

Conspectus
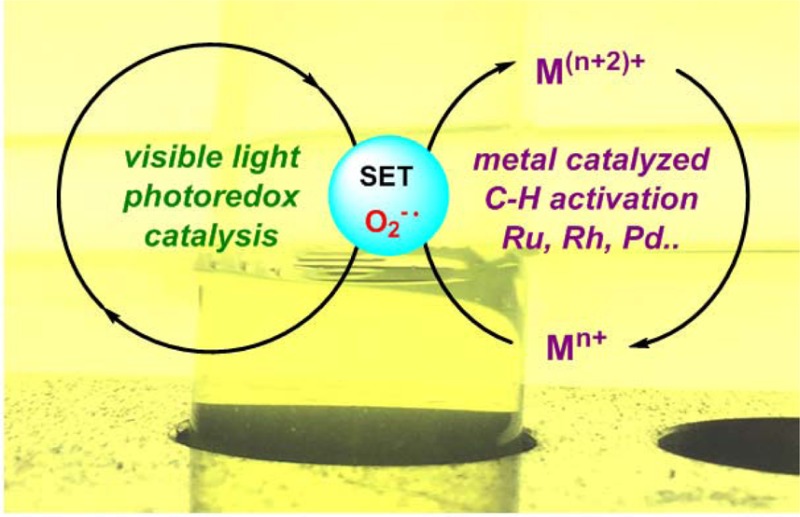
The development of efficient catalytic systems for direct aromatic C–H bond functionalization is a long-desired goal of chemists, because these protocols provide environmental friendly and waste-reducing alternatives to classical methodologies for C–C and C–heteroatom bond formation. A key challenge for these transformations is the reoxidation of the in situ generated metal hydride or low-valent metal complexes of the primary catalytic bond forming cycle. To complete the catalytic cycle and to regenerate the C–H activation catalyst, (super)stoichiometric amounts of Cu(II) or Ag(I) salts have often been applied. Recently, “greener” approaches have been developed by applying molecular oxygen in combination with Cu(II) salts, internal oxidants that are cleaved during the reaction, or solvents or additives enabling the metal hydride reoxidation. All these approaches improved the environmental friendliness but have not overcome the obstacles associated with the overall limited functional group and substrate tolerance. Hence, catalytic processes that do not feature the unfavorable aspects described above and provide products in a streamlined as well as economically and ecologically advantageous manner would be desirable.
In this context, we decided to examine visible light photoredox catalysis as a new alternative to conventionally applied regeneration/oxidation procedures. This Account summarizes our recent advances in this expanding area and will highlight the new concept of merging distinct redox catalytic processes for C–H functionalizations through the application of visible light photoredox catalysis. Photoredox catalysis can be considered as catalytic electron-donating or -accepting processes, making use of visible-light absorbing homogeneous and heterogeneous metal-based catalysts, as well as organic dye sensitizers or polymers. As a consequence, photoredox catalysis is, in principle, an ideal tool for the recycling of any given metal catalyst via a coupled electron transfer (ET) process.
Here we describe our first successful endeavors to address the above challenges by combining visible light photoredox catalysis with different ruthenium, rhodium, or palladium catalyzed C–H activations. Since only small amounts of the oxidant are generated and are immediately consumed in these transformations, side reactions of substrates or products can be avoided. Thus, usually oxidant-sensible substrates can be used, which makes these methods highly suitable for complex molecular structure syntheses. Moreover, mechanistic studies shed light on new reaction pathways, intermediates, and in situ generated species. The successful development of our dual catalysis concept, consisting of combined visible light photoredox catalysis and metal catalyzed C–H functionalization, provides many new opportunities for further explorations in the field of C–H functionalization.
1. Introduction
The C–H olefination reaction, the Fujiwara–Moritani reaction, was discovered in the late 1960s when catalytic amounts of Pd(II) salts in combination with stoichiometric amounts of Cu(II) salts promoted the addition of arenes to acrylates (Scheme 1).1,2
Scheme 1. First Report on Pd-Catalyzed Olefination Reactions by Fujiwara and Moritani.

Cu(II) salts were needed in the reaction because the resulting zerovalent Pd-metal complexes had to be reoxidized by electron transfer (ET) to complete a full catalytic cycle (Scheme 2, type I). Not only Pd-catalyzed C–H functionalization reactions require regeneration of the active species,3−6 but related catalytic reactions using rhodium7−9 or ruthenium10,11 complexes constitute further examples that require Cu(II) or Ag(I) salts for the same reason. Systems involving peroxides, peroxyesters, polyoxometallates, and benzoquinone as oxidants have also been developed and successfully applied as alternatives to Cu(II) and Ag(I) salts in olefination reactions.12,13
Scheme 2. Overview of Oxidant-Dependent C–H Olefination Reactions.

Since the pioneering work of Fujiwara and Moritani, not only have improvements in the reaction scope and directing groups been reported,14 but also greener approaches using molecular oxygen together with lower amounts of external oxidants have been described.15−17 Quite recently, internal oxidants (Scheme 2, type II) have been applied to reoxidize the metal-hydride species by internal N–O bond cleavage of the directing groups (e.g., cleavage of N–OMe bonds in Weinreb amides).18,19 Additionally, efforts have been directed toward re-evaluating the concept of directing groups, and use of substrates without such moieties have been evaluated.20 Moreover, halogenated solvents and additives proved to be effective oxidants for rhodium-catalyzed C–H functionalization reactions (Scheme 2, type III). By cleavage of the C–Hal bond and subsequent protonation of the aromatic solvent or additive, reoxidation of the catalyst could be achieved.21,22
Although many years of research have passed since its first discovery, detailed mechanistic understanding of the C–H activation process has not been fully achieved and is therefore still a topic of current research.23−32 Combining the mechanistic aspects known to date, the following general mechanism can be proposed (Scheme 3): after insertion of the catalyst MX2 (A) into the aromatic C–H bond, which often requires a directing group on the substrate, a metal–aryl complex B is formed. Coordination of an olefin and subsequent insertion into the M–aryl bond yields the metal complex C from which, after β-hydride elimination, a metal hydride or, depending on the reaction conditions, a zerovalent metal complex D is obtained. In order to recycle the metal catalyst, (super)stoichiometric amounts of Cu(II) or Ag(I) salts are necessary for the reoxidation to the active catalyst A.
Scheme 3. General Mechanism for Metal-Catalyzed C–H Functionalization Reactions.

Although photoredox catalysis has been intensively employed for electron transfer reactions,33−43 limited examples are known in which the photoredox process was used to alter the nature of a catalyst oxidation state. Recent examples include palladium-,44,45 gold-,46−51 and nickel-catalyzed52−56 reactions.
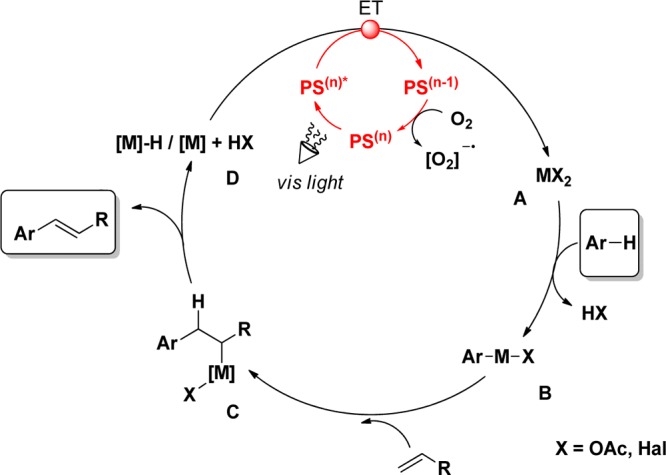
Based on our initial results in the area of photoredox-catalyzed SET processes,57,58 we became interested in developing protocols based on the combination of photoredox catalysis with metal-catalyzed C–H functionalizations. Driven by the observation that most C–H olefination reactions need stoichiometric amounts of oxidants for catalyst regeneration, we decided to examine such transformations from a photochemical point of view. The oxidants undergo a redox process with the low-valent metal complex D that is formed in situ after reductive elimination of the product (Scheme 4). The oxidant thereby transfers electrons onto this intermediary species while being reduced itself. Because this reaction essentially represents an electron transfer in the same manner in which photoredox catalysts undergo electron transfer to acceptor and donor molecules, substitution of stoichiometric amounts of external oxidants with a catalytic electron transfer reagent, such as a photoredox catalyst or photosensitizer (PS), seemed plausible.
Scheme 4. General mechanism for Metal-Catalyzed C–H Functionalization Reactions Using Combined Photoredox Catalysis.
2. Combined Metal and Photoredox Catalysis
2.1. Merging Rhodium and Photoredox Catalysis in C–H Functionalizations
The need for catalytic transformations and our endeavor to identify more elegant and effective reagents and catalysts led us to review well-established oxidant-dependent C–H olefination reactions. A rhodium-catalyzed C–H functionalization that provides ortho-olefinated Weinreb amides, which are versatile building blocks in organic synthesis, was taken as a model reaction.59 Indeed, the application of catalytic amounts of a photoredox catalyst provided very good yields in the rhodium-catalyzed olefination, showcasing the successful replacement strategy for the stoichiometric oxidant. Moreover, the results were comparable to those attained in previous reports (Scheme 5).60
Scheme 5. Rh-Catalyzed Olefination in the Presence of Photoredox Catalysis.

In the course of the reaction, the oxygen-atom of the Weinreb carbonyl serves as a directing group for the ligand-poor five-membered rhodacycle that is obtained after the first aryl C–H activation. According to the generally reported metal-catalyzed reaction mechanism (Scheme 3), a rhodium hydride D is formed that in the absence of an external oxidant represents the termination of this reaction. Superstoichiometric amounts of, for example, Cu(II) salts have been reported to oxidize the resulting hydride and enable the restart of the catalytic cycle. Given the fact that photoredox catalysts have been previously reported as being electron acceptors for a variety of different purposes, a visible-light driven catalytic process was thought to be a good alternative to commonly used Cu(II) salts. Application of 1 mol % of [Ru(bpy)3](PF6)2 was sufficient for a high yielding catalyst regeneration, and the previously used reaction temperature could be lowered from 120 to 80 °C, without loss of reactivity.
In general, it was possible not only to transform a broad range of frequently used Weinreb amides bearing many functional groups for subsequent transformations but also to extend the scope of the reaction to functionalized secondary and primary amides and to obtain products in good to very good yields (Table 1). Moreover, not only acrylates were tolerated in the olefination reaction, but additionally vinyl silanes and sulfones could be successfully applied.
Table 1. Scope of the Rh- and PR-Catalyzed Olefination Reaction.

The feasibility of secondary amides let us conclude that the amide nitrogen works as a directing group for the ortho-olefination. Furthermore, the successful use of various dialkylamides demonstrated that the N–OMe group was not acting as an internal oxidant as previously reported (Scheme 2, type II).19 In addition, the use of halogenated arenes as oxidants was ruled out since no cleavage of C–Cl bonds (Scheme 2, type III) could be detected when deuterated chlorobenzene was applied in the reaction. In order to evaluate the potential involvement of singlet oxygen species in the reaction mechanism, further control reactions were performed in deuterated solvent since it is known that the solubility and reactivity of singlet oxygen varies with such a switch in the solvent. Comparable yields obtained in the nondeuterated/deuterated chlorobenzene as solvent led to the conclusion that singlet oxygen species are not involved in the reaction pathway.61−63
However, the recycling of the photocatalyst in the presence of air is achieved via a redox process in which superoxide anions are formed. Superoxides, as well as decomposition products, including hydroxyl radicals, belong to the class of highly reactive oxygen species (ROS), which in turn could be responsible for the regeneration of the rhodium catalyst. Therefore, control experiments using 1 equiv of potassium superoxide (KO2) as a superoxide source and 1 equiv of 18-crown-6 were performed under the exclusion of air and oxygen.64 Again the product was formed in 36% yield, demonstrating that superoxide allows the reoxidation of the intermediary rhodium species to close the catalytic cycle. However, the handling of KO2, an explosive material, at elevated temperatures is rather uncomfortable. Thus, the newly developed photoredox catalysis protocol enables the in situ preparation of the reactive oxidant under mild and safe conditions.
Finally, we also conducted experiments with stoichiometric amounts of the photocatalyst under the exclusion of air and oxygen. Interestingly, in this case we obtained the desired product, indicating that a direct electron transfer from the photocatalyst allows the recycling of the active rhodium catalyst as well.
Based on these observations, the following mechanism was proposed (Scheme 6): initial C–H activation of the Weinreb amide leads to the five-membered rhodacycle A, in which the carbonyl oxygen of the Weinreb amide coordinates to the Rh(III) center to stabilize the ligand-poor metal complex.
Scheme 6. Proposed Mechanism of Rh-Catalyzed Olefination in the Presence of Photoredox Catalysis.
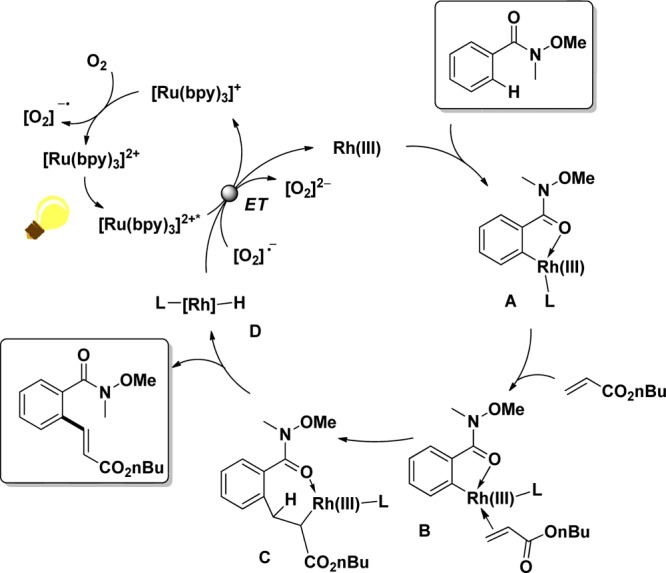
Coordination of the acrylate (complex B) and subsequent insertion into the Caryl–Rh bond forms intermediate C from which β-hydride elimination occurs to liberate the corresponding olefinated product and Rh–H species D. Instead of the Cu(II) oxidant, the photoredox-based process allows the recycling of the initial Rh(III) complex through direct electron transfer from either (i) the visible-light excited Ru(II)* or (ii) the superoxide anion or derived reactive oxygen species, which are formed in the regeneration of the Ru(II) photocatalyst.
Following our work on heterogeneous photoredox catalysis,57,65 we then applied various heterogeneous electron-transfer photosensitizers using the reaction conditions developed. Among the evaluated heterogeneous semiconductors, tungsten trioxide proved to be the best choice, delivering the desired product with a comparable yield of 84% (Scheme 7).66 Thus, by using a stoichiometric amount of a photoredox catalyst, the regeneration of the active species was established. Additionally, application of a heterogeneous photosensitizer allowed the reoxidation of the metal catalyst without formation of inorganic waste. This concept allowed the recycling of tungsten trioxide with reapplication in subsequent cycles.
Scheme 7. Application of Semiconductors for the Rh-Catalyzed Olefination Reaction.

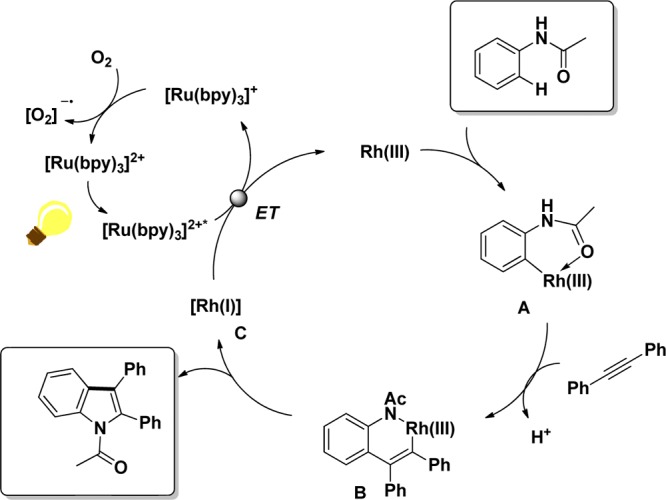
Having gained insights into the combined photoredox/rhodium catalysis, we were interested to test whether a cyclization with alkynes would also be feasible (Schemes 8 and 9). In principle, the low-valent Rh(I) species should be reoxidized in the same manner as previously shown for the Rh-hydride intermediate. To test our hypothesis, we chose the cyclization of anilides with alkynes as a proof-of-concept reaction. The transformation was previously reported by Fagnou and co-workers using a combined Rh- and Cu-based system and is of interest since the resulting indole structures play an important role in pharmaceutical chemistry.67,68 We were pleased to see that also in this case the stoichiometric amounts of copper oxidants can be replaced by catalytic amounts of a photoredox catalyst (1 mol %).69 A subsequent mechanistic investigation led us to propose that the Rh(I) species C, delivered by reductive elimination of intermediate B, is reoxidized to Rh(III) with simultaneous reduction of the excited Ru(II)* photocatalyst (Scheme 9).
Scheme 8. Rh-Catalyzed Cyclization in the Presence of Photoredox Catalysis.

Scheme 9. Combined Rhodium- and Photoredox-Catalyzed Cyclization Reaction of Acetanilide and Diphenylacetylene.
Further mechanistic studies including KIE experiments showed that the photoredox process proceeds independently from the C–H activation step (KIE = 4.5). This also confirmed our hypothesis that photoredox catalysis does not generally change the nature of C–H functionalizations and serves only as substitution for the oxidant.
The functional group tolerance is surprisingly broad, and substrates bearing halides, CF3-groups, as well as esters, could be applied effectively. Furthermore, nonsymmetric alkynes could be applied. We were also able to extend the dual catalysis methodology to pyrrole derivatives as demonstrated in Table 2.
Table 2. Scope of the Combined Rhodium- and Photoredox-Catalyzed Cyclization Reaction of Anilides and Alkynes.

2.2. Combined Palladium and Photoredox Catalysis
Having proven for the first time that photoredox catalysis is a suitable alternative for stoichiometric Cu(II) or Ag(I) additives, we went on to study more frequently used and widely applied catalytic systems. Our attention was drawn to oxidative Pd(II) chemistry, which is one of the most intensively investigated areas of C–H functionalization.3−6 As illustrated in the Introduction, these methodologies also have the disadvantage that external oxidants are required in large amounts. Hence, the newly established photoredox-based catalysis concept should allow for the successful extension to Pd(II)/Cu(II) olefination reactions (Scheme 10).
Scheme 10. Photoredox Catalysis in Palladium-Catalyzed Cyclization Reaction.

In the presence of catalytic amounts of the [Ir(ppy)2(bpy)](PF6) photocatalyst, high yields were obtained under the dual catalytic system. Again the yields were comparable to the Cu(II) based system,70,71 and control reactions revealed the crucial influence of the oxygen species that are also generated in the rhodium-photoredox system.72 Therefore, we tested KO2 in the cyclization reaction and found that it acts as an oxidant. Thus, the superoxide anion can be added in the form of potassium salt or formed in situ by photoredox catalysis.
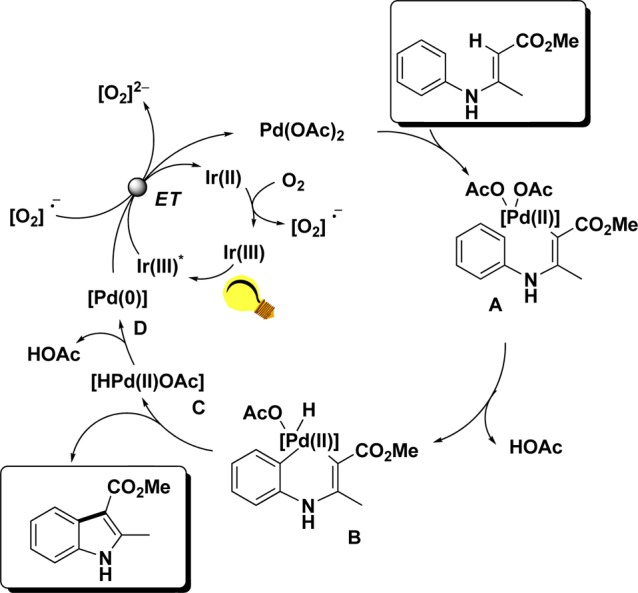
Based on these results, a more complex reaction mechanism was proposed that included two different, simultaneously operating, reoxidation modes (Scheme 11): activation of both olefinic (intermediate A) and aromatic (intermediate B) C–H bonds by a Pd(II) catalyst leads to Pd(II) hydride B, which delivers the corresponding indole product and simultaneously forms Pd(II)-hydride C. Reductive elimination of acetic acid now releases a Pd(0)-complex (D), which by means of photoredox catalysis can be reoxidized to the active Pd(II) catalyst. As illustrated in the case of Rh, reduction of molecular oxygen produces superoxide anions that allow the completion of the photoredox cycle by means of Ir(II) to Ir(III) oxidation. In agreement with the previous mechanism, superoxide anions are capable of reoxidation of the Pd(0) species D to Pd(II).
Scheme 11. Plausible Mechanism for the Combined Pd- and Photoredox-Catalyzed Olefination Reaction.
As illustrated for the rhodium photoredox catalytic system, we also aimed for the realization of a heterogeneous recycling protocol for this methodology (Scheme 12). In this case, bismuth vanadate proved to be superior over other heterogeneous semiconductors delivering the cyclized indole in very good yield (81%).66
Scheme 12. Pd-Semiconductor-Catalyzed Olefination Reaction.

In this case, catalytic amounts of bismuth vanadate could be applied. The scope of the reaction was broad and different substituents on the aromatic and ester moieties were generally well accepted (see Table 3). Interestingly, the reaction showed acceptance of photoredox-active groups, such as benzylic or chlorinated carbon atoms. Moreover, nonprotected indoles were obtained as synthetically valuable products.
Table 3. Scope of the Palladium- and Visible-Light-Photoredox-Catalyzed Cyclization Reaction.

The results demonstrate that not only in rhodium- but also in palladium-catalyzed C–H functionalizations, the reoxidation of the transition metal can be achieved by a homogeneous or heterogeneous visible light photoredox catalysis process. While potassium superoxide can act as oxidant, the yields are inferior, and decomposition products are observed. In contrast, in the photoredox process only small amounts of reactive oxygen species are formed, and under these milder reaction conditions, better yields are observed.
2.3. Merging Ruthenium and Visible Light Photoredox Catalysis in ortho-C–H Olefinations
Finally, our attention focused on another commonly applied catalytic system for C–H functionalization. Based on Ackermann’s work73 with pyridines as directing groups in the ortho-olefination of phenyl ethers, we started our investigations for a photoredox-based reoxidation protocol. Similar to the previous cases, comparable yields were obtained when catalytic amounts (3 mol %) of [Ir(ppy)2(bpy)]PF6 were applied as the photoredox catalyst to the reaction (Scheme 13).74
Scheme 13. Combined Ruthenium- and Photoredox-Catalyzed Olefination Reaction of Phenol Ethers.

In general, similar trends were observed. Mechanistically, a Ru(II)-dimer is activated by addition of a silver salt, delivering the corresponding monomer A that can undergo C–H activation by chelating to the pyridyl directing group (B) (Scheme 14). Subsequent insertion of an acrylic ester leads to the formation of species C from which β-hydride elimination can occur. In the absence of acid, the Ru(II)-hydride D obtained forms a Ru(0) intermediate E that needs an external oxidant in order to re-enter the catalytic cycle. As demonstrated in the Pd catalysis, the photoexcited Ir(III)* catalyst, as well as the superoxide radicals formed in situ, allows the reoxidation to Ru(II) A.
Scheme 14. Proposed Mechanism for the Ruthenium- and Photoredox-Catalyzed Olefination Reaction.
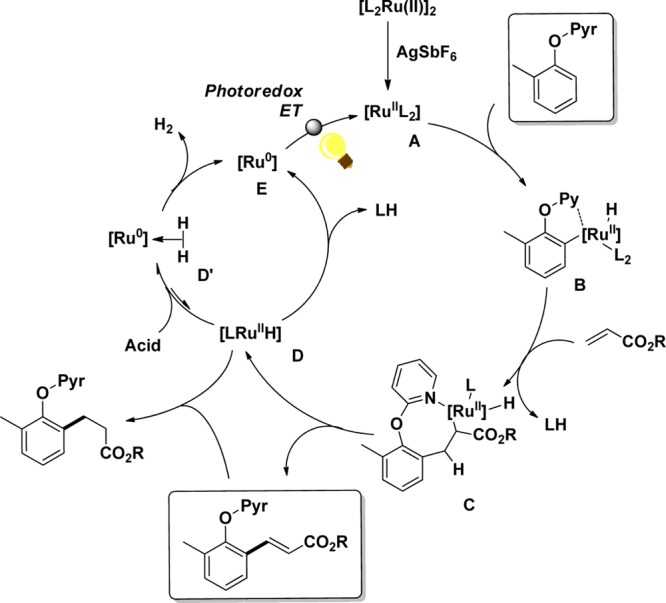
However, we observed the formation of not only the desired olefinated product but also the corresponding hydrogenated aliphatic ester. After careful studies, we found that acidic additives with carefully chosen pKa values allow the selective formation of the olefinated product. In line with previous reports,75−77 we conclude that the well-described Ru(0)-hydrogen species (D′) can be formed when Ru(II)-hydride complexes (intermediate D) are treated with moderately acidic additives, such as acetic acid. Because acids preferably shift this equilibrium toward the Ru-hydrogen complex (D′), the reported hydrogenation of unsaturated esters by Ru(II)-hydride complexes is efficiently suppressed.78 The Ru-hydrogen complexes (D′) are then transformed into the corresponding Ru(0)-complex E by releasing molecular hydrogen.79 Subsequently the photoredox-catalyzed regeneration can take place. This hypothesis was confirmed by varying the ruthenium and photoredox catalyst ratio. When more photoredox catalyst was applied in the reaction, increasing the depletion of Ru–H D, lower amounts of hydrogenated product were observed. In contrast, when more ruthenium catalyst was provided for the C–H activation, the amount of Ru-hydride D increased and more of the hydrogenated product was obtained.
In order to get insights into the kinetics of the described transformation, each reoxidation process, namely, the electron transfer from the photoredox catalyst and the superoxide anion, as well as the initial catalytic process, were analyzed individually (Figure 1). The following plot illustrates that the processes with either KO2 (2 equiv) or photoredox catalyst (200 mol %) under an argon atmosphere proceeded much slower compared with the standard reaction conditions (3 mol % photoredox catalyst under air) and led to lower yields after 24 h, indicating that under catalytic conditions a dramatic increase in reactivity is achieved.
Figure 1.

Kinetic plot for oxidant dependent, combined ruthenium- and photoredox-catalyzed olefination reaction of phenol ethers: (■) 3 mol % [Ir(ppy)2(bpy)]PF6 was applied as oxidant under aerobic conditions; (▲) 2 equiv of KO2 was applied as oxidant under inert conditions; (○) 200 mol % [Ir(ppy)2(bpy)]PF6 was applied as oxidant under inert conditions.
As described above, the same transformation was also achieved when the homogeneous photoredox catalyst was replaced with bismuth vanadate as heterogeneous semiconductor photosensitizer without loss of selectivity and reactivity (Scheme 15).66
Scheme 15. Heterogeneous-Catalyzed Olefination Reaction of Phenol Ethers Using a Semiconducting Photosensitizer.

Application of the developed dual catalysis methodology demonstrated a good variety of substitution patterns and generally good to very good yields were obtained (Table 4). Again, addition of acetic acid improved the selectivity in favor of the olefinated products by shifting the equilibrium toward the Ru-hydrogen complex.
Table 4. Scope of the Combined Ruthenium- and Photoredox-Catalyzed Cyclization Reaction.

3. Conclusion and Perspective
During the past few years we, and others, were able to demonstrate that visible light photoredox catalysis can be a valuable tool for single electron transfer reactions, as well as for the generation of reactive oxygen species including superoxide anions. The latter observations led us to question whether a photoredox-catalyzed process could be applied to substitute (super)stoichiometric amounts of external oxidants in transition metal catalyzed C–H functionalizations. As shown in this Account, our initial work focused on the three most representative metals (Rh, Pd, Ru) for C–H olefination reactions. These transformations were previously achieved if large amounts of Cu(II) salts were applied. However, we successfully demonstrated that either visible light homogeneous photoredox catalysts or heterogeneous semiconductor-based catalysts are perfectly suitable substitutes. Mechanistic studies revealed that the photoredox process is independent from the C–H activation reaction. Moreover, we were able to show that the photoredox catalysts generate and carefully release superoxide radicals that can independently work as oxidants as demonstrated by the use of potassium superoxide. While more detailed mechanistic studies need to be undertaken, the successful development of our dual catalysis concept, consisting of combined visible light photoredox catalysis and metal catalyzed C–H functionalization, provides many new opportunities for further explorations.
Acknowledgments
The research leading to the results reported has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement No. 617044 (SunCatChem).
Biographies
David C. Fabry was born in Neuss (Germany) and studied chemistry at the RWTH-Aachen University. He concluded his master thesis under the supervision of Prof. Michael Willis (Oxford University, U.K.). Currently he is performing his doctoral studies under the supervision of Prof. Magnus Rueping (RWTH-Aachen University, Germany). His research interests focus on the development of continuous flow systems and novel combinations of metal-catalyzed reactions with photoredox catalysis.
Magnus Rueping studied at the Technical University of Berlin, Trinity College Dublin, and ETH Zürich. In 2002, he obtained his Ph.D. from the ETH and subsequently conducted postdoctoral studies at Harvard University. In August 2004, he was directly appointed to a C3-associate professorship, the Degussa Endowed Professorship of Synthetic Organic Chemistry at Goethe University Frankfurt. After four years in Frankfurt, he accepted a Chair and Full Professorship of Organic Chemistry at RWTH Aachen University and was recently appointed Professor of Chemical Science at KAUST.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Photoredox Catalysis in Organic Chemistry”.
References
- Moritani I.; Fujiwara Y. Aromatic Substitution of Styrene-Palladium Chloride Complex. Tetrahedron Lett. 1967, 8, 1119–1122. 10.1016/S0040-4039(00)90648-8. [DOI] [Google Scholar]
- Fujiwara Y.; Moritani I.; Danno S.; Asano R.; Teranishi S. Aromatic Substitution of Olefins. VI. Arylation of Olefins with Palladium(II) Acetate. J. Am. Chem. Soc. 1969, 91, 7166–7169. 10.1021/ja01053a047. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Wang J.; Mao F.; Kwong F. Y. Palladium-Catalyzed Cross-Dehydrogenative Functionalization of C(Sp2)-H Bonds. Chem. - Asian J. 2014, 9, 26–47. 10.1002/asia.201300990. [DOI] [PubMed] [Google Scholar]
- Yoshikai N.; Wei Y. Synthesis of Pyrroles, Indoles, and Carbazoles Through Transition-Metal-Catalyzed C-H Functionalization. Asian J. Org. Chem. 2013, 2, 466–478. 10.1002/ajoc.201300016. [DOI] [Google Scholar]
- Broggini G.; Beccalli E. M.; Fasana A.; Gazzola S. Palladium-Catalyzed Dual C-H or N-H Functionalization of Unfunctionalized Indole Derivatives with Alkenes and Arenes. Beilstein J. Org. Chem. 2012, 8, 1730–1746. 10.3762/bjoc.8.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald R. I.; Liu G.; Stahl S. S. Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications. Chem. Rev. 2011, 111, 2981–3019. 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G.; Wang F.; Li X. C-C, C-O and C-N Bond Formation via Rhodium(III)-Catalyzed Oxidative C-H Activation. Chem. Soc. Rev. 2012, 41, 3651–3678. 10.1039/c2cs15281a. [DOI] [PubMed] [Google Scholar]
- Colby D. A.; Tsai A. S.; Bergman R. G.; Ellman J. A. Rhodium Catalyzed Chelation-Assisted C-H Bond Functionalization Reactions. Acc. Chem. Res. 2012, 45, 814–825. 10.1021/ar200190g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wencel-Delord J.; Dröge T.; Liu F.; Glorius F. Towards Mild Metal-Catalyzed C-H Bond Activation. Chem. Soc. Rev. 2011, 40, 4740–4761. 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]
- De Sarkar S.; Liu W.; Kozhushkov S. I.; Ackermann L. Weakly Coordinating Directing Groups for Ruthenium(II)-Catalyzed C-H Activation. Adv. Synth. Catal. 2014, 356, 1461–1479. 10.1002/adsc.201400110. [DOI] [Google Scholar]
- Ackermann L. Carboxylate-Assisted Ruthenium-Catalyzed Alkyne Annulations by C-H/Het-H Bond Functionalizations. Acc. Chem. Res. 2014, 47, 281–295. 10.1021/ar3002798. [DOI] [PubMed] [Google Scholar]
- Chen X.; Engle K. M.; Wang D.-H.; Yu J.-Q. Palladium(II)-Catalyzed C-H Activation/C-C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem., Int. Ed. 2009, 48, 5094–5115. 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bras J.; Muzart J. Intermolecular Dehydrogenative Heck Reactions. Chem. Rev. 2011, 111, 1170–1214. 10.1021/cr100209d. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Lu W. Towards Ideal Synthesis: Alkenylation of Aryl C-H Bonds by a Fujiwara–Moritani Reaction. Chem. - Eur. J. 2014, 20, 634–642. 10.1002/chem.201303670. [DOI] [PubMed] [Google Scholar]
- Konnick M. M.; Stahl S. S. Reaction of Molecular Oxygen with a Pd II-Hydride to Produce a Pd II-Hydroperoxide: Experimental Evidence for an HX-Reductive-Elimination Pathway. J. Am. Chem. Soc. 2008, 130, 5753–5762. 10.1021/ja7112504. [DOI] [PubMed] [Google Scholar]
- Popp B. V.; Stahl S. S. Insertion of Molecular Oxygen Into a Palladium-Hydride Bond: Computational Evidence for Two Nearly Isoenergetic Pathways. J. Am. Chem. Soc. 2007, 129, 4410–4422. 10.1021/ja069037v. [DOI] [PubMed] [Google Scholar]
- Stahl S. S. Palladium Oxidase Catalysis: Selective Oxidation of Organic Chemicals by Direct Dioxygen-Coupled Turnover. Angew. Chem., Int. Ed. 2004, 43, 3400–3420. 10.1002/anie.200300630. [DOI] [PubMed] [Google Scholar]
- Huang H.; Ji X.; Wu W.; Jiang H. Transition Metal-Catalyzed C-H Functionalization of N-Oxyenamine Internal Oxidants. Chem. Soc. Rev. 2015, 44, 1155–1171. 10.1039/C4CS00288A. [DOI] [PubMed] [Google Scholar]
- Rakshit S.; Grohmann C.; Besset T.; Glorius F. Rh(III)-Catalyzed Directed C-H Olefination Using an Oxidizing Directing Group: Mild, Efficient, and Versatile. J. Am. Chem. Soc. 2011, 133, 2350–2353. 10.1021/ja109676d. [DOI] [PubMed] [Google Scholar]
- Kuhl N.; Hopkinson M. N.; Wencel-Delord J.; Glorius F. Beyond Directing Groups: Transition-Metal-Catalyzed C-H Activation of Simple Arenes. Angew. Chem., Int. Ed. 2012, 51, 10236–10254. 10.1002/anie.201203269. [DOI] [PubMed] [Google Scholar]
- Wencel-Delord J.; Nimphius C.; Wang H.; Glorius F. Rhodium(III) and Hexabromobenzene - a Catalyst System for the Cross-Dehydrogenative Coupling of Simple Arenes and Heterocycles with Arenes Bearing Directing Groups. Angew. Chem., Int. Ed. 2012, 51, 13001–13005. 10.1002/anie.201205734. [DOI] [PubMed] [Google Scholar]
- Barrett A. G. M.; Itoh T.; Wallace E. M. η6-Benzene)(η5-ethyltetramethyl-cyclopentadienyl)rhodium (III) hexafluorophosphate: A reagent for catalytic phenol oxidative coupling. Tetrahedron Lett. 1993, 34, 2233–2234. 10.1016/S0040-4039(00)77581-2. [DOI] [Google Scholar]
- Zhang X.-S.; Chen K.; Shi Z.-J. Transition Metal-Catalyzed Direct Nucleophilic Addition of C-H Bonds to Carbon-Heteroatom Double Bonds. Chem. Sci. 2014, 5, 2146–2159. 10.1039/c3sc53115e. [DOI] [Google Scholar]
- Suess A. M.; Ertem M. Z.; Cramer C. J.; Stahl S. S. Divergence Between Organometallic and Single-Electron-Transfer Mechanisms in Copper(II)-Mediated Aerobic C-H Oxidation. J. Am. Chem. Soc. 2013, 135, 9797–9804. 10.1021/ja4026424. [DOI] [PubMed] [Google Scholar]
- Ackermann L.; Kozhushkov S. I.; Yufit D. S. Ruthenium-Catalyzed Hydroarylation of Methylenecyclopropanes Through C-H Bond Cleavage: Scope and Mechanism. Chem. - Eur. J. 2012, 18, 12068–12077. 10.1002/chem.201200406. [DOI] [PubMed] [Google Scholar]
- Boisvert L.; Goldberg K. I. Reactions of Late Transition Metal Complexes with Molecular Oxygen. Acc. Chem. Res. 2012, 45, 899–910. 10.1021/ar2003072. [DOI] [PubMed] [Google Scholar]
- Campbell A. N.; Stahl S. S. Overcoming the “Oxidant Problem”: Strategies to Use O2 as the Oxidant in Organometallic C-H Oxidation Reactions Catalyzed by Pd (and Cu). Acc. Chem. Res. 2012, 45, 851–863. 10.1021/ar2002045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendlandt A. E.; Suess A. M.; Stahl S. S. Copper-Catalyzed Aerobic Oxidative C-H Functionalizations: Trends and Mechanistic Insights. Angew. Chem., Int. Ed. 2011, 50, 11062–11087. 10.1002/anie.201103945. [DOI] [PubMed] [Google Scholar]
- Powers D. C.; Ritter T. Bimetallic Redox Synergy in Oxidative Palladium Catalysis. Acc. Chem. Res. 2012, 45, 840–850. 10.1021/ar2001974. [DOI] [PubMed] [Google Scholar]
- Lin Z. Interplay Between Theory and Experiment: Computational Organometallic and Transition Metal Chemistry. Acc. Chem. Res. 2010, 43, 602–611. 10.1021/ar9002027. [DOI] [PubMed] [Google Scholar]
- Ke Z.; Cundari T. R. Palladium-Catalyzed C-H Activation/C-N Bond Formation Reactions: DFT Study of Reaction Mechanisms and Reactive Intermediates. Organometallics 2010, 29, 821–834. 10.1021/om900895t. [DOI] [Google Scholar]
- Boutadla Y.; Davies D. L.; Macgregor S. A.; Poblador-Bahamonde A. I. Mechanisms of C-H Bond Activation: Rich Synergy Between Computation and Experiment. Dalton Trans. 2009, 5820–5831. 10.1039/b904967c. [DOI] [PubMed] [Google Scholar]
- Garlets Z. J.; Nguyen J. D.; Stephenson C. R. J. The Development of Visible-Light Photoredox Catalysis in Flow. Isr. J. Chem. 2014, 54, 351–360. 10.1002/ijch.201300136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reckenthäler M.; Griesbeck A. G. Photoredox Catalysis for Organic Syntheses. Adv. Synth. Catal. 2013, 355, 2727–2744. 10.1002/adsc.201300751. [DOI] [Google Scholar]
- Xuan J.; Lu L. Q.; Chen J. R.; Xiao W. J. Visible-Light-Driven Photoredox Catalysis in the Construction of Carbocyclic and Heterocyclic Ring Systems. Eur. J. Org. Chem. 2013, 2013, 6755–6770. 10.1002/ejoc.201300596. [DOI] [Google Scholar]
- Hu J.; Wang J.; Nguyen T. H.; Zheng N. The Chemistry of Amine Radical Cations Produced by Visible Light Photoredox Catalysis. Beilstein J. Org. Chem. 2013, 9, 1977–2001. 10.3762/bjoc.9.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L.; Xia W. Photoredox Functionalization of C-H Bonds Adjacent to a Nitrogen Atom. Chem. Soc. Rev. 2012, 41, 7687–7697. 10.1039/c2cs35203f. [DOI] [PubMed] [Google Scholar]
- Xuan J.; Xiao W. J. Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. 10.1002/anie.201200223. [DOI] [PubMed] [Google Scholar]
- Hoffmann N. Homogeneous Photocatalytic Reactions with Organometallic and Coordination Compounds - Perspectives for Sustainable Chemistry. ChemSusChem 2012, 5, 352–371. 10.1002/cssc.201100286. [DOI] [PubMed] [Google Scholar]
- Narayanam J. M. R.; Stephenson C. R. J. Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev. 2011, 40, 102–113. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]
- Tucker J. W.; Stephenson C. R. J. Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. J. Org. Chem. 2012, 77, 1617–1622. 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]
- Levin M. D.; Kim S.; Toste F. D. Photoredox Catalysis Unlocks Single-Electron Elementary Steps in Transition Metal Catalyzed Cross-Coupling. ACS Cent. Sci. 2016, 2, 293–301. 10.1021/acscentsci.6b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeldt S. R.; Sanford M. S. Combining Transition Metal Catalysis with Radical Chemistry: Dramatic Acceleration of Palladium - Catalyzed C-H Arylation with Diaryliodonium Salts. Adv. Synth. Catal. 2012, 354, 3517–3522. 10.1002/adsc.201200738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyani D.; McMurtrey K. B.; Neufeldt S. R.; Sanford M. S. Room-Temperature C-H Arylation: Merger of Pd-Catalyzed C-H Functionalization and Visible-Light Photocatalysis. J. Am. Chem. Soc. 2011, 133, 18566–18569. 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkinson M. N.; Sahoo B.; Glorius F. Dual Photoredox and Gold Catalysis: Intermolecular Multicomponent Oxyarylation of Alkenes. Adv. Synth. Catal. 2014, 356, 2794–2800. 10.1002/adsc.201400580. [DOI] [Google Scholar]
- Sahoo B.; Hopkinson M. N.; Glorius F. Combining Gold and Photoredox Catalysis: Visible Light-Mediated Oxy- and Aminoarylation of Alkenes. J. Am. Chem. Soc. 2013, 135, 5505–5508. 10.1021/ja400311h. [DOI] [PubMed] [Google Scholar]
- Hopkinson M. N.; Sahoo B.; Li J. L.; Glorius F. Dual Catalysis Sees the Light: Combining Photoredox with Organo-, Acid, and Transition-Metal Catalysis. Chem. - Eur. J. 2014, 20, 3874–3886. 10.1002/chem.201304823. [DOI] [PubMed] [Google Scholar]
- Shu X.-Z.; Zhang M.; He Y.; Frei H.; Toste F. D. Dual Visible Light Photoredox and Gold-Catalyzed Arylative Ring Expansion. J. Am. Chem. Soc. 2014, 136, 5844–5847. 10.1021/ja500716j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlahuext-Aca A.; Hopkinson M. N.; Garza-Sanchez R. A.; Glorius F. Alkyne Difunctionalization by Dual Gold/Photoredox Catalysis. Chem. - Eur. J. 2016, 22, 5909–5913. 10.1002/chem.201600710. [DOI] [PubMed] [Google Scholar]
- Tlahuext-Aca A.; Hopkinson M. N.; Sahoo B.; Glorius F. Dual Gold/Photoredox-Catalyzed C(sp)–H Arylation of Terminal Alkynes with Diazonium Salts. Chem. Sci. 2016, 7, 89–93. 10.1039/C5SC02583D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellis J. C.; Primer D. N.; Molander G. A. Dual Catalysis. Single-Electron Transmetalation in Organoboron Cross-Coupling by Photoredox/Nickel Dual Catalysis. Science 2014, 345, 433–436. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Merging Photoredox with Nickel Catalysis: Coupling of Carboxyl Sp3-Carbons with Aryl Halides. Science 2014, 345, 437–440. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble A.; McCarver S. J.; MacMillan D. W. C. Merging Photoredox and Nickel Catalysis: Decarboxylative Cross-Coupling of Carboxylic Acids with Vinyl Halides. J. Am. Chem. Soc. 2015, 137, 624–627. 10.1021/ja511913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu L.; Lipshultz J. M.; MacMillan D. W. C. Merging Photoredox and Nickel Catalysis: The Direct Synthesis of Ketones by the Decarboxylative Arylation of α-Oxo Acids. Angew. Chem., Int. Ed. 2015, 54, 7929–7933. 10.1002/anie.201501908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy J. J.; Melchiorre P. Light opens pathways for nickel catalysis. Nature 2015, 524, 297–298. 10.1038/nature15200. [DOI] [PubMed] [Google Scholar]
- Rueping M.; Koenigs R. M.; Poscharny K.; Fabry D. C.; Leonori D.; Vila C. Dual Catalysis: Combination of Photocatalytic Aerobic Oxidation and Metal Catalyzed Alkynylation Reactions – C-C Bond Formation Using Visible Light. Chem. - Eur. J. 2012, 18, 5170–5174. 10.1002/chem.201200050. [DOI] [PubMed] [Google Scholar]
- Rueping M.; Zoller J.; Fabry D. C.; Poscharny K.; Koenigs R. M.; Weirich T. E.; Mayer J. Light-Mediated Heterogeneous Cross Dehydrogenative Coupling Reactions: Metal Oxides as Efficient, Recyclable, Photoredox Catalysts in C-C Bond-Forming Reactions. Chem. - Eur. J. 2012, 18, 3478–3481. 10.1002/chem.201103242. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Li C.; Li Y.; Yin F.; Wang X. S. Rhodium-Catalyzed C-H Olefination of Aryl Weinreb Amides. Adv. Synth. Catal. 2013, 355, 1724–1728. 10.1002/adsc.201300174. [DOI] [Google Scholar]
- Fabry D. C.; Zoller J.; Raja S.; Rueping M. Combining Rhodium and Photoredox Catalysis for C-H Functionalizations of Arenes: Oxidative Heck Reactions with Visible Light. Angew. Chem., Int. Ed. 2014, 53, 10228–10231. 10.1002/anie.201400560. [DOI] [PubMed] [Google Scholar]
- Ogilby P. R.; Foote C. S. Chemistry of Singlet Oxygen. 42. Effect of Solvent, Solvent Isotopic Substitution, and Temperature on the Lifetime of Singlet Molecular Oxygen. J. Am. Chem. Soc. 1983, 105, 3423–3430. 10.1021/ja00349a007. [DOI] [Google Scholar]
- Ogilby P. R.; Foote C. S. Chemistry of Singlet Oxygen. 34. Unexpected Solvent Deuterium Isotope Effects on the Lifetime of Singlet Molecular Oxygen (1Δg). J. Am. Chem. Soc. 1981, 103, 1219–1221. 10.1021/ja00395a041. [DOI] [Google Scholar]
- Merkel P. B.; Nilsson R.; Kearns D. R. Deuterium Effects on Singlet Oxygen Lifetimes in Solutions. New Test of Singlet Oxygen Reactions. J. Am. Chem. Soc. 1972, 94, 1030–1031. 10.1021/ja00758a072. [DOI] [Google Scholar]
- Fabry D. Ph.D. Dissertation, 2016, RWTH Aachen, Verlagsgruppe Mainz GmbH. [Google Scholar]
- Fabry D. C.; Ronge M. A.; Rueping M. Immobilization and Continuous Recycling of Photoredox Catalysts in Ionic Liquids for Applications in Batch Reactions and Flow Systems: Catalytic Alkene Isomerization by Using Visible Light. Chem. - Eur. J. 2015, 21, 5350–5354. 10.1002/chem.201406653. [DOI] [PubMed] [Google Scholar]
- Fabry D. C.; Zoller J.; Rueping M.. C-H Functionalization Reactions with Visible Light and Heterogeneous Photocatalysts as Catalytic Oxidants. Manuscript in preparation.
- Stuart D. R.; Bertrand-Laperle M.; Burgess K. M. N.; Fagnou K. Indole Synthesis via Rhodium Catalyzed Oxidative Coupling of Acetanilides and Internal Alkynes. J. Am. Chem. Soc. 2008, 130, 16474–16475. 10.1021/ja806955s. [DOI] [PubMed] [Google Scholar]
- Stuart D. R.; Alsabeh P.; Kuhn M.; Fagnou K. Rhodium(III)-Catalyzed Arene and Alkene C-H Bond Functionalization Leading to Indoles and Pyrroles. J. Am. Chem. Soc. 2010, 132, 18326–18339. 10.1021/ja1082624. [DOI] [PubMed] [Google Scholar]
- Kim H. J.; Fabry D. C.; Mader S.; Rueping M.. Assistance of Photoredox Catalysis in C-H Activation for the Synthesis of Nitrogen Containing Heterocycles. Manuscript in preparation.
- Neumann J. J.; Rakshit S.; Dröge T.; Würtz S.; Glorius F. Exploring the Oxidative Cyclization of Substituted N-Aryl Enamines: Pd-Catalyzed Formation of Indoles From Anilines. Chem. - Eur. J. 2011, 17, 7298–7303. 10.1002/chem.201100631. [DOI] [PubMed] [Google Scholar]
- Würtz S.; Rakshit S.; Neumann J. J.; Dröge T.; Glorius F. Palladium-Catalyzed Oxidative Cyclization of N-Aryl Enamines: From Anilines to Indoles. Angew. Chem., Int. Ed. 2008, 47, 7230–7233. 10.1002/anie.200802482. [DOI] [PubMed] [Google Scholar]
- Zoller J.; Fabry D. C.; Ronge M. A.; Rueping M. Synthesis of Indoles Using Visible Light: Photoredox Catalysis for Palladium-Catalyzed C-H Activation. Angew. Chem., Int. Ed. 2014, 53, 13264–13268. 10.1002/anie.201405478. [DOI] [PubMed] [Google Scholar]
- Ma W.; Ackermann L. Ruthenium(II)-Catalyzed C-H Alkenylations of Phenols with Removable Directing Groups. Chem. - Eur. J. 2013, 19, 13925–13928. 10.1002/chem.201301988. [DOI] [PubMed] [Google Scholar]
- Fabry D. C.; Ronge M. A.; Zoller J.; Rueping M. C-H Functionalization of Phenols Using Combined Ruthenium and Photoredox Catalysis: in Situ Generation of the Oxidant. Angew. Chem., Int. Ed. 2015, 54, 2801–2805. 10.1002/anie.201408891. [DOI] [PubMed] [Google Scholar]
- Silantyev G. A.; Filippov O. A.; Tolstoy P. M.; Belkova N. V.; Epstein L. M.; Weisz K.; Shubina E. S. Hydrogen Bonding and Proton Transfer to Ruthenium Hydride Complex CpRuH(Dppe): Metal and Hydride Dichotomy. Inorg. Chem. 2013, 52, 1787–1797. 10.1021/ic301585k. [DOI] [PubMed] [Google Scholar]
- Creutz C.; Chou M. H.; Hou H.; Muckerman J. T. Hydride Ion Transfer From Ruthenium(II) Complexes in Water: Kinetics and Mechanism. Inorg. Chem. 2010, 49, 9809–9822. 10.1021/ic101124q. [DOI] [PubMed] [Google Scholar]
- Belkova N. V.; Dub P. A.; Baya M.; Houghton J. Kinetics and Thermodynamics of Proton Transfer to Cp*Ru(Dppe)H: via Dihydrogen Bonding and (H2-H2)-Complex to the Dihydride. Inorg. Chim. Acta 2007, 360, 149–162. 10.1016/j.ica.2006.07.106. [DOI] [Google Scholar]
- Wiles J. A.; Bergens S. H. The First Structure Determination of a Diastereomeric Hydrido-Olefin Putative Intermediate in Catalytic Enantioselective Hydrogenation. Organometallics 1999, 18, 3709–3714. 10.1021/om990482r. [DOI] [Google Scholar]
- Ryan O. B.; Tilset M.; Parker V. D. Oxidation of the Ruthenium Hydride Complex (.Eta.5-C5H5)Ru(CO) (PPh3)H: Generation of a Dihydrogen Complex by Oxidatively Induced Intermolecular Proton Transfer. Organometallics 1991, 10, 298–304. 10.1021/om00047a064. [DOI] [Google Scholar]