

Abstract
Iron-catalyzed cross-couplings with simple ferric salts have been known since the 1970s, pioneered by Kochi for cross-coupling using alkylmagnesium nucleophiles including MeMgBr. While Kochi observed the formation of a S = 1/2 iron species in reactions of simple ferric salts with MeMgBr proposed to be an iron(I) species, the identity of this species has remained undefined for nearly 40 years. Herein, we report the isolation and characterization of [MgCl(THF)5][Fe8Me12], which combined with EPR and MCD studies is shown to be consistent with Kochi’s S = 1/2 species. Reaction studies with β-bromostyrene demonstrate that this species alone displays minimal reactivity but, when combined with additional MeMgBr, leads to rapid and selective formation of cross-coupled product.
In the 1970s, Kochi demonstrated that simple ferric salts are effective in the cross-coupling of alkylmagnesium nucleophiles and alkenyl halides.1–6 That work inspired significant research in the following decades on the development of iron-catalyzed cross-coupling methods, demonstrating the ability of iron to effectively promote a variety of cross-couplings, including Kumada, Negishi, and Suzuki–Miyaura reactions.7–12 Despite the advances in iron-based cross-coupling methodologies, the mechanism of these reactions, including the identification of the active iron species responsible for catalysis, has largely remained undefined.
While recent studies have begun to define the iron active species and mechanisms in iron-bisphosphine catalyzed cross-couplings,9,13,14 the nature of the iron active species and mechanisms of catalysis in cross-couplings with simple iron salts have remained poorly understood. Early mechanistic studies by Kochi and co-workers identified the formation of an S = 1/2 iron species in reaction of simple ferric salts and MeMgBr,5,6 which led to a proposed reaction mechanism involving an iron(I) active species and an FeI/FeIII mechanistic cycle (Scheme 1).6 However, the identity of this S = 1/2 species has never been determined and remains one of the longest-standing mysteries in transition-metal catalyzed cross-coupling.
Scheme 1.
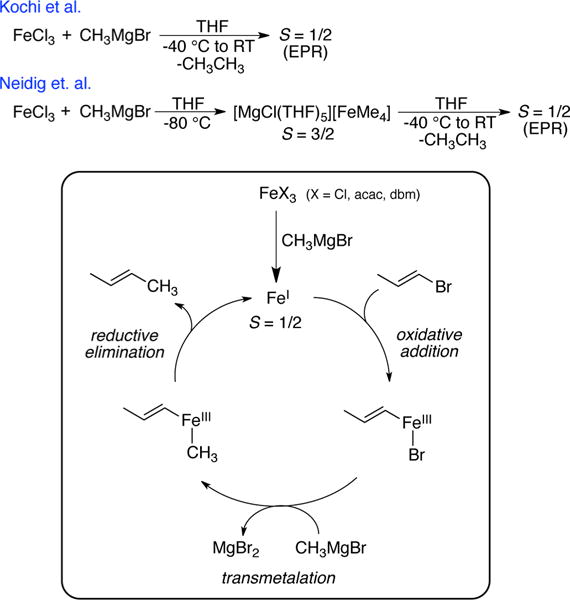
Spectroscopic Studies and Kochi’s Mechanistic Proposal for Iron-Catalyzed Cross-Couplings with MeMgBr and Simple Ferric Salts
Toward the goal of identifying the iron species formed in situ in this chemistry, Fürstner and co-workers reported a homoleptic tetramethyliron(II) ferrate complex, [(Me4Fe)(MeLi)] [Li(OEt2)]2, synthesized from reaction of MeLi with FeCl3 or FeCl2 in Et2O.15,16 This complex exhibits a color change from red to yellow as well as reactivity toward activated electrophiles when dissolved in THF16 and was also found to be active in both stoichiometric and catalytic ring-opening/cross-coupling reactions with MeMgBr.17 However, an iron(II) center is not capable of generating the S = 1/2 electron paramagnetic resonance (EPR) signal observed by Kochi, except in cases where the iron(II) center is part of a mixed-valence multinuclear system. More recent studies from our group identified the formation of the homoleptic tetraalkyliron(III) ferrate complex [MgCl(THF)5][FeMe4] from the reaction of FeCl3 with MeMgBr in THF at low temperature (Scheme 1).18 Upon warming, this distorted square-planar S = 3/2 species converts to the S = 1/2 species originally observed by Kochi and co-workers, with concomitant formation of ethane, consistent with its intermediacy in the reduction pathway of FeCl3 to generate the reduced S = 1/2 species.
Herein we report the isolation and characterization of [MgCl(THF)5][Fe8Me12] (1) from reaction of FeCl3 with MeMgBr in THF, identified via EPR and near-infrared (NIR) magnetic circular dichroism (MCD) as Kochi’s previously observed S = 1/2 species. This novel iron cluster is demonstrated to be reactive with electrophile and effective for the generation of cross-coupled product in the presence of additional MeMgBr. These results demonstrate the importance of small iron clusters in cross-coupling catalysis, which represents a new paradigm with regard to the types of iron species operative in this chemistry.
Our previous studies of the thermal decomposition of [MgCl(THF)5][FeMe4] indicated that warming toward room temperature was required for reductive elimination of ethane to occur with concomitant formation of Kochi’s S = 1/2 species. Combined with Kochi’s previous observation that this S = 1/2 species is ultimately unstable in solution at RT and decays within 15 min,4 we envisioned that initial reaction of FeCl3 with MeMgBr at low temperature followed by warming (to promote the formation of the S = 1/2 species) and then rapid cooling (to disfavor its decomposition) would provide a pathway for the isolation of this species. Reaction of FeCl3 with 5 equiv of MeMgBr at −80 °C followed by warming to 0 °C for 5 min and then immediate cooling to −80 °C yielded a yellow-brown solution from which dark brown single crystals of 1 suitable for X-ray diffraction (XRD) could be obtained upon layering of the THF solution with pentane at −80 °C.
The single-crystal XRD structure of this highly air- and temperature-sensitive complex contains a MgX(THF)5+ cation, X = 0.92 Cl and 0.08 Br, and a distorted heterocubane Fe8Me12− anion with faces that are diamond-shaped (Figure 1). To the best of our knowledge, only five other Fe8 cubic structures are reported in the Cambridge Structural Database,19–23 and all are near-perfect cubes with square faces and μ4-bridging sulfido ligands capping the faces. Their Fe–Fe distances range from 2.64 to 2.81 Å, which differ considerably from those in 1, which range from 2.4188(15) to 2.4514(15) Å. Due to the diamond-shaped faces, the Fe–Fe distances in 1 across the short face diagonals range from 2.7988(14) to 2.8295(14) Å. Atoms Fe1, Fe2, Fe3, and Fe4, which sit at the vertices of an approximate tetrahedron, are each ligated facially by three iron atoms and three methyl groups, with Fe–C bond lengths ranging from 2.135(9) to 2.175(8) Å. Atoms Fe5, Fe6, Fe7, and Fe8, which also sit at the vertices of a distorted tetrahedron that interpenetrates the other, are each ligated facially by three iron atoms and also have close contact to methyl groups at Fe–C distances ranging from 2.311(8) to 2.366(9) Å. Unfortunately, the methyl group hydrogen atoms could not be located unambiguously in the difference Fourier map; hence, they ultimately were not modeled. However, it is worth noting that models that either minimze H⋯H close contacts or orient the methyl groups to favor agostic interactions with the more distant of the two iron centers might be possible (see Supporting Information (SI)). There are reported structural precedents in which an asymmetric bridging methyl group participates in one Fe–C σ bond and one C–H⋯Fe η2 agostic interaction.24 Unfortunately, other experiments that usually offer insight into the presence of such agostic interactions, including vibrational analysis, have not been viable to date due to the high thermal sensitivity and paramagnetic nature of this material. Lastly, the electronic structure of 1 is likely complex due to the direct Fe–Fe bonding and formally mixed-valence nature of the cluster. While calculations of this complex are beyond traditional DFT methods, detailed studies of the electronic structure of 1 are anticipated to be of significant interest within the theoretical community.
Figure 1.
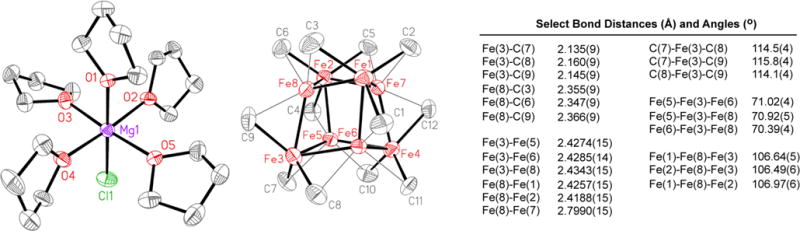
X-ray crystal structure of [MgCl(THF)5][Fe8Me12] (1) with selected bond lengths and angles. Thermal ellipsoids are shown at 50% probability. The methyl hydrogens are excluded as their positions could not be unambiguously defined in the difference Fourier map, and the hydrogens on the cation are omitted for clarity. The halide on the Mg cation species is disordered with both Cl and Br coordination (Cl is the major species).
While 1 represents a novel iron–methyl cluster formed upon reaction of FeCl3 with MeMgBr in THF, it was important to spectroscopically characterize 1 in solution for comparison to the S = 1/2 species formed in situ as previously observed by Kochi.5 The 5 K EPR sspectrum of 1 in THF (Figure 2, top), prepared from dissolution of crystals of 1 in THF at −80 °C to prevent thermal decomposition, contains a broad S = 1/2 signal, analogous to that previously observed by Kochi5,6 and confirmed by our group to form in situ upon reaction of FeCl3 with 5 or 20 equiv of MeMgBr (Figure 2, bottom). A similar broad S = 1/2 EPR signal was also reported by Kochi for the analogous reaction with Fe(acac)3.5,6 EPR spin quantitation indicates that the S = 1/2 species represents (within error) effectively all of the iron formed in solution. While the EPR data are consistent with 1 being the previously unidentified S = 1/2 species, such broad EPR signals alone are insufficient to definitively prove that these species are identical. NIR MCD is a much higher resolution probe of geometric and electronic structure, and the NIR MCD spectrum of 1 in THF/2-Me-THF (Figure 3, top), prepared from dissolution of crystals of 1 in 1:1 THF:2-MeTHF at −80 °C to prevent thermal decomposition, is consistent with the analogous spectrum of the in situ generated S = 1/2 species from FeCl3 (Figure 3, middle) or Fe(acac)3 (Figure 3, bottom) in 1:1 THF:2-MeTHF, demonstrating that 1 is Kochi’s previously identified S = 1/2 species. The solid-state EPR and NIR MCD spectra of 1 (see SI) are consistent with the solution data, displaying only differences in signal broadness (EPR) or individual transition intensities (MCD) indicative of slight differences in the distributions of bond lengths, angles, and/or close contact interactions in solution versus the solid state. Lastly, while the Mössbauer spectrum of 1 could not be obtained due to low crystal yields, the 80 K Mössbauer spectrum of the in situ-formed S = 1/2 species using Fe(acac)3 is described by a broad doublet signal (see SI).
Figure 2.
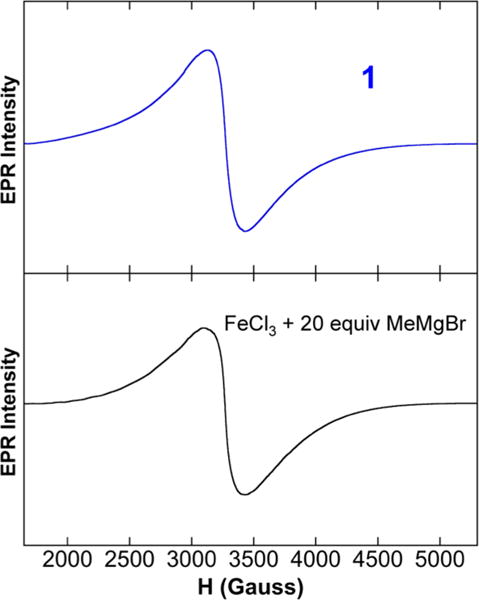
5 K EPR spectra of 1 (top) and Kochi’s in situ-formed S = 1/2 species (bottom).
Figure 3.
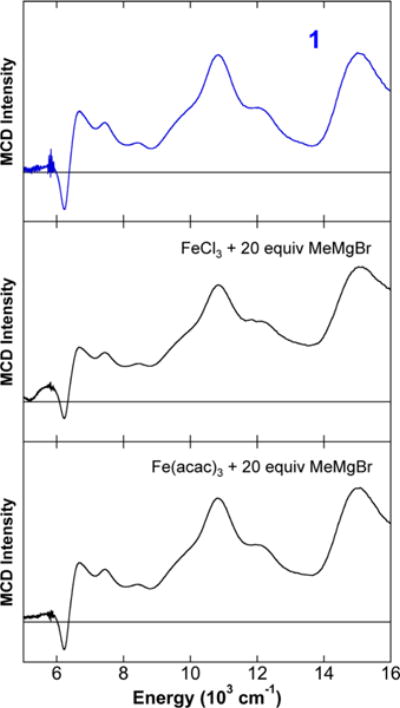
5 K, 7 T NIR MCD spectra of 1 (top) and Kochi’s in situ-formed S = 1/2 iron species from reaction of 20 equiv of MeMgBr with FeCl3 (middle) or Fe(acac)3 (bottom). All spectra were collected in 1:1 THF:2-MeTHF.
Beyond the important demonstration that the in situ-formed S = 1/2 species is Fe8Me12−, the most critical question is whether this species can serve as an effective and reactive species for forming cross-coupled product. Hence, studies of the reaction of 1 with electrophile were performed in order to evaluate the ability of this cluster to form cross-coupled product. β-Bromostyrene was selected as the electrophile for this study due to the formation of non-gaseous cross-coupled product for robust GC analysis and its ability to serve as an effective electrophile in cross-couplings with ferric salts (see SI for catalytic data). Prior to performing the reaction studies, it was first necessary to evaluate the lifetime of 1 in THF at RT. Crystals of 1 previously held at −80 °C were added to a known volume of RT THF, and, as a function of time, aliquots were freeze-trapped for EPR analysis. As shown in Figure 4A, 1 slowly decomposes in RT THF with a decay of ~5% per minute over the course of 5 min (Figure 4A inset). Thus, reactions with β-bromostyrene performed within 2–3 min would have limited decomposition of 1. For electrophile reactions, crystals of 1 were dissolved in a known volume of RT THF, an EPR sample was freeze-trapped 40 s after dissolution to determine the amount of 1 present by spin-quantitated EPR, and then a known amount of β-bromostyrene was added. After addition of β-bromostyrene, an aliquot of the reaction was chemically quenched after 40 s for analysis by GC. Using the spin-quantitated EPR of 1 prior to electrophile addition, it was determined that these reactions involved ~15 mM 1 and ~0.5 equiv of β-bromostyrene for each run. While cross-coupled product does form in this reaction, only ~5% of β-methylstyrene (with respect to β-bromostyrene) was found to form after 40 s of reaction (Scheme 2). No significant increase in product was observed at extended reaction time (~6% after 120 s), and no additional side products were observed.
Figure 4.
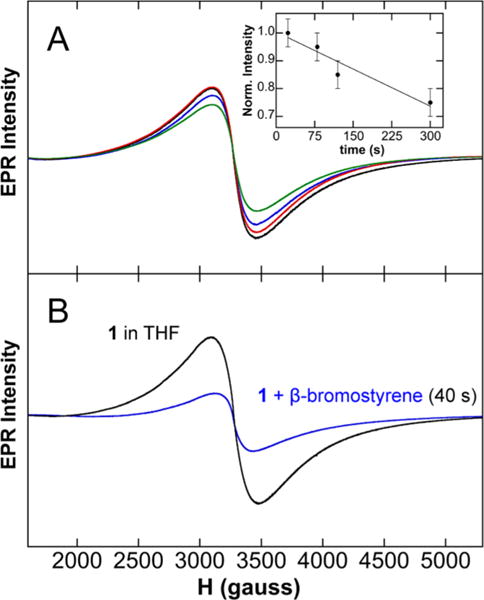
EPR studies of (A) the thermal stability of 1 in THF at RT and (B) the changes in the amount of 1 in solution in reaction studies with electrophile. The inset in (A) shows a linear fit of the decay of 1 in RT THF over the course of 5 min.
Scheme 2.
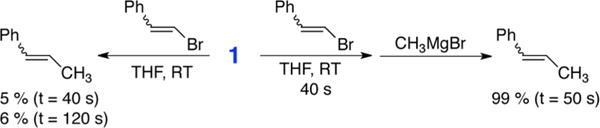
Reactions of 1 with β-Bromostyrene
Despite the minimal formation of cross-coupled product following 40 s of direct reaction of 1 with β-bromostyrene, EPR analysis of the reaction freeze-trapped 40 s after electrophile addition indicated significant consumption of 1 upon addition of electrophile (Figure 4B), though no new EPR-active species are observed to be generated. Interestingly, in Kochi’s original studies, it was suggested that reaction of the iron active species with electrophile might first form an initial intermediate species which then required reaction with additional MeMgBr to form cross-coupled product.6 To test this hypothesis for 1, an experiment was also performed where more MeMgBr (1.25 equiv with respect to β-bromostyrene) was added 40 s after the addition of β-bromostyrene, and the reaction was quenched following an additional 50 s of reaction time. With the addition of MeMgBr, essentially complete conversion of the electrophile to selectively form cross-coupled product was observed (99% yield with respect to electrophile) (Scheme 2). Furthermore, EPR of the reaction solution freeze-trapped 50 s after MeMgBr addition demonstrates the near-quantitative re-formation of 1 in situ (see SI). Overall, these results are consistent with 1 being an effective reactive species for the formation of cross-coupled product in the presence of additional MeMgBr. While challenging, future studies will be directed toward the identification of the initial intermediate species formed upon reaction of 1 with electrophile in order to further probe the underlying mechanism of this reaction.
In summary, the S = 1/2 species formed in situ in reactions of simple ferric salts and MeMgBr, originally observed by EPR in the 1970s by Kochi, has been isolated and identified as [MgCl(THF)5][Fe8Me12]. While the direct reaction of this species with electrophile generates minimal cross-coupled product, reaction with electrophile followed by addition of MeMgBr leads to rapid reaction to selectively form cross-coupled product. Importantly, the identification of an iron cluster as a reactive species in iron-catalyzed cross-coupling represents a new paradigm in catalyst structure for such reactions, contrasting previous proposals centered on mononuclear iron species. Future studies toward elucidating the underlying mechanism on the reactions of 1 with electrophile will further develop our understanding of iron-catalyzed cross-coupling reactions using ferric salts.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Insitutes of Health (R01GM111480 to M.L.N.) and by an Alfred P. Sloan Fellowship to M.L.N.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b03760.
Experimental methods and supplementary data including EPR, MCD, Mössbauer, and X-ray crystal details of 1 (PDF)
X-ray crystallographic data for 1 (CIF)
Notes
The authors declare no competing financial interest.
References
- 1.Tamura M, Kochi JK. J Am Chem Soc. 1971;93:1487–1489. [Google Scholar]
- 2.Tamura M, Kochi JK. J Organomet Chem. 1971;31:289–309. [Google Scholar]
- 3.Tamura M, Kochi JK. Bull Chem Soc Jpn. 1971;44:3063–3073. [Google Scholar]
- 4.Neumann SM, Kochi JK. J Org Chem. 1975;40:599–606. [Google Scholar]
- 5.Kwan CL, Kochi JK. J Am Chem Soc. 1976;98:4903–4912. [Google Scholar]
- 6.Smith RS, Kochi JK. J Org Chem. 1976;41:502–509. [Google Scholar]
- 7.Bauer I, Knolker H-J. Chem Rev. 2015;115:3170–3387. doi: 10.1021/cr500425u. [DOI] [PubMed] [Google Scholar]
- 8.Sherry BD, Furstner A. Acc Chem Res. 2008;41:1500. doi: 10.1021/ar800039x. [DOI] [PubMed] [Google Scholar]
- 9.Bedford RB. Acc Chem Res. 2015;48:1485–1493. doi: 10.1021/acs.accounts.5b00042. [DOI] [PubMed] [Google Scholar]
- 10.Cassani C, Bergonzini G, Wallentin C-J. ACS Catal. 2016;6:1640–1648. [Google Scholar]
- 11.Mako TL, Byers JA. Inorg Chem Front. 2016 doi: 10.1039/C5QI00295H. [DOI] [Google Scholar]
- 12.Jana R, Pathak TP, Sigman MS. Chem Rev. 2011;111:1417–1492. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daifuku SL, Al-Afyouni MH, Snyder BER, Kneebone JL, Neidig ML. J Am Chem Soc. 2014;136:9132–9143. doi: 10.1021/ja503596m. [DOI] [PubMed] [Google Scholar]
- 14.Daifuku SL, Kneebone JL, Snyder BER, Neidig ML. J Am Chem Soc. 2015;137:11432–11444. doi: 10.1021/jacs.5b06648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furstner A, Krause H, Lehmann CW. Angew Chem Int Ed. 2006;45:440–444. doi: 10.1002/anie.200502859. [DOI] [PubMed] [Google Scholar]
- 16.Furstner A, Martin R, Krause H, Seidel G, Goddard R, Lehmann CW. J Am Chem Soc. 2008;130:8773–8787. doi: 10.1021/ja801466t. [DOI] [PubMed] [Google Scholar]
- 17.Sun C-L, Furstner A. Angew Chem Int Ed. 2013;52:13071–13075. doi: 10.1002/anie.201307028. [DOI] [PubMed] [Google Scholar]
- 18.Al-Afyouni MH, Fillman KL, Brennessel WW, Neidig ML. J Am Chem Soc. 2014;136:15457–15460. doi: 10.1021/ja5080757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saak W, Pohl S. Angew Chem Int Ed Engl. 1984;23:907–908. [Google Scholar]
- 20.Pohl S, Opitz U. Angew Chem Int Ed Engl. 1993;32:863–864. [Google Scholar]
- 21.Pohl S, Barklage W, Saak W, Opitz U. J Chem Soc Chem Commun. 1993:1251–1252. [Google Scholar]
- 22.Goh C, Segal BM, Huang J, Long JR, Holm RH. J Am Chem Soc. 1996;118:11844–11853. [Google Scholar]
- 23.Marsh RE, Clemente DA. Inorg Chim Acta. 2007;360:4017–4024. [Google Scholar]
- 24.Dawkins GM, Green M, Orpen AG, Stone FGA. J Chem Soc Chem Commun. 1982:41–43. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.