

Abstract
Gefitinib, an EGFR tyrosine kinase inhibitor, is used as FDA approved drug in breast cancer and non-small cell lung cancer treatment. However, this drug has certain side effects and complications for which the underlying molecular mechanisms are not well understood. By systems biology based in silico analysis, we identified off-targets of gefitinib that might explain side effects of this drugs. The crystal structure of EGFR-gefitinib complex was used for binding pocket similarity searches on a druggable proteome database (Sc-PDB) by using IsoMIF Finder. The top 128 hits of putative off-targets were validated by reverse docking approach. The results showed that identified off-targets have efficient binding with gefitinib. The identified human specific off-targets were confirmed and further analyzed for their links with biological process and clinical disease pathways using retrospective studies and literature mining, respectively. Noticeably, many of the identified off-targets in this study were reported in previous high-throughput screenings. Interestingly, the present study reveals that gefitinib may have positive effects in reducing brain and bone metastasis, and may be useful in defining novel gefitinib based treatment regime. We propose that a system wide approach could be useful during new drug development and to minimize side effect of the prospective drug.
Lung cancer is the most common cancer as evident from a comprehensive global report that also showed ∼1.8 million new cases reported in 20121. It has been one of the leading causes of cancer-related mortality worldwide (19.4% of all cancers). Additionally, it is more prominent in developing countries (58%) than in developed countries1. The abnormal activation of epidermal growth factor receptor (EGFR) tyrosine kinase is responsible for promoting various tumor types, including lung cancer and breast cancer either via an increase in the levels of extracellular ligand, hetero-dimerization of EGFR or its mutational activation2,3. The most common EGFR mutations reported so far in the case of non-small cell lung cancer (NSCLC) are the deletion of exon 19 and substitution mutation (L858R) at exon 21, leading to constitutive tyrosine kinase activity independent of ligand binding4.
Considering the role of EGFR in tumor progression, targeting it for NSCLC treatment is an effective approach. In this direction, various small molecule tyrosine kinase inhibitors, such as erlotinib, gefitinib, lapatinib have been developed and are being used as US Food and Drug Administration (FDA) approved drugs in breast cancer and NSCLC treatment regime5. Gefitinib has emerged as a novel therapeutic molecule impairing the tyrosine kinase activity of EGFR effectively. This impairment leads to blockage of downstream signaling and thus inhibits the tumor proliferation activity of EGFR6,7,8. This drug is administered orally at a dosage of 250–500 mg/day and is implemented as first-, second- and third -line therapy in cases of NSCLC9.
Gefitinib has certain side effects such as nausea, vomiting, diarrhea and interstitial lung disease10. These adverse side effects may be accounted by inhibition of either EGFR and/or drug off-targets11. Therefore, analyzing the off-targets of this drug will prove to be effective to reveal the true scenario of Gefitinib: aids and ills and will help in rational modifications of this drug to minimize the side effects. Additionally, for successful establishment of highly efficient drug-based therapies, early identification of adverse drug effects can be a crucial step as up to 40% of drug failures occur during development, adverse events in pre-clinical trials and pharmacokinetics12.
Identification of drug off-targets by an in vitro counter screening of compounds against numerous receptors and enzymes is expensive and time-consuming13,14. In contrast, in silico analysis of drug off-targets is safe, time-efficient, economical and provides a deeper understanding of the molecular mechanisms of protein-drug interactions. It has been shown that based on the establishment of the structure-activity relationship of small molecules, an in silico off-target identification can be obtained15,16. Various structure-based tools for comparing binding sites of small ligands of distantly related proteins have been developed17.
In the present study, we identified gefitinib off-targets using structure-based systems biology approach. We could confirm the binding of identified off-targets with gefitinib using a reverse docking approach. Additionally, through comparative re-docking analyses of identified off-targets with their respective experimentally characterised ligands (ligands that were present in the crystal structure of the protein) and gefitinib, we observed that a few identified off-targets may bind more efficiently with gefitinib compared to their previously reported and experimentally validated ligands. Furthermore, literature survey and data mining has clearly shown several of the identified off-targets were validated in previously reported in vitro studies. Together, these observations clearly suggest that off-targets of gefitinib identified in this study might be true off targets and could be involved in the molecular mechanism underlying the possible side effects of this drug. Interestingly, our study suggested not only negative side effects but also positive roles of gefitinib. Additionally, we could identify non-human off-targets that may be used for effective treatment of pathogen-based diseases.
Results
Prediction of gefitinib off-targets through Molecular interaction field (MIF) similarity search
To carry out the molecular interaction fields (MIFs) similarity search, the crystal structure of EGFR kinase with gefitinib (PDB id: 4WKQ) was used as query structure. This structure showed the binding of different molecules; viz., gefitinib (IRE), 2-(N-morpholino)-Ethanesulfonic acid and sodium ion in the native state. The binding pocket of EGFR kinase around the gefitinib was calculated at the distance of 3 Å and it was found to be defined by residues Leu718, Gly719, Ala743, Ile744, Lys745, Glu762, Met766, Leu788, Thr790, Gln791, Leu792, Met793, Pro794, Phe795, Gly796, Arg841, Asn842, Leu844, Thr854, Asp855 and Phe856 (Fig. 1A,B). Gefitinib interacts with Met793 in the EGFR binding pocket via H-bond formation with the nitrogen atom of the quinazoline ring and Van der Waals interactions.
Figure 1. Identification of the Gefitinib binding pocket in EGFR kinase domain.
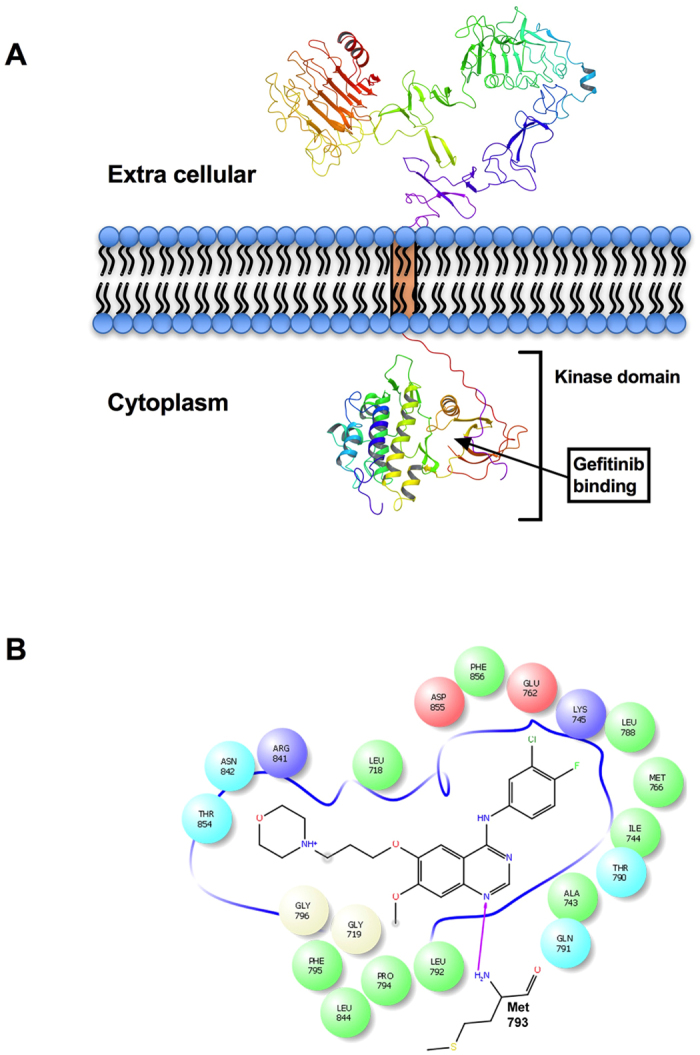
(A) Schematic illustration of EGFR receptor on the cell membrane. (B) The gefitinib binding pocket was calculated at the distance of 3 Å using Maestro. The identified ligand binding pocket is shown in 2D. Residues are colored according to their properties (red, negatively charged; blue, positively charged; cyan, polar; green, hydrophobic; and white, neutral). The H-bond is shown with magenta arrow.
The MIFs of ligand binding cavities of proteins listed in sc-PDB database (containing 8077 protein structures) were calculated using six properties; H-bond donor/acceptor, aromatic, hydrophobic and positively/negatively charged interactions and compared with query MIF (i.e. Gefitinib binding pocket of EGFR). All the analyzed protein structures were ranked and arranged according to the Tanimoto scores (designated as MIF ranking; Supplementary Table S1). In total, 128 protein structures were found to have Tanimoto scores of ≥0.35 value. These were considered as putative off-targets of Gefitinib, and were selected for further analysis. These selected protein structures represent 50 proteins (Table 1). These hits belong to following species; human (41), rat (3), Xenopus laevis, Pseudomonas putida, Toxoplasma gondii, Cryptosporidium parvum, E. coli, Betula pendula and Zea mays (1 each). For subsequent analysis, we focused on the 41 identified-human proteins (Table 1). Most of the putative off-target hits (107) belong to protein kinase family (pfam ID: PF00069 and PF07714). The remaining hits belong to pfam domain Ephrin type-A receptor 2 transmembrane domain, EF-hand domain, Cartilage oligomeric matrix protein, transforming growth factor beta type I GS-motif, MAATS-type transcriptional repressor, Dihydroorotate dehydrogenase, Protein Kinase C terminal domain etc. The pfam ids of above domains are listed in Table 1. Furthermore, the known functions and subcellular localization of the selected proteins are shown in Table 1.
Table 1. The details of off-targets of gefitinib identified through MIF similarity serach analysis.
| S. No. | Gene names | PDB ID | UniprotKB ID | Pfam ID | Organism | Functions | Locations |
|---|---|---|---|---|---|---|---|
| 1 | BMPR1B | 3MDY | O00238 | PF08515 | Human | positive regulation of chondrocyte differentiation | cell membrane |
| 2 | SRC SRC1 | 1YOL, 1Y57 3EN5, 3EN7, 3EN4 | P12931 P00523 | PF07714 | Human Chicken | regulation of cytoskeletal organization, activation of EGF-mediated calcium- chloride channel | cell membrane, mitochondrial inner membrane, nucleus, cytoplasm |
| 3 | FGFR2 BEK KGFR KSAM | 3RI1, 1OEC | P21802 | PF07714 | Human | regulates the cell proliferation, differentiation, migration, apoptosis, and embryonic development | cell membrane, Golgi apparatus, cytoplasmic vesicles |
| 4 | ACVRL1 ACVRLK1 ALK1 | 3MY0 | P37023 | PF08515 | Human | regulator of normal blood vessel development | cell membrane |
| 5 | MAPK8 JNK1 PRKM8 SAPK1 SAPK1C | 3ELJ | P45983 | PF00069 | Human | regulates the circadian clock | cytoplasm, nucleus |
| 6 | ALK | 2XBA | Q9UM73 | ALK_HUMAN | Human | genesis and differentiation of the nervous system | cell membrane |
| 7 | Comp | 1FBM | P35444 | COMP_RAT | Rat | regulates structural integrity of cartilage | extra cellular matrix |
| 8 | AURKA AIK AIRK1 ARK1 AURA AYK1 BTAK IAK1 STK15 STK6 | 3LAU, 3UOH | O14965 | PF00069 | Human | regulates cell cycle progression | cytoplasm, cytoskeleton, microtubule organizing center, centrosome |
| 9 | DMPK DM1PK MDPK | 2VD5 | Q09013 | DMPK_HUMAN | Human | maintains skeletal muscle structure and function | cell membrane, ER membrane, mitochondrial outer membrane |
| 10 | SLK KIAA0204 STK2 | 2J51 | Q9H2G2 | PF00069 | Human | mediates apoptosis and actin stress fiber dissolution | cytoplasm |
| 11 | ERBB4 HER4 | 2R4B | Q15303 | PF07714 | Human | regulates development of the heart, the central nervous system and the mammary gland, promote apoptosis | cell membrane, nucleus and mitochondria |
| 12 | ttgR T1E_0244 | 2UXH | Q9AIU0 | TTGR_PSEPT | Pseudomonas putida (strain DOT-T1E) | responsible for antibiotics resistance | not known |
| 13 | TNK2 ACK1 | 3EQR | Q07912 | ACK1_HUMAN | Human | involved in trafficking and clathrin-mediated endocytosis | cell membrane, nucleus, endosome |
| 14 | EGFR ERBB ERBB1 HER1 | 3UG2, 3LZB | P00533 | EGFR_HUMAN | Human | activates several signal cascades | cell membrane, ER membrane nucleus membrane, Golgi membrane, endosome |
| 15 | STK25 SOK1 YSK1 | 2XIK | O00506 | STK25_HUMAN | Human | response against environmental stress | cytoplasm, Golgi apparatus |
| 16 | LCK | 3KMM, 3KXZ, 3ACK, 3MPM, 3AC3, 3AC2, 3AD4 | P06239 | LCK_HUMAN | Human | responsible for selection and maturation of developing T-cells | cytoplasm, cell membrane |
| 17 | IGF1R | 2ZM3 | P08069 | IGF1R_HUMAN | Human | involved in cell growth and survival control | cell membrane |
| 18 | CHEK1 CHK1 | 2HOG, 2E9N, 4FTI, 4FSR, 2XF0, 2HXL, 4FTQ, 2HY0, 4FT3, 4FTN, 2BRB, 2BRH, 2XEY, 4FTO, 2QHM, 2E9U, 4FT0, 3UN9, 4FTC, 2BR1 | O14757 | CHK1_HUMAN | Human | required for cell cycle arrest and activation of DNA repair | nucleus, cytoplasm |
| 19 | PRKCI DXS1179E | 1ZRZ | P41743 | KPCI_HUMAN | Human | Plays a protective role against apoptotic stimuli | cytoplasm, membrane, nucleus, endosome |
| 20 | MAP3K9 MLK1 PRKE1 | 3DTC | P80192 | M3K9_HUMAN | Human | role in the cascades of cellular responses | intracellular |
| 21 | FGFR1 BFGFR CEK FGFBR FLG FLT2 HBGFR | 3TT0, 4F64 | P11362 | FGFR1_HUMAN | Human | essential for regulation of embryonic development, cell proliferation, differentiation and migration | cytoplasm, cell membrane, nucleus, cytoplasmic vesicles |
| 22 | MAPK14 CSBP CSBP1 CSBP2 CSPB1 MXI2 SAPK2A | 1DI9, 3FLW, 3KF7, 3ROC, 3IW7, 3FLZ, 3MVM, 2ZB1, 2RG6, 1WBW | Q16539 | MK14_HUMAN | Human | essential component of the MAP kinase signal transduction pathways responsible for activating the cascades of cellular responses | cytoplasm. nucleus |
| 23 | KIT SCFR | 3G0E | P10721 | KIT_HUMAN | Human | responsible for regulation of hematopoiesis, stem cell maintenance, gametogenesis, mast cell development, and in melanogenesis | cytoplasm, cell membrane |
| 24 | STK17B DRAK2 | 3LM0 | O94768 | ST17B_HUMAN | Human | positive regulator of apoptosis | cell membrane, nucleus |
| 25 | aurkb-a airk2-a | 2VRX | Q6DE08 | AUKBA_XENLA | Xenopus laevis | key regulator of mitosis | nucleus, chromosomes |
| 26 | ACK2 | 1ZOH | P28523 | CSK2A_MAIZE | Zea mays (Maize) | utilizes acidic proteins | not known |
| 27 | ABL1 ABL JTK7 | 3UE4, 3KF4 (Mouse) | P00519 | ABL1_HUMAN | Human | responsible for cell growth and survival | cytoplasm, nucleus, mitochondria |
| 28 | MAPK10 JNK3 JNK3A PRKM10 SAPK1B | 2O2U, 4Z9L, 3CGO, 2O0U, 3TTJ, 2ZDT | P53779 | MK10_HUMAN | Human | involved in neuronal proliferation, differentiation, migration and programmed cell death | cytoplasm. nucleus, mitochondria, membrane |
| 29 | MAPKAPK2 | 3KA0 | P49137 | MAPK2_HUMAN | Human | involved in cytokine production, endocytosis, reorganization of the cytoskeleton, chromatin remodeling and DNA damage response | cytoplasm, nucleus |
| 30 | BTK AGMX1 ATK BPK | 3PJ1, 3PJ2, 3OCT | Q06187 | BTK_HUMAN | Human | Vital for B lymphocyte development, differentiation and signaling | cytoplasm. nucleus, cell membrane |
| 31 | ACVR1 ACVRLK2 | 3MTF, 3OOM | Q04771 | ACVR1_HUMAN | Human | involved for left-right pattern formation during embryogenesis | membrane |
| 32 | SYK | 1XBC | P43405 | KSYK_HUMAN | Human | regulates innate and adaptive immunity, cell adhesion, osteoclast maturation, platelet activation and vascular development | cytoplasm, cell membrane |
| 33 | EPHB4 HTK MYK1 TYRO11 | 2VX1, 2VWU, 2×9F, 2VWX, 2VWW | P54760 | EPHB4_HUMAN | Human | important in tumor angiogenesis | cell membrane |
| 34 | CDPK1 | 3T3U, 3NYV, 3MWU, 3MA6 | Q9BJF5A3FQ16 | Q9BJF5_TOXGO A3FQ16_CRYPI | Toxoplasma gondii Cryptosporidium parvum (strain Iowa II) | binds with metals and nucleotides | not known |
| 35 | CDK6 CDKN6 | 3NUX | Q00534 | CDK6_HUMAN | Human | controls cell cycle and differentiation | cytoplasm. nucleus |
| 36 | DHODH | 3F1Q | Q02127 | PYRD_HUMAN | Human | catalyzes the conversion of dihydroorotate to orotate | mitochondrial inner membrane |
| 37 | CDK2 CDKN2 | 3EZV, 2UZD, 2IW6, 3R6X, 1DI8, 2VTQ, 2VU3, 2W17, 3R1Q, 1GIJ | P24941 | CDK2_HUMAN | Human | control of the cell cycle; essential for meiosis, but non-essential for mitosis | cytoplasm. nucleus, endosome |
| 38 | PIM1 | 3MA3, 2O65, 3R04, 3T9I, 3VBY, 3UMX, 4ENY, 3CY3, 3JY0 | P11309 | PIM1_HUMAN | Human | involved in cell survival and cell proliferation, provide advantage in tumorigenesis | cytoplasm. nucleus, cell membrane |
| 39 | STK10 LOK | 4BC6 | O94804 | STK10_HUMAN | Human | regulates lymphocyte migration | cell membrane |
| 40 | Mapk1 Erk2 Mapk Prkm1 | 2Z7L | P63086 | MK01_RAT | Rat | plays important role in the MAPK/ERK cascade | cytoplasm, nucleus |
| 41 | PRKACA PKACA | 3AMB, 1SZM | P17612 P00517 | KAPCA_HUMAN KAPCA_BOVIN | Human Bovine | involved in the regulation of platelets | cytoplasm. nucleus, cell membrane, mitochondria |
| 42 | ITK EMT LYK | 3T9T, 3V8T | Q08881 | ITK_HUMAN | Human | involved in regulation of the adaptive immune response | cytoplasm |
| 43 | MET | 3ZZE | P08581 | MET_HUMAN | Human | mediates entry of the pathogen into cells | membrane |
| 44 | CHK2 | 2YCQ, 2YIT | O96017 | CHK2_HUMAN | Human | required for checkpoint-mediated cell cycle arrest | nucleus |
| 45 | TTK | 3HMO | P33981 | TTK_HUMAN | Human | associated with cell proliferation | membrane, spindle |
| 46 | NTRK1 | 4AOJ | P04629 | NTRK1_HUMAN | Human | involved in the development and the maturation of the central and peripheral nervous systems | cell membrane, early endosome membrane, late endosome membrane |
| 47 | TTR | 1KGJ | P02767 | TTHY_RAT | Rat | transports thyroxine from the bloodstream to the brain | secreted |
| 48 | HSD17B1 | 3DHE | P14061 | DHB1_HUMAN | Human | has 20-alpha-HSD activity | cytoplasm |
| 49 | METF | 3FSU | P0AEZ1 | METF_ECOLI | E.coli | converts homocysteine to methionine | cytosol |
| 50 | BETVIA | 4A84 | P15494 | BEV1A_BETPN | Betula pendula | steroid carrier protein | cytoplasm |
As anticipated, the top ranked structure in MIF search analysis was found to be that of mutated EGFR kinase domain (G719S/T790M) in complex with gefitinib. This data indicates that the method used for binding pocket similarity analysis is appropriate and quite accurate. The other top-ranked structures such as Serine/threonine-protein kinase Chk1 (CHEK1; MIF ranked 2) in complex with 3-(Indol-2-yl) indazoles and Mitogen activated protein kinase-14 (MAPK14; MIF ranked 3) bound with an inhibitor 4-[3-methylsulfanylanilino]-6,7-dimethoxyquinazoline (PDB IDs: 2HOG and 1DI9, respectively) show high similarity to EGFR binding site and might the true off-target proteins.
The superimposed structures obtained from the detailed MIF analysis of query (EGFR) and top ranked off-targets (CHEK1 and MAPK14) are shown for five probes (color spheres; Fig. 2). The probes of query and off-target proteins are shown by bigger and smaller spheres, respectively. There are abundant hydrophobic probes surrounding ligands of both query protein and off-targets (cyan spheres in Fig. 2A,B). H-donor probes (blue sphere) of query and off-targets surround the nitrogen (N)-containing C-rings of the ligands. In case of MAPK14, H-donor residues surrounded O-31 and N-containing C-ring same as N-containing C-rings and F present in side group in gefitinib. Positive and negative probes (green and magenta spheres, respectively) surround N-containing C-ring and S-21 containing C-ring in CHEK1 and MAPK14 bound ligands respectively. This indicates that binding pockets of EGFR and the identified off-targets are very similar in nature and therefore it could be argued that gefitinib might be able to bind with these off-targets efficiently and modulate their functions.
Figure 2. Binding site structure similarity between query protein and off-targets CHEK1 and MAPK14.

The superimposed structures of query and off-target proteins in complex with respective ligands are shown (A,B; top, left panel). The similarity of binding pockets in query (green) and off-targets (cyan) are shown (A,B; top, right panel). The hydrophobic, donor, acceptor, negative and positive binding site probes are shown separately and are represented by cyan, blue, red, magenta and green colored spheres, respectively (A,B; middle and lower panel). Large spheres represent the query binding probe while smaller ones represent the off-targets binding probe.
Identification and characterization of binding pockets of off-targets
For binding pocket estimation, we considered only pocket in which the ligand of respective off-target protein was bound. We defined the pocket at the threshold distance of 4 Å based on ligand proximity that is limited to short interactions. The defined pocket with a score ≥0.5 was considered as highly druggable pocket. After estimation of pockets in 128 off-targets, 120 pockets were found to have druggable probability greater than 0.5. Only 8 pockets had values of less than 0.5 (Supplementary Table S2). Since all the identified-pockets were from the crystal structure and have at least one bound ligand, the predicated druggability value of less than 0.5 could not be ignored. The PockDrug server also calculated 66 physicochemical properties (such as hydrophobicity, polarity, aromaticity etc.) of the pockets and the selected parameters are shown in Supplementary Table S2. The binding pocket volumes ranged from 257.99 to 1766.88.
In silico confirmation of off-targets using ligand-protein docking
To further investigate the off-targets identified through MIF similarity searches, we used a reverse docking approach. The docking of gefitinib with each of 128 off-target structures was performed. The docking score was calculated for each gefitinib binding pose that ranges from −1.224 to −12.025, and was subjected to local refinement, binding energy calculations (MM-GBSA method). The 128 structures were re-ranked according to the MM-GBSA binding energy (G-rank; according to lowest binding energies; Supplementary Table S3). Notably, the mutant EGFR kinase domain bound with gefitinib (G179S/T790M; PDB id 3UG2), that was ranked first in MIF similarity search, also showed efficient docking score and binding energy (−6.818 and −77.11 kcal/mol, respectively). The ligand interacts with Met793 and Asp800 via H-bond and Glu791 residues via polar contacts (Fig. 3A). Seven human off-targets (i.e. MAPK10, PIM-1, DHODH, ERBB-4, HSD17B1, CHK2, CHK1) were found to bind with gefitinib with equal or better binding energy than EGFR (Supplementary Table S3). The binding energy for these off-target ranges from −103.446 to −94.712 kcal/mol. The residues involved in binding of gefitinib with these off-targets are shown in Fig. 3B–H.
Figure 3.

Molecular interactions of gefitinib with selected human off-targets; (A) mutant EGFR kinase domain; (B) PIM- 1, (C) MAPK10; (D) CHEK1; (E) DHODH; (F) ERBB4; (G) CHK2 and (H) HSD17B1. PDB codes are shown in brackets. The H-bond and polar interactions are represented by black line and green lines respectively.
Comparison of binding efficiency of identified off- targets with gefitinib and reported ligands
To compare and verify the data, we also performed reversed docking of identified off-targets with their previously reported ligands that were already present in the crystal structure of the off-targets. The docking score and binding energies of these bound ligands are shown in Supplementary Table S3. For determining the difference in binding efficiency of gefitinib ((∆Ggef) and the respective bound ligand (∆Glig) with the off-target structures, the ratio of binding energies (∆Ggef/∆Glig) was calculated and plotted against average binding energies (Fig. 4). The off-targets having ∆Ggef/∆Glig value ≥ 1.5 were considered to be having significant binding efficiency towards gefitinib compared to their respective reported ligand (Fig. 4, red spheres). In contrast to the bound ligand, gefitinib showed more efficient binding with 15 human and 1 non-human off-target structures (Fig. 4; Table 2). This observation is in agreement with various in vitro studies that have reported ligands present in the co-crystals of some of these off-target structures had less efficient binding (IC50) compared to other inhibitors used in respective in vitro studies (Table 2). This indicates that gefitinib might also be a potent inhibitor of the identified off-targets.
Figure 4. Comparison of the binding energies of gefitinib (∆Ggef) and the respective bound ligand (∆Glig) with the off-target structures.
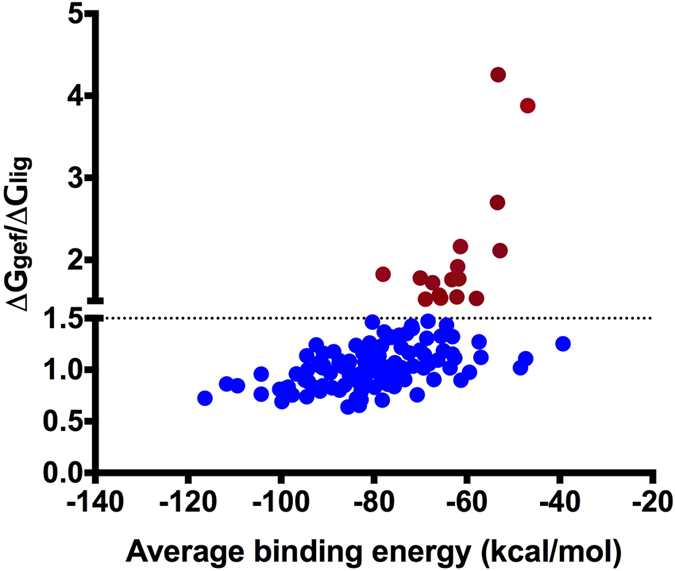
The ratio of gefitinib and reported ligand binding energies (∆Ggef/∆Glig) were plotted against the average binding energy of both. The dotted line shows the threshold ratio 1.5. The spheres with read colour show the significant ratio above the threshold value.
Table 2. The list of characterized off-targets having significant binding efficiency towards gefitinib compared to their respective reported ligands.
| S. No. | Name | PDB | Ligand present in co-crystal | gefitinib MM-GBSA binding energy | known ligands MM-GBSA binding energy | ∆Ggef/∆Glig | IC50 (nM) of known ligand |
|---|---|---|---|---|---|---|---|
| 1 | Cyclin-dependent kinase 2 | 2UZD | C85 | −86.307 | −20.274 | 4.257028707 | ND |
| 2 | Cyclin-dependent kinase 2 | 2VTQ | LZA | −74.582 | −19.222 | 3.880033295 | ND |
| 3 | Fibroblast growth factor receptor 2 | 3RI1 | 3RH | −77.977 | −28.889 | 2.699193465 | 180 |
| 4 | Ephrin type-B receptor 4 | 2×9F | UNN | −83.961 | −38.83 | 2.16227144 | 140 |
| 5 | STE20-like serine/threonine-protein kinase | 2J51 | DKI | −71.754 | −33.967 | 2.112462096 | 140 |
| 6 | Mitogen-activated protein kinase 14 | 1WBW | LI4 | −81.464 | −42.498 | 1.916890207 | 2 |
| 7 | HTH-type transcriptional regulator TtgR | 2UXH | QUE | −100.838 | −55.277 | 1.824230693 | ND |
| 8 | Serine/threonine-protein kinase pim-1 | 3MA3 | 01I | −89.67 | −50.377 | 1.779978959 | 61 |
| 9 | Bone morphogenetic protein receptor type-1B | 3MDY | LDN | −78.885 | −44.586 | 1.769277352 | 5000 (Kd) |
| 10 | Activin receptor type-1 | 3MTF | A3F | −80.671 | −45.83 | 1.760222562 | 614 (Kd) |
| 11 | Mitogen-activated protein kinase 10 | 2O2U | 738 | −85.273 | −49.501 | 1.722652068 | 400 (Ki) |
| 12 | Cyclin-dependent kinase 2 | 2IW6 | QQ2 | −80.586 | −51.35 | 1.569347614 | 44000 |
| 13 | Insulin-like growth factor 1 receptor | 2ZM3 | 575 | −75.531 | −48.801 | 1.547734678 | 22900 |
| 14 | Cyclin-dependent kinase 2 | 2W17 | I19 | −79.496 | −51.79 | 1.534968141 | 60 |
| 15 | Serine/threonine-protein kinase Chk1 | 2BRH | DFW | −70.09 | −45.808 | 1.530082082 | 9.7 |
| 16 | Tyrosine-protein kinase ITK/TSK | 3V8T | 477 | −83.127 | −54.685 | 1.520106062 | 0.3 |
Retrospective studies of identified off-targets
Various studies have been published on the comprehensive analysis of kinase inhibitors including gefitinib for their selectivity18,19. The quantitative inhibition data for gefitinib were extracted from previous studies as well as the curated databases DSigDB and ChEMBL18,19,20,21. These data were compared with the data obtained from the present study. The results from the above comparison validated the proteins ERBB4, PIM1, MAPK10, MAPK14, ALK, LCK, BTK, ABL1, SRC, STK10, TNK2, KIT, IGF1R, SLK, CHK2, MET, STK17B and SYK as true off-targets of gefitinib (Table 3). These data confirm the specificity of the in silico prediction of gefitinib off-targets. Additionally, we also found DHOH, HSD17B1, BMPR1B, NTRK1, ACVRL1, and TTK and proteins as new off-targets of gefitinib that were not included in previous reports (Table 3). Furthermore, we curated the published quantitative inhibition data to identify other off-targets. In total, 22 off-targets that were identified in previous in vitro studies however were not found among top 128 hits in our study (Supplementary Table S4). These proteins were also included for further analysis to assess the effects of gefitinib on molecular pathways and diseases.
Table 3. Binding energies of identified off-targets with gefitinib and their comparison with data previously reported in high throughput in vitro studies.
| S. No. | Name | Gene Name | mmGBSA DG binding energy (kcal/mol) | Anastassiadis et al. (% activity) | Davis et al. (Kd) | Apsel et al. (IC50) | ChEMBL (Ki) |
| 1 | Mitogen-activated protein kinase10 | MAPK10, JNK3 | −103.446 | 96.64 | 3200 | — | 794.33 |
| 2 | Serine/Threonine-protein kinase pim-1 | PIM1 | −102.399 | 86.45 | — | — | 1995.26 |
| 3 | Dihydroorotate dehydrogenase (quinone) mitochondrial | DHODH | −100.487 | — | — | — | — |
| 4 | Receptor tyrosine-protein kinase erbB-4 | ERBB4, HER4 | −100.195 | 24.15 | 410 | — | 158.49 |
| 5 | Estradiol 17-beta-dehydrogenase 1 | HSD17B1 | −97.635 | — | — | — | — |
| 6 | Serine/Threonine-protein kinase Chk2 | CHEK2, CHK2 | −94.826 | 83.95 | 800 | — | 630.96 |
| 7 | Serine/Threonine-protein kinase Chk1 | CHEK1, CHK1 | −94.712 | 95.27 | — | — | 6309.57 |
| 8 | Tyrosine-protein kinase Lck | LCK | −93.312 | 68.44 | 630 | — | 398.11 |
| 9 | Hepatocyte growth factor receptor | MET | 92.659 | — | 3500 | — | — |
| 10 | Mitogen activated protein kinase 14 | MAPK14 | −91.953 | 74.59 | ND | — | 501.19 |
| 11 | tyrosine-protein kinase BTK | BTK | −90.291 | 60.56 | — | — | 1258.93 |
| 12 | Tyrosine-protein kinase ABL1 | ABL1, JTK7 | −89.891 | 86.35 | 230 | 1200 | 630 |
| 13 | Mitogen-activated protein kinase kinase kinase 9 | MAP3K9, MLK1 | −89.843 | 109.52 | — | — | — |
| 14 | Activin receptor type-1 | ACVR1, ALK2 | −88.685 | 105.63 | — | — | 3981.07 |
| 15 | Epidermal growth factor receptor | EGFR, ERBB, HER1 | −88.354 | 2.97 | 1 | — | 0.4 |
| 16 | High affinity nerve growth factor receptor | NTRK1 | −87.784 | — | — | — | — |
| 17 | Serine/Threonine-protein kinase 25 | STK25 | −86.644 | 97.38 | — | — | — |
| 18 | proto-oncogene tyrosine protein kinase Src | SRC | −86.531 | 79 | 3800 | 1100 | 1995.26 |
| 19 | Cyclin-dependent kinase 2 | CDK2, CDKN2 | −86.307 | 100.65 | — | — | 6309.57 |
| 20 | Serine/Threonine-protein kinase 17B | STK17B, DRAK2 | −84.931 | — | 3800 | — | — |
| 21 | protein kinase C iota type | PRKCI | −84.923 | — | — | — | 5011.87 |
| 22 | Ephrin type-B receptor 4 | EPHB4, HTK, MYK1 | −83.961 | 70.89 | 2500 | 1000 | |
| 23 | Tyrosine-protein kinaseITK/TSK | ITK, EMT, LYK | −83.127 | 99.94 | — | — | — |
| 24 | Myotonin protein kinase | DMPK, MDPK | −81.558 | 90.09 | 6900 | — | — |
| 25 | Serine/Threonine-protein kinase10 | STK10, LOK | −80.265 | 38.59 | 470 | — | — |
| 26 | Aurora kinase A | AURKA, STK15, STK6 | −74.879 | 95.27 | — | — | 3162.28 |
| 27 | Bone morphogenetic protein receptor type-1B | BMPR1B | −78.885 | — | — | — | — |
| 28 | Serine/Threonine-protein kinase receptor R3 | ACVRL1, ALK1 | −78.538 | 96.86 | — | — | — |
| 29 | ALK tyrosine kinase receptor | ALK | −78.228 | 98.75 | — | — | 1258.93 |
| 30 | Fibroblast growth factor receptor 2 | FGFR2, BEK | −77.977 | 98.73 | — | — | — |
| 31 | mitogen activated protein kinase 8 | MAPK8, JNK1 | −77.826 | 93.07 | — | — | — |
| 32 | FIbroblast growth factor receptor 1 | FGFR1 | −76.2 | 96.4 | − | — | 3981.07 |
| 33 | MAP kinase-activated protein kinase 2 | MAPKAPK2 | −75.845 | 97.5 | — | — | |
| 34 | Insulin-like growth factor 1 receptor | IGF1R | −75.531 | 92.33 | — | — | 1258.93 |
| 35 | Activated CDC42 kinase1 | TNK2, ACK1 | −74.789 | 72.8 | — | — | |
| 36 | Mast/stem cell growth factor receptor Kit | KIT, SCFR | −73.003 | 96.4 | 1800 | — | — |
| 37 | STE20-like serine/threonine-protein kinase | SLK, STK2 | −71.754 | 83.71 | 920 | — | 398.11 |
| 38 | cAMP-dependent protein kinase catalytic subunit alpha | PRKACA, PKACA | −69.932 | — | — | — | 5011.87 |
| 39 | Mitogen-activated protein kinase 1 (Rat; identical to Human) | MAPK1, ERK2 | −69.262 | 103.1 | — | — | |
| 40 | Tyrosine-protein kinase SYK | SYK | −65.782 | 104.18 | — | — | 1584.89 |
| 41 | Dual specificity protein kinase TTK | TTK | −64.705 | ||||
| 42 | Cyclin-dependent kinase 6 | CDK6, CDKN6 | −63.744 | 98.69 | — | — | — |
Biological pathways analysis
Biological processes were predicted on the basis of gene ontology and the pathways were ranked according to the p-value calculated using Genomatrix software. In total, 971 pathways were found to be significantly correlated with the input off-target genes (Supplementary Table S5). During this analysis, cellular processes such as protein phosphorylation and related pathways were detected in top 50 hits. The cellular proliferation pathways such as cell growth and apoptosis were strongly associated with gefitinib off-targets. Other biological processes, such as cell differentiation, cell communication, stress response, developmental and metabolic process were also found as top hits according to the p-value. The signaling pathways such as MAPK cascade, immune-response-regulating signalling pathway, serine/threonine kinase pathways and neurotrophin TRK receptor signalling pathway were the major pathways that could be correlated with the major reported side effects (Supplementary Fig. S1)22.
Associated disease analysis
Clinical diseases prediction is crucial to explain the clinical outcome of the side effects of gefitinib. Sixty clinical diseases were predicted using Genomatrix curated database that were significantly correlated with the gefitinib off-target proteins (Supplementary Table S6). These diseases were group in following broad categories: (i) different cancer types (ii) blood disorders, (iii) bone diseases and (iv) reproductive disorders (Fig. 5). Other discrete diseases/abnormalities of pituitary, endocrine system, hypothalamic, gastrointestinal and bone-marrow were also predicted. These results suggest that gefitinib might have side effects that play a major role in above mentioned diseases.
Figure 5. Prediction of clinical diseases that might be modulated by gefitinib-induced side effects.

Discussion
In the present study, we have carried out a comprehensive analysis of gefitinib off-targets using a systems biology approach; most of the identified off-targets could be validated by retrospective analysis of previously reported studies. In addition, we could also identify a few new off-targets such as DHODH, BMPR1B, NTRK1 and HSD17B1. Together, these observations could be useful for defining the molecular basis of gefitinib-induced side effects and might help in rational improvement of the drug for better treatment.
In our analysis, the mutant EGFR kinase domain in complex with gefitinib interacts with gefitinib through the use of the same residues as the wild type EGFR. Characteristically, the wild type EGFR complexed with an imidazo[2,1-b]thiazole inhibitor (PDBID 3LZB) also showed efficient binding energy with gefitinib. However, the interacting residues were found to be different and had the lowest binding energy. This suggests that the pocket of EGFR kinase domain may adapt to different conformations for interaction with gefitinib.
Interestingly, the top ranked off-targets showed highest binding site similarity but not efficient binding energy. For example, the EGFR to gefitinib docking shows binding energy −88.354 kcal/mol and gefitinib acts as a strong EGFR inhibitor (∼97% inhibition; Ki 0.4 nM (Table 3). Another off-target ERBB4 showed the maximum affinity with gefitinib but was found to be lower ranked in MIF analyses. Notably, ERBB4 is efficiently inhibited by gefitinib (∼76% inhibition; Kd 410 nM) (Table 3). Additionally, other proteins e.g. DHODH and BMPR1B etc. (that were not reported in previous in vitro analyses) also showed efficient binding energy with gefitinib. These observations suggest that reverse docking might be a suitable approach for confirming the binding affinities. Previously, it has also been shown that the binding energy calculated on docked poses was useful for predicting the binding affinity of the ligand to the receptor23.
During our analyses, a few non-humans off-targets such as src (Gallus gallus domesticus), ttgr (Pseudomonas putida), aurkb-a (Xenopus laevis), ack2 (Zea mays), cdpk1 (Toxoplasma gondii/Cryptosporidium parvum) and erk2 (Rattus rattus) were also found to exhibit efficient binding energy with gefitinib. Amongst the non-human off-targets, ttgr (PDB id: 2UXH), a helix turn helix type transcriptional regulator and antibiotic binding repressor of Pseudomonas putida was found to exhibit highly efficient binding with gefitinib (binding energy −100.838 kcal/mol). This observation suggests that gefitinib could be used in potential combination therapy for treatment of antibiotic resistant strains of Pseudomonas putida.
Previously reported in vitro, in vivo and clinical studies have suggested that gefitinib may induce side effects like pro-apoptosis and cell-cycle inhibition possibly via interacting with off-targets24. A recent study has demonstrated that gefitinib is able to induce cardiac hypertrophy through differential expression of apoptotic and oxidative stress genes25. In contrast, a few previous studies have also demonstrated positive effects of gefitinib such as bone pain relief during bone metastasis and brain metastasis26,27,28. However, the underline molecular basis of such effects is poorly understood. In the present study, we identified that gefitinib off-targets are associated with different biological pathways that may explain the molecular mechanism of such positive and negative effects of gefitinib. The identified off-targets (ACVR1, DHODH, BTK, FGFR1, EGFR, FGFR2 and CHEK2) are linked with bone diseases. Similarly, an earlier report demonstrating drug resistance against EGFR therapy in rectal diseases and non-small cell lung cancer through dysregulation of EGFR endocytosis can be explained via identified off-targets (LYN, SRC, ABL2, ABL1, SYK, TNK2, MAPKAPK2, GAK and MAPK1) that are involved in endocytosis process.
This study has found the off-targets binding to gefitinib suggesting the molecular mechanisms of the side-effects of this drug. The biological processes, regulated by off-targets are interesting to focus in future studies. Notably, this study also suggested positive roles of gefitinib in the treatment regime. System-wide in silico approaches may facilitate the identification of side effects of preclinical and commercial drugs onto the target and off-targets. This strategy may have important applications for rational improvement of drug design and development.
Methods
Druggable proteome data set
Sc-PDB database v.2013 (http://cheminfo.u-strasbg.fr/scPDB/) was used as source of druggable proteome for off-target analysis. The data set contains 8077 structures with druggable binding sites that represent 3678 proteins and 5608 different HET ligands29.
Binding site similarity search
To compare physicochemical similarities in binding pockets of different proteins, MIF within binding sites’ volumes and pairwise MIF similarities between binding sites were calculated using the IsoMIF Finder (http://bcb.med.usherbrooke.ca/isomif)30. In this study, the crystal structure of EGFR kinase with Gefitinib (PDB id 4WKQ; 1.85 Å) was selected as query protein. The binding pocket at the distance of 3 Å around gefitinib was cropped using GetCleft tool30 and subsequently used for MIF similarities search against Sc-PDB database. The parameters used for analysis were as follows: grid spacing 1.5 Å and geometric distance threshold 3.0 Å. Structures were ranked by Tanimoto score and designated as MIF rank. Top 128 hits of gefitinib binding targets were selected for further studies. The details of 128 PDB structures with references were listed in Supplementary Table S8.
Pocket estimation and characterization
The pockets of top 128 hits were estimated based on ligand proximity within a fixed distance threshold from the bound ligand. To extract the residues localized within threshold distance; “PockDrug-Server” (http://pockdrug.rpbs.univ-paris-diderot.fr) was used. The PDB files were uploaded on the server and “prox” method was selected to estimate the pocket using threshold distance at 4 Å. The ligand information in HET code was also given during prediction31.
Protein-Ligand docking
The potential off-targets-identified from MIF similarity search were further processed for binding analysis of gefitinib and previously characterized respective ligands. The Glide 6.9 ligand-receptor docking program (Schrödinger 10.4; Schrödinger Inc, USA) was used for docking of gefitinib to each off-target structure. The ligand library of gefitinib was prepared by LigPrep tool from Schrödinger program with OPLS-2005 force field. Receptor grid was generated in the vicinity of bound ligand of each identified-off target crystal structure using Glide-Receptor Grid generation tool with default parameters. Ligand docking was performed with extra precision (XP) Glide docking module. The binding energies of docking poses were calculated using MM-GBSA method (Prime, Schrödinger Inc, USA) with default parameters.
Literature mining and retrospective studies
PubMed and Google Scholar were used to search publications and research studies relevant to gefitinib. These reports were analyzed for determining the selectivity of gefitinib. We also searched DSigDB database, a collection of small molecules including drugs based compounds and their quantitative inhibition data, for the analysis of gefitinib inhibition32.
To analyze the biological pathways corresponding to identified off-targets, Genomatrix software (Genomatrix, Munich, Germany) with Gene Ranker and GePS (Pathway system) modules were used. The identified off-targets were used as input query, network pathways were constructed and the identified pathways were ranked according to p-value.
Additional Information
How to cite this article: Verma, N. et al. Identification of gefitinib off-targets using a structure-based systems biology approach; their validation with reverse docking and retrospective data mining. Sci. Rep. 6, 33949; doi: 10.1038/srep33949 (2016).
Supplementary Material
Acknowledgments
We are thankful to Prof. A K Gupta for critical discussion during manuscript preparation. This work was supported by CURAJ grant for departmental research activity.
Footnotes
Author Contributions N.V., D.B., J.P. and P.G. conceived the idea for the work and designed the experiments. N.V., A.K.R., V.K. and K.R.C. performed the experiments. D.B., J.P. and P.G. contributed invaluable suggestions during the entire experiments. N.V., A.K.R. and P.G. contributed to the analysis and interpretation of the results. All authors contributed to the writing of the manuscript.
References
- Ferlay J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–E386 (2015). [DOI] [PubMed] [Google Scholar]
- Wells A. EGF receptor. Int. J. Biochem. Cell Biol. 31, 637–643 (1999). [DOI] [PubMed] [Google Scholar]
- De Luca A. et al. The role of the EGFR signaling in tumor microenvironment. J. Cell. Physiol. 214, 559–567 (2008). [DOI] [PubMed] [Google Scholar]
- Lynch T. J. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 350, 2129–2139 (2004). [DOI] [PubMed] [Google Scholar]
- Traxler P. et al. AEE788: a dual family epidermal growth factor receptor/ErbB2 and vascular endothelial growth factor receptor tyrosine kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res . 64, 4931–4941 (2004). [DOI] [PubMed] [Google Scholar]
- Sirotnak F. M. Studies with ZD1839 in preclinical models. Semin. Oncol. 30, 12–20 (2003). [DOI] [PubMed] [Google Scholar]
- Ciardiello F. et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin. Cancer Res . 6, 2053–2063 (2000). [PubMed] [Google Scholar]
- Ciardiello F. & Tortora G. A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin. Cancer Res . 7, 2958–2970 (2001). [PubMed] [Google Scholar]
- Tiseo M., Bartolotti M., Gelsomino F. & Bordi P. Emerging role of gefitinib in the treatment of non-small-cell lung cancer (NSCLC). Drug Des. Devel. Ther . 4, 81–98 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A. F. M. M., Korashy H. M. & Kassem M. G. in Profiles of Drug Substances, Excipients and Related Methodology Vol. 39 (ed. Brittain Harry G. ) 239–264 (Academic Press, 2014). [DOI] [PubMed] [Google Scholar]
- Cohen M. H. et al. United States Food and Drug Administration Drug Approval summary: Gefitinib (ZD1839; Iressa) tablets. Clin. Cancer Res. 10, 1212–1218 (2004). [DOI] [PubMed] [Google Scholar]
- Kennedy T. Managing the drug discovery/development interface. Drug Discov. Today 2, 436–444 (1997). [Google Scholar]
- Whitebread S., Hamon J., Bojanic D. & Urban L. Keynote review: in vitro safety pharmacology profiling: an essential tool for successful drug development. Drug Discov. Today 10, 1421–1433 (2005). [DOI] [PubMed] [Google Scholar]
- Xie L., Xie L., Kinnings S. L. & Bourne P. E. Novel Computational Approaches to Polypharmacology as a Means to Define Responses to Individual Drugs. Annu. Rev. Pharmacol. Toxicol. 52, 361–379 (2012). [DOI] [PubMed] [Google Scholar]
- Fliri A. F., Loging W. T., Thadeio P. F. & Volkmann R. A. Analysis of drug-induced effect patterns to link structure and side effects of medicines. Nat. Chem. Biol. 1, 389–397 (2005). [DOI] [PubMed] [Google Scholar]
- Chang R. L., Xie L., Xie L., Bourne P. E. & Palsson B. O. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLoS Comput. Biol. 6, e1000938 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalencas X. & Mestres J. Identification of Similar Binding Sites to Detect Distant Polypharmacology. Mol. Inform . 32, 976–990 (2013). [DOI] [PubMed] [Google Scholar]
- Anastassiadis T., Deacon S. W., Devarajan K., Ma H. & Peterson J. R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1039–1045 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I. et al. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 (2011). [DOI] [PubMed] [Google Scholar]
- Bento A. P. et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res . 42, D1083–D1090 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apsel B. et al. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 4, 691–699 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M., Letunic I., Jensen L. J. & Bork P. The SIDER database of drugs and side effects. Nucleic Acids Res . 44, D1075–D1079 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L. The Many Roles of Computation in Drug Discovery. Science 303, 1813–1818 (2004). [DOI] [PubMed] [Google Scholar]
- Blumenberg M. Differential transcriptional effects of EGFR inhibitors. PLoS ONE 9, e102466 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korashy H. M. et al. Molecular Mechanisms of Cardiotoxicity of Gefitinib in Vivo and in Vitro Rat Cardiomyocyte: Role of Apoptosis and Oxidative Stress. Toxicol. Lett (2016). [DOI] [PubMed] [Google Scholar]
- Normanno N. et al. Gefitinib inhibits the ability of human bone marrow stromal cells to induce osteoclast differentiation: implications for the pathogenesis and treatment of bone metastasis. Endocr. Relat. Cancer 12, 471–482 (2005). [DOI] [PubMed] [Google Scholar]
- Nakamichi S. et al. Successful EGFR-TKI rechallenge of leptomeningeal carcinomatosis after gefitinib-induced interstitial lung disease. Jpn. J. Clin. Oncol. 43, 422–425 (2013). [DOI] [PubMed] [Google Scholar]
- Zampa G. et al. Prolonged control of bone metastases in non-small-cell lung cancer patients treated with gefitinib. Lung Cancer 60, 452–454 (2008). [DOI] [PubMed] [Google Scholar]
- Desaphy J., Bret G., Rognan D. & Kellenberger E. sc-PDB: a 3D-database of ligandable binding sites–10 years on. Nucleic Acids Res . 43, D399–D404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier M. & Najmanovich R. Detection of Binding Site Molecular Interaction Field Similarities. J. Chem. Inf. Model. 55, 1600–1615 (2015). [DOI] [PubMed] [Google Scholar]
- Hussein H. A. et al. PockDrug-Server: a new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res . 43, W436–W442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo M. et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics 31, 3069–3071 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.