENV type 4-H241, and DENV type 4-D85-019, were obtained from BEI Resources while DENV type 2-16681 was provided by Dr. Qianjun Li (University of Alabama). All viruses were propagated in the C6/36 cells, and virus titers were calculated by plaque assay on BHK21 cells (ATCC).
Differentiation of Human Pluripotent Stem Cells to Hepatocyte-like Cells
To initiate differentiation, we collected hPSCs as small clumps from a plate containing 80%–90% confluent stem cells and transferred them into a 12-well plate coated with Matrigel at the confluence of 8%–10% in 1 mL of mTeSR1 in each well. After 1–2 days of culture, the medium was replaced by chemically defined basal medium (DMEM/F12 [Mediatech]) containing 1.8% BSA [Millipore], 0.055 mM β-mercaptoethanol [Life Technologies], 1× L-glutamine [Life Technologies], and 1× B27 supplement [Life Technologies]) supplemented with 100 ng/mL Activin A (R&D Systems), 25 ng/mL Wnt 3a (R&D), 10 μM LY294002 (Sigma), and 10 ng/mL fibroblast growth factor 2 (FGF2) (R&D) for 24 hr, followed by 100 ng/mL Activin A and 10 ng/mL FGF2 for 48 hr to specify definitive endoderm (DE). DE was then induced into hepatoblast for 3 days using 1 μM retinoic acid (Sigma), 1 μM SB431542 (Sigma), 10 ng/mL BMP4 (R&D), and 10 ng/mL FGF2. Subsequently, hepatic progenitors were differentiated for 6 days into early HLCs using 50 ng/mL HGF (Peprotech), 30 ng/mL FGF4 (Peprotech), and 50 ng/mL epidermal growth factor (EGF) (Peprotech), and further differentiated using 10 ng/mL oncostatin M (R&D) for 3 days. The HLCs were then grown in Clonetics HCM Hepatocyte Culture Medium (Lonza) containing “Singlequots” omitting EGF and hydrocortisone with changes of fresh medium every 2 days.
DENV Infection
For DENV infection, cells were incubated with virus at the indicated MOI for 6 hr before the medium was changed to the one appropriate for the specific cell type and differentiation stage. At 48 or 72 hr post infection, cells were harvested for western blotting or immunofluorescence analyses. The same amount of “spent” medium of uninfected C6/36 cells was added to the cells as the negative control. To inhibit DENV infection, we incubated 1,000 U/mL IFN-α2a (Sigma), 1 μM mycophenolic acid (Sigma), 5 μg/mL chloroquine (Sigma), or 0.1% DMSO (Sigma) with HLCs at day 15 for 8 hr before DENV infection. For the study of IFN response induced by DENV, HLCs were treated with 0.1% DMSO (Sigma), 1 μM Janus kinase inhibitor (JAKi) (Millipore), or 2 μM TANK-binding kinase 1 inhibitor (TBKi) (Millipore) for 3 hr. Cells were then infected with DENV for 48 hr after incubation with or without 1,000 U/mL of IFN-α2a for 8 hr. For the cell death assay, day-15 HLCs were pretreated with 1 μM JAKi or 50 μM Z-Vad (Santa Cruz Biotechnology) for 3 hr, followed by DENV infection for 24–144 hr.
RNA-Seq Data Analyses and Real-Time qRT-PCR
Total RNA were isolated from the differentiated cells of day 5, day 10, and day 12 with or without DENV infection by the miRNeasy Mini kit (Qiagen) and then were sent to the Clinical Microarray Core at UCLA and Genomics Research Center at the University of Rochester for RNA-seq. The TruSeq RNA Sample Preparation Kit V2 (Illumina) was used for next-generation sequencing library construction as per manufacturer's protocols. In brief, mRNA was purified from 100 ng of total RNA with oligo-dT magnetic beads and fragmented. First-strand cDNA synthesis was performed with random hexamer priming followed by second-strand cDNA synthesis. End repair and 3′ adenylation was then performed on the double-stranded cDNA. Illumina adaptors were ligated to both ends of the cDNA, purified by gel electrophoresis, and amplified with PCR primers specific to the adaptor sequences to generate amplicons of approximately 200–500 bp in size. The amplified libraries were hybridized to the Illumina single-end flow cell and amplified using the cBot (Illumina) at a concentration of 8 pM per lane. Single-end reads of 100 nt were generated for each sample and aligned to the hg19 reference genome with Tophat2 (Kim et al., 2013). Gene-level counts were calculated using reads with alignment qualities >10 using featureCounts (Liao et al., 2014), and differential expression was tested with DESeq2 (Love et al., 2014). All the RNA-seq data were deposited in the NCBI Sequence Read Archive with the accession number SRA: SRP078315. For RT-PCR, the isolated total RNA was cleaned up by RNA Clean Kit (Zymo) and converted to first-strand cDNA by SuperScript III kit (Life Technologies). Real-time qPCR was then performed to analyze the expression of DENV cofactors and receptors and DENV RNA levels.
NS1 ELISA
HLCs at day 15 were pretreated for 8 hr with or without 1,000 U/mL of IFN-α2a, followed by infection with DENV at an MOI of 2 for 6 hr. The inoculum was then removed and the cells washed stringently with PBS five times before fresh medium was added. The supernatants were collected each day from day 16 to day 21 for measuring nonstructural protein 1 (NS1) in the supernatant, using a DENV NS1 Antigen ELISA Kit (Abnova). After each change of the spent medium, cells were washed stringently as described above.
Immunofluorescence Staining
Cells grown on coverslips were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 10 min, and blocked with PBGB (PBS containing 10% normal goat serum, and 1% BSA) at room temperature for 2 hr. Subsequently, cells were incubated with primary antibodies at 4°C overnight. After three washes with PBS, fluorescein isothiocyanate or tetramethylrhodamine isothiocyanate-conjugated secondary antibody (Sigma) was added and incubated at room temperature for 2 hr. The stained coverslips were mounted in Vectashield medium (Vector Laboratories) after three washes with PBS. Images were processed by Adobe Photoshop.
MTT Assay
Cell viability was measured by a colorimetric MTT assay (Life Technologies). Before the assay, the culture medium was changed to the phenol red-free Clonetics HCM Hepatocyte Culture Medium. After incubation with MTT or DMSO, the optical absorbance was read at 540 nm.
TUNEL Assay
Cell samples were fixed and stained using the In Situ Cell Death Detection kit (Roche Applied Science) according to the manufacturer's instructions. For the positive control, 0.5 μM doxorubicin (Sigma) was incubated with HLCs for 24 hr.
ROS Detection
ROS production in the DENV-infected cells was detected using CM-H2DCFDA General Oxidative Stress Indicator kit (Life Technologies). After DENV infection of day-15 HLCs for 48 hr, cells were washed twice with PBS and incubated with 1 μM CM-H2DCFDA probe for 30 min at 37°C. Following three washes with PBS, oxidation of the probe in the DENV-infected cells was detected by monitoring the increase in green fluorescence with a fluorescence microscope. To inhibit ROS production, we used 10 μM ammonium pyrrolidinedithiocarbamate (PDTC) (Abcam) to treat HLCs during DENV infection.
Statistical Analysis
All the data are presented as mean ± SD. Three independent experiments were performed. p Values were calculated by two-tailed Student's t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Author Contributions
J.L. and H.T. designed the study. J.L. conducted most of the experiments. D.V. performed the bioinformatics analysis. Y.C. designed the primers and did the RNA purification. H.T. and J.L. wrote the manuscript.
Acknowledgments
We would like to thank Dr. Charles Rice for Huh7.5 cells and Dr. Qianjun Li for DENV2. We thank Brian Washburn, Christy Hammack, and Sarah Ogden for technical assistance, and members of the Tang laboratory for helpful comments. This study was supported by NIH/NIAID grant R21 AI119530 to H.T.
Published: August 18, 2016
Footnotes
Supplemental Information includes five figures and five tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.07.012.
Supplemental Information
References
- Acosta E.G., Kumar A., Bartenschlager R. Revisiting dengue virus-host cell interaction: new insights into molecular and cellular virology. Adv. Virus Res. 2014;88:1–109. doi: 10.1016/B978-0-12-800098-4.00001-5. [DOI] [PubMed] [Google Scholar]
- Ashour J., Laurent-Rolle M., Shi P.Y., Garcia-Sastre A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009;83:5408–5418. doi: 10.1128/JVI.02188-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsitis S.J., Coloma J., Castro G., Alava A., Flores D., McKerrow J.H., Beatty P.R., Harris E. Tropism of dengue virus in mice and humans defined by viral nonstructural protein 3-specific immunostaining. Am. J. Trop. Med. Hyg. 2009;80:416–424. [PubMed] [Google Scholar]
- Beatty P.R., Puerta-Guardo H., Killingbeck S.S., Glasner D.R., Hopkins K., Harris E. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci. Transl. Med. 2015;7:304ra141. doi: 10.1126/scitranslmed.aaa3787. [DOI] [PubMed] [Google Scholar]
- Bhatt S., Gething P.W., Brady O.J., Messina J.P., Farlow A.W., Moyes C.L., Drake J.M., Brownstein J.S., Hoen A.G., Sankoh O., Myers M.F. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackley S., Kou Z., Chen H., Quinn M., Rose R.C., Schlesinger J.J., Coppage M., Jin X. Primary human splenic macrophages, but not T or B cells, are the principal target cells for dengue virus infection in vitro. J. Virol. 2007;81:13325–13334. doi: 10.1128/JVI.01568-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass A.L., Huang I.C., Benita Y., John S.P., Krishnan M.N., Feeley E.M., Ryan B.J., Weyer J.L., van der Weyden L., Fikrig E., Adams D.J. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Hernandez A., Thepparit C., Suksanpaisan L., Smith D.R. Dengue virus entry into liver (HepG2) cells is independent of hsp90 and hsp70. J. Med. Virol. 2007;79:386–392. doi: 10.1002/jmv.20786. [DOI] [PubMed] [Google Scholar]
- Che P., Tang H., Li Q. The interaction between claudin-1 and dengue viral prM/M protein for its entry. Virology. 2013;446:303–313. doi: 10.1016/j.virol.2013.08.009. [DOI] [PubMed] [Google Scholar]
- Diamond M.S., Zachariah M., Harris E. Mycophenolic acid inhibits dengue virus infection by preventing replication of viral RNA. Virology. 2002;304:211–221. doi: 10.1006/viro.2002.1685. [DOI] [PubMed] [Google Scholar]
- Farias K.J., Machado P.R., da Fonseca B.A. Chloroquine inhibits dengue virus type 2 replication in vero cells but not in C6/36 cells. ScientificWorldJournal. 2013;2013:282734. doi: 10.1155/2013/282734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gissen P., Arias I.M. Structural and functional hepatocyte polarity and liver disease. J. Hepatol. 2015;63:1023–1037. doi: 10.1016/j.jhep.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green A.M., Beatty P.R., Hadjilaou A., Harris E. Innate immunity to dengue virus infection and subversion of antiviral responses. J. Mol. Biol. 2014;426:1148–1160. doi: 10.1016/j.jmb.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M.G., Harris E. Dengue. Lancet. 2015;385:453–465. doi: 10.1016/S0140-6736(14)60572-9. [DOI] [PubMed] [Google Scholar]
- Halstead S.B. Dengue. Lancet. 2007;370:1644–1652. doi: 10.1016/S0140-6736(07)61687-0. [DOI] [PubMed] [Google Scholar]
- Heaton N.S., Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe. 2010;8:422–432. doi: 10.1016/j.chom.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgard P., Stockert R. Heparan sulfate proteoglycans initiate dengue virus infection of hepatocytes. Hepatology. 2000;32:1069–1077. doi: 10.1053/jhep.2000.18713. [DOI] [PubMed] [Google Scholar]
- Huy N.T., Van Giang T., Thuy D.H., Kikuchi M., Hien T.T., Zamora J., Hirayama K. Factors associated with dengue shock syndrome: a systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2013;7:e2412. doi: 10.1371/journal.pntd.0002412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessie K., Fong M.Y., Devi S., Lam S.K., Wong K.T. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J. Infect. Dis. 2004;189:1411–1418. doi: 10.1086/383043. [DOI] [PubMed] [Google Scholar]
- Jopling C.L., Yi M., Lancaster A.M., Lemon S.M., Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lin J.C., Lin S.C., Chen W.Y., Yen Y.T., Lai C.W., Tao M.H., Lin Y.L., Miaw S.C., Wu-Hsieh B.A. Dengue viral protease interaction with NF-kappaB inhibitor alpha/beta results in endothelial cell apoptosis and hemorrhage development. J. Immunol. 2014;193:1258–1267. doi: 10.4049/jimmunol.1302675. [DOI] [PubMed] [Google Scholar]
- Liu X., Yu X., Zack D.J., Zhu H., Qian J. TiGER: a database for tissue-specific gene expression and regulation. BMC Bioinformatics. 2008;9:271. doi: 10.1186/1471-2105-9-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marianneau P., Cardona A., Edelman L., Deubel V., Despres P. Dengue virus replication in human hepatoma cells activates NF-kappaB which in turn induces apoptotic cell death. J. Virol. 1997;71:3244–3249. doi: 10.1128/jvi.71.4.3244-3249.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques R.E., Guabiraba R., Del Sarto J.L., Rocha R.F., Queiroz A.L., Cisalpino D., Marques P.E., Pacca C.C., Fagundes C.T., Menezes G.B., Nogueira M.L. Dengue virus requires the CC-chemokine receptor CCR5 for replication and infection development. Immunology. 2015;145:583–596. doi: 10.1111/imm.12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean J.E., Wudzinska A., Datan E., Quaglino D., Zakeri Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011;286:22147–22159. doi: 10.1074/jbc.M110.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meertens L., Carnec X., Lecoin M.P., Ramdasi R., Guivel-Benhassine F., Lew E., Lemke G., Schwartz O., Amara A. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe. 2012;12:544–557. doi: 10.1016/j.chom.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanari Y., Atsuzawa K., Usuda N., Watashi K., Hishiki T., Zayas M., Bartenschlager R., Wakita T., Hijikata M., Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- Modhiran N., Watterson D., Muller D.A., Panetta A.K., Sester D.P., Liu L., Hume D.A., Stacey K.J., Young P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015;7:304ra142. doi: 10.1126/scitranslmed.aaa3863. [DOI] [PubMed] [Google Scholar]
- Morgan M.J., Liu Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison J., Laurent-Rolle M., Maestre A.M., Rajsbaum R., Pisanelli G., Simon V., Mulder L.C., Fernandez-Sesma A., García-Sastre A. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 2013;9:e1003265. doi: 10.1371/journal.ppat.1003265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Jordan J.L., Laurent-Rolle M., Ashour J., Martinez-Sobrido L., Ashok M., Lipkin W.I., Garcia-Sastre A. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 2005;79:8004–8013. doi: 10.1128/JVI.79.13.8004-8013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olagnier D., Peri S., Steel C., van Montfoort N., Chiang C., Beljanski V., Slifker M., He Z., Nichols C.N., Lin R., Balachandran S. Cellular oxidative stress response controls the antiviral and apoptotic programs in dengue virus-infected dendritic cells. PLoS Pathog. 2014;10:e1004566. doi: 10.1371/journal.ppat.1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham A.M., Langlois R.A., TenOever B.R. Replication in cells of hematopoietic origin is necessary for Dengue virus dissemination. PLoS Pathog. 2012;8:e1002465. doi: 10.1371/journal.ppat.1002465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povoa T.F., Alves A.M., Oliveira C.A., Nuovo G.J., Chagas V.L., Paes M.V. The pathology of severe dengue in multiple organs of human fatal cases: histopathology, ultrastructure and virus replication. PLoS One. 2014;9:e83386. doi: 10.1371/journal.pone.0083386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y., Li Y., Zhang Y., Zhang L., Wang Z., Zhang X., Gui L., Huang J. IFI6 inhibits apoptosis via mitochondrial-dependent pathway in dengue virus 2 infected vascular endothelial cells. PLoS One. 2015;10:e0132743. doi: 10.1371/journal.pone.0132743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelandt P., Obeid S., Paeshuyse J., Vanhove J., Van Lommel A., Nahmias Y., Nevens F., Neyts J., Verfaillie C.M. Human pluripotent stem cell-derived hepatocytes support complete replication of hepatitis C virus. J. Hepatol. 2012;57:246–251. doi: 10.1016/j.jhep.2012.03.030. [DOI] [PubMed] [Google Scholar]
- Rothman A.L. Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nat. Rev. Immunol. 2011;11:532–543. doi: 10.1038/nri3014. [DOI] [PubMed] [Google Scholar]
- Roy A., Sarkar D., Chakraborty S., Chaudhuri J., Ghosh P., Chakraborty S. Profile of hepatic involvement by dengue virus in dengue infected children. N. Am. J. Med. Sci. 2013;5:480–485. doi: 10.4103/1947-2714.117313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed M., Andreo U., Chung H.Y., Espiritu C., Branch A.D., Silva J.M., Rice C.M. SEC14L2 enables pan-genotype HCV replication in cell culture. Nature. 2015;524:471–475. doi: 10.1038/nature14899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta J., Sharma V. Dengue and its effects on liver. World J. Clin. Cases. 2015;3:125–131. doi: 10.12998/wjcc.v3.i2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsa M.M., Mondotte J.A., Iglesias N.G., Assuncao-Miranda I., Barbosa-Lima G., Da Poian A.T., Bozza P.T., Gamarnik A.V. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009;5:e1000632. doi: 10.1371/journal.ppat.1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W., Dorner M., Feulner M., Imanaka N., Murphy M.Y., Ploss A., Rice C.M. Dengue reporter viruses reveal viral dynamics in interferon receptor-deficient mice and sensitivity to interferon effectors in vitro. Proc. Natl. Acad. Sci. USA. 2012;109:14610–14615. doi: 10.1073/pnas.1212379109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz R.E., Trehan K., Andrus L., Sheahan T.P., Ploss A., Duncan S.A., Rice C.M., Bhatia S.N. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2012;109:2544–2548. doi: 10.1073/pnas.1121400109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomai A., Schwartz R.E., Ramanan V., Bhatta A., de Jong Y.P., Bhatia S.N., Rice C.M. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc. Natl. Acad. Sci. USA. 2014;111:12193–12198. doi: 10.1073/pnas.1412631111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si-Tayeb K., Noto F.K., Nagaoka M., Li J., Battle M.A., Duris C., North P.E., Dalton S., Duncan S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva B.M., Sousa L.P., Gomes-Ruiz A.C., Leite F.G., Teixeira M.M., da Fonseca F.G., Pimenta P.F., Ferreira P.C., Kroon E.G., Bonjardim C.A. The dengue virus nonstructural protein 1 (NS1) increases NF-kappaB transcriptional activity in HepG2 cells. Arch. Virol. 2011;156:1275–1279. doi: 10.1007/s00705-011-0969-0. [DOI] [PubMed] [Google Scholar]
- Suksanpaisan L., Cabrera-Hernandez A., Smith D.R. Infection of human primary hepatocytes with dengue virus serotype 2. J. Med. Virol. 2007;79:300–307. doi: 10.1002/jmv.20798. [DOI] [PubMed] [Google Scholar]
- Touboul T., Hannan N.R., Corbineau S., Martinez A., Martinet C., Branchereau S., Mainot S., Strick-Marchand H., Pedersen R., Di Santo J., Weber A. Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development. Hepatology. 2010;51:1754–1765. doi: 10.1002/hep.23506. [DOI] [PubMed] [Google Scholar]
- van Zijl F., Zulehner G., Petz M., Schneller D., Kornauth C., Hau M., Machat G., Grubinger M., Huber H., Mikulits W. Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol. 2009;5:1169–1179. doi: 10.2217/fon.09.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.J., Grouard-Vogel G., Sun W., Mascola J.R., Brachtel E., Putvatana R., Louder M.K., Filgueira L., Marovich M.A., Wong H.K., Blauvelt A. Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 2000;6:816–820. doi: 10.1038/77553. [DOI] [PubMed] [Google Scholar]
- Wu X., Robotham J.M., Lee E., Dalton S., Kneteman N.M., Gilbert D.M., Tang H. Productive hepatitis C virus infection of stem cell-derived hepatocytes reveals a critical transition to viral permissiveness during differentiation. PLoS Pathog. 2012;8:e1002617. doi: 10.1371/journal.ppat.1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H., Zhong G., Xu G., He W., Jing Z., Gao Z., Huang Y., Qi Y., Peng B., Wang H., Fu L. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.