

Summary
The challenges to effective drug delivery to brain tumors are twofold: (1) there is a lack of non-invasive methods of local delivery and (2) the blood-brain barrier limits systemic delivery. Intranasal delivery of therapeutics to the brain overcomes both challenges. In mouse model of malignant glioma, we observed that a small fraction of intranasally delivered neural stem cells (NSCs) can migrate to the brain tumor site. Here, we demonstrate that hypoxic preconditioning or overexpression of CXCR4 significantly enhances the tumor-targeting ability of NSCs, but without altering their phenotype only in genetically modified NSCs. Modified NSCs deliver oncolytic virus to glioma more efficiently and extend survival of experimental animals in the context of radiotherapy. Our findings indicate that intranasal delivery of stem cell-based therapeutics could be optimized for future clinical applications, and allow for safe and repeated administration of biological therapies to brain tumors and other CNS disorders.
Graphical Abstract
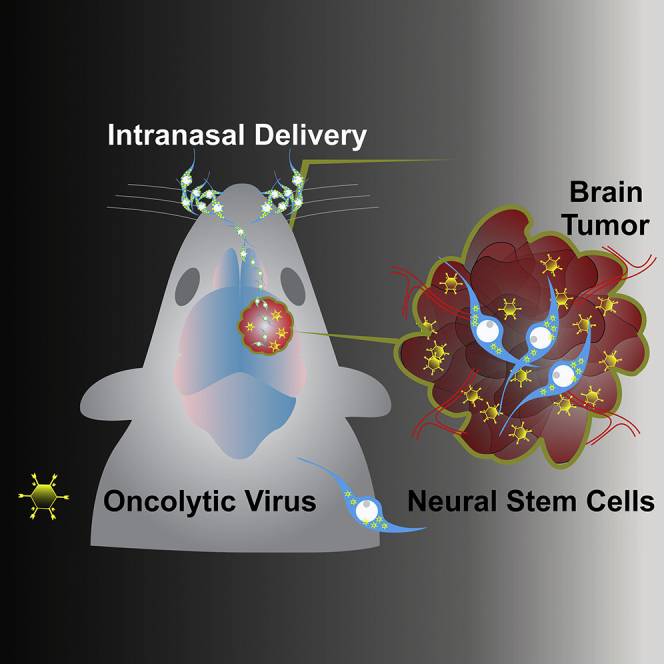
Highlights
-
•
Intranasal delivery of NSCs is a promising platform for glioma therapy
-
•
Hypoxia or CXCR4 overexpression enhances NSC migration to glioma
-
•
Oncolytic viruses loaded in CXCR4-enhanced NSCs improve animal survival
-
•
Non-invasive intranasal NSC-based therapies warrant clinical translation
In this report, Balyasnikova and colleagues demonstrate that hypoxic preconditioning or overexpression of CXCR4 in neural stem cells enhances intranasal delivery of oncolytic virus to irradiated glioma, resulting in survival benefits for experimental animals. These findings indicate that intranasal stem cell-based oncolytic virotherapy could be optimized for future clinical applications in the context of brain tumors.
Introduction
Malignant gliomas (MGs), a group of high-grade primary adult brain tumors, are classically known for being highly aggressive and incurable. With an average median survival of only 14.6 months following the current standard-of-care treatment regimen, MGs are in desperate need of novel and effective treatment strategy (Stupp et al., 2005). The blood-brain barrier (BBB) renders several highly effective systemic chemotherapeutic agents ineffective in the setting of MGs (Lesniak and Brem, 2004, Lesniak et al., 2001). Along with presence of the BBB there are several other intrinsic tumor characteristics that orchestrate the eventual failure of the current standard of care, such as inherent resistance of tumor cells to chemotherapeutic agents (Decleves et al., 2006, Tian et al., 2015), complex interplay between radiation and hypoxia that results in radioresistance (Dahan et al., 2014, Moeller et al., 2004, Vordermark et al., 2004), and the fluid phenotype of cancer cells that seamlessly transition between differentiated and dedifferentiated forms following chemotherapy (Auffinger et al., 2014, Safa et al., 2015).
The particular anatomy of the olfactory and trigeminal neural pathways connects the nasal mucosa directly with the brain and the perivascular pathway by circumventing the BBB (Jiang et al., 2015). Using intranasal delivery, several therapeutic agents, such as small molecules, proteins, hormones, and nanoparticles, have been shown to successfully reach the brain (Andrade, 2015, Elnaggar et al., 2015, Feng et al., 2012, Kim et al., 2012). The intranasal route has also been explored for delivering larger cell-based carriers such as mesenchymal stem cells (MSCs) in the setting of ischemic brain injury and Parkinson's disease (Danielyan et al., 2011, van Velthoven et al., 2010). Reitz et al. (2012) demonstrated that intranasally delivered neural stem cells (NSCs) localize to the intracranial human or murine glioma xenografts in mouse models. Furthermore, our group has shown that therapeutic MSCs and NSCs when delivered to the nasal cavity not only travel to intracranial tumors in mice, but also prolong the animals' survival (Balyasnikova et al., 2014, Gutova et al., 2015). Further validation of intranasal delivery of various therapeutics to the brain is of particular interest in the context of MG.
One of the emerging front-runners of experimental therapeutic options for targeting MG is virotherapy whereby oncolytic viruses (OVs), such as CRAd-S-pK7 (Ulasov et al., 2007), selectively infect tumor cells and induce a specific anti-glioma cytotoxic response. We have shown that stem cells not only can efficiently deliver OVs to experimental glioma (Ahmed et al., 2011b, Ahmed et al., 2013, Morshed et al., 2015), but also delay the premature viral neutralization by the host's immune system (Ahmed et al., 2010). Intranasal delivery of OVs loaded in stem cell carriers allows for a non-invasive and repeatable treatment regimen, although to date the feasibility of such an approach has not investigated.
As MGs are known for the presence of prominent intratumoral areas of hypoxia (Dey et al., 2014, Keunen et al., 2011, Pistollato et al., 2010, Womeldorff et al., 2014), hypoxia-driven glioma-tropic NSCs (Zhao et al., 2008) can provide an optimal cell-based carrier for OV delivery. We had previously demonstrated that hypoxia can enhance the motility of NSCs, and CXCR4 overexpression is the main mechanism contributing to this phenomenon. The expression of SDF-1, a CXCR4 ligand, is significantly associated with the hypoxic environment of MG (Zhao et al., 2008). We and others have also previously shown that SDF-1 expression is significantly higher in glioma tissue after radiation therapy (Balyasnikova et al., 2014, Zhao et al., 2008). Therefore, in this study we investigated whether CXCR4 overexpression on the surface of NSCs either by hypoxic preconditioning or genetic modification of NSCs allows for enhanced migratory properties, and the therapeutic outcome of intranasal delivery of OV-loaded CXCR4-expressing NSCs in glioma-bearing mice.
We hypothesized that intranasal delivery of CXCR4-expressing NSCs loaded with CRAd-S-pK7 will lead to tumor-specific delivery of OV and increased survival in the mouse model of glioma. Our results show that CXCR4-enhanced NSCs possess higher motility toward SDF-1 gradient in vitro, delivered OV to glioma xenograft more efficiently and provided a survival advantage to irradiated mice bearing aggressive intracranial tumors. Moreover, genetically modified NSCs would also retain their stemness and genetic stability, making them a desirable cell carrier for the intranasal delivery of therapeutics to the MG. These studies establish an effective strategy to significantly improve stem cell-based oncolytic virotherapy via selective enhancement of tumor tropism signaling. Further refinement and expansion strategies beyond the CXCR4/SDF-1 signaling axis should also be explored to optimize the clinical translation value of this approach.
Results
Hypoxic Preconditioning Enhances the Tropism of NSCs toward Glioma
The natural tumor tropism of stem cells is the foundation for using NSCs as a therapeutic conduit to accomplish targeted delivery of therapeutics (Ahmed et al., 2011a, Ahmed et al., 2013, Balyasnikova et al., 2014). The hypoxic environment of glioma is thought to contribute to the mechanism of NSC tropism (Zhao et al., 2008).
The migratory preference of NSCs toward glioma cell lines were explored and defined for both NNSCs and HNSCs via several in vitro assays. All assays consistently supported the notion that HNSCs migrate faster toward glioma cells and travel longer distances in a given time. In Figures 1A and 1B, a transwell migration assay using tumor cells grown as tumorspheres at the bottom of the dish shows significantly more GFP+ HNSCs that preferentially migrated across the porous cell-culture inserts compared with GFP+ NNSCs (9.9% ± 0.8% of relative presence for HNSCs versus 0.4% ± 0.1% for NNSCs in GBM43 cultures, n = 5, ∗∗∗p < 0.001; 64.9% ± 4.8% of relative presence for HNSCs versus 37.8% ± 8.0% for NNSCs in U87MG mCherry cultures, n = 5, ∗p < 0.05). A similar outcome was observed for collagen hydrogel cultures, where the separation between tumor cells and the seeded GFP+ NSCs was achieved by a layer of gelated hydrogel (Figure 1C). Within 12 hr, significantly more GFP+ HNSCs migrated to the bottom of the dish across the gel layer (14 ± 2 HNSCs per visual field versus 8 ± 1 NNSCs for GBM43; 18 ± 2 HNSCs per visual field versus 9 ± 1 NNSCs for U87MG mCherry; n = 7, ∗p < 0.05). The speed of migration toward glioma cells by GFP+ NSCs was also determined to be faster (less time lapsed for the travel between cell patches created by ibidi 2D silicone chambers) based on live cell microscopy using the Olympus VivaView microscope, in that HNSCs take significantly less time to travel to the U87MG mCherry cells compared with NNSCs (Figures S1A–S1C, 120 ± 12 min for HNSCs versus 220 ± 11 min for NNSCs; n = 6, ∗p < 0.001). Thus, these data clearly indicate that hypoxic preconditioning significantly increases tropism of NSCs toward patient-derived GBM43 and U87MG cells growing as tumorspheres or as attached cells, in agreement with previous observations (Zhao et al., 2008).
Figure 1.

Hypoxia Pretreatment Enhances Tropism of NSC toward Human GBM Cells
(A and B) GFP+ NSCs treated with hypoxia (HNSCs; 1% O2; 24 hr) or control NSCs (NNSCs) were compared for ability to migrate toward tumorspheres. HNSCs effectively cross the culture insert membrane and accumulate within tumorspheres of patient-derived GBM43 glioma cells visualized with DAPI nuclear stain (blue) (A) and U87MG mCherry cells (B). Right panels represent quantification of the relative presence of GFP+ NSCs, indicating significantly higher numbers of GFP+HNSCs than NNSCs inside the tumorspheres by 24 hr of co-culture (n = 5, Student's t test; ∗∗∗p < 0.001 for GBM43, ∗p < 0.05 U87MG). Scale bars, 100 μm.
(C) Collagen hydrogel migration assay further demonstrates the enhanced tumor tropism by NSCs after hypoxia pretreatment; Confocal z-stack 3D reconstruction shows GFP+ NSCs (green) seeded on top of the collagen hydrogel layer migrating to the bottom of the culture dishes in both GBM43blue (blue cells, top panels) and U87MG mCherry adherent cultures (red cells, bottom panel) by 12 hr. Quantification of migrated GFP+ NSCs by microscopy visual field (right) confirms the increased “homing” by HNSCs in both the GBM43blue and U87MG mCherry cells after hypoxia pretreatment (n = 7; ∗∗p < 0.01, Student's t test, error bars with SEM).
Higher Surface Expression of CXCR4 in HNSCs and CXCR4NSCs Drives Migration of NSCs toward SDF-1 Gradient In Vitro
We previously demonstrated that brain radiation therapy (XRT) increases the expression of SDF-1 in U87MG xenograft tumor (Balyasnikova et al., 2014), suggesting that the CXCR4/SDF-1 signaling axis could potentially be utilized to optimize tumor tropism of NSCs toward irradiated glioma tissue. Here we show that hypoxia-treated NSCs show about a 2-fold increase in the surface expression of CXCR4 in HNSCs over NNSCs (Figure 2A; n = 3, ∗p < 0.0001) as determined by flow cytometry. Although CXCR7, another receptor interacting with SDF-1, shows hypoxia-induced surface expression in NSCs, the increase in protein expression in HNSCs was <10% when compared with NNSC controls (Figure S1C). Genetic modification of NSCs via transduction with lentivirus encoding for CXCR4, significantly increased mRNA CXCR4 expression in CXCR4NSCs in comparison with cells transduced with control lentivirus VCNSCs (n = 3, ∗p < 0.0001), as determined by qRT-PCR (Figure 2B). Nearly all transduced NSCs were expressing CXCR4 on the cell surface as shown by flow cytometry (Figure 2C), which is about 4 and 2.5 times higher than in NNSCs and HNSCs, respectively. These results demonstrate that CXCR4 is a potential receptor responsible for increased migration of NSCs toward glioma tumors secreting SDF-1.
Figure 2.

Analysis of the Surface CXCR4 Receptor Expression in NSCs
(A) Hypoxia pretreatment increases the surface CXCR4 receptor expression on NSCs by over 2-fold in comparison with NSCs growing under normoxic conditions (flow cytometry).
(B) Overexpression of CXCR4 in genetically modified NSCs demonstrated by analysis of mRNA levels in vector control (VCNSCs) and CXCR4NSCs (n = 3; ∗∗∗p < 0.001, Student's t test, error bars with SEM).
(C) Flow cytometry confirms that majority of CXCR4NSCs are positive for the surface CXCR4 expression, representing nearly 5- and 2.5-fold higher CXCR4 expression levels than in non-modified NSCs growing under normoxic and hypoxic conditions, respectively.
The higher CXCR4 expression in HNSCs and CXCR4NSCs correlated with the increase in chemotaxis toward SDF-1 gradient in 3D migration assays (Figure 3A). Specifically, HNSCs demonstrated a significantly increased migration distance in response to SDF-1 stimulation (10 ng) upon 24 hr of hypoxia treatment (79 ± 19 pixels; n = 30 cells) compared with NNSCs (23 ± 2 pixels; n = 30; ∗p < 0.001). Moreover, CXCR4NSCs showed significantly improved migration over HNSCs within 24 hr toward the SDF-1-containing reservoir (114 ± 28 pixels; n = 30, ∗p < 0.001). The presence of blocking antibody against CXCR4 immunoglobulin G, however, abolished the migration toward the SDF-1 source for both HNSCs and CXCR4NSCs (Figure 3B), suggesting a key dependence on CXCR4 in SDF-1-induced NSC migration.
Figure 3.

Migration of HNSCs and CXCR4NSCs Depends on SDF-1 Gradient
(A) Using the 3D chemotaxis assay μ chamber slides, NSCs treated with hypoxic culture, as well as NSCs overexpressing CXCR4 receptors, demonstrate significantly longer chemotactic migration distance toward SDF-1 containing medium chamber (n = 30 cells; ∗∗∗p < 0.001, Student's t test).
(B) The SDF-1-dependent chemotaxis was abolished in both HNSCs and CXCR4NSCs as demonstrated by blocking with anti-CXCR4 monoclonal antibody (n = 30; ∗∗∗p < 0.001, Student's t test, error bars with SEM).
Hypoxia induces a phenotypic shift toward neural progenitors in NSCs, whereas genetic modification of NSCs to overexpress CXCR4 does not change their phenotype.
To ensure that 1% hypoxic culture for 24 hr does not negatively affect the therapeutic potentials of NSCs, we conducted an analysis of mRNA and protein expression for markers of stemness and differentiation by qRT-PCR, western blotting, and immunocytochemistry (ICC). The mRNA expression for SOX2, hNESTIN, MUSASHI (markers of neural stemness), βIII-TUBULIN, and GFAP (markers of differentiation) were assessed by qRT-PCR (Figure 4A). Our results demonstrate significant changes in stemness markers after 24 hr of treatment with hypoxia, where hNESTIN mRNA levels significantly increased (443 ± 36-fold increase over normoxia; n = 3, ∗∗∗p < 0.001), whereas SOX2 and MUSASHI were significantly decreased (0.04 ± 0.04- and 0.33 ± 0.11-fold respectively; n = 3, ∗∗∗p < 0.001) in comparison with normoxia, indicating a possible phenotypic shift toward the more motile neural progenitor stages of NSCs. Moreover, the neural differentiation marker βIII-TUBULIN was significantly reduced (0.45 ± 0.10-fold; n = 3, ∗∗∗p < 0.001) in comparison with normoxia. Worth noting is that the astroglial marker GFAP (data not shown) mRNA was undetectable under either normoxia or hypoxia treatments. ICC staining for the definitive astroglial differentiation marker S100B showed complete absence in NSCs under either condition, confirming that hypoxic preconditioning did not induce NSC differentiation toward astrocytes (Figure S2C). ICC for stemness and differentiation marker expressions supports the qRT-PCR results (Figures S2A–S2C). These observations confirm that, while hypoxic preconditioning allows NSCs to largely maintain their stem cell profiles, a phenotypic shift toward hNESTIN+ neural progenitor stages with reduced terminal differentiation potentials is the predominant outcome for 24-hr hypoxia exposure, in agreement with the observed increase in motility and tumor tropism.
Figure 4.

Phenotypic Characterization of NSCs after Hypoxic Culture or CXCR4 Receptor Genetic Enhancement Strategies
(A) Compared with normoxic culture, hypoxic pretreatment for 24 hr led to significant mRNA changes in stemness markers such as human NESTIN (hNESTIN), SOX2, and MUSASHI (n = 3; ∗∗∗p < 0.001, Student's t test).
(B) In contrast, genetic enhancement of CXCR4 receptors demonstrate comparable mRNA levels of stemness markers, and early-stage neural progenitor marker βIII TUBULIN (n = 3; p > 0.05, Student's t test).
(C) The analysis of hNESTIN, SOX2, and βIII TUBULIN via western blotting demonstrates that protein expression corresponds to mRNA expression and shows no changes after genetic modification of NSCs to overexpress CXCR4 receptor (n = 3; p > 0.05, Student's t test, error bars with SEM).
(D) Cytogenetic analysis of NSCs indicates that CXCR4NSCs retain their normal karyotype after genetic enhancement of CXCR4 receptor, similarly to VCNSCs.
Consistent with knowledge that hypoxia induces upregulation of HIF1α, we observed the increase in HIF1α expression in hypoxia-treated NSCs, which was accompanied by activation of signaling as judged by an increase in pAKT (Figures S2D–S2F). Similar induction of pAKT was accomplished by treatment of NSCs with SDF-1. Both SDF-1 and hypoxia-induced pAKT were partially blocked by the CXCR4 receptor antagonist AMD3100, thus indicating a CXCR4-dependent mechanism of activation of downstream signaling in hypoxia-treated cells. Hypoxia-induced signaling within the NSCs can involve complex signaling networks that are known to regulate the apoptosis, differentiation, and migratory/metastatic behaviors of many normal or cancerous cell types (Barriga et al., 2013, Guan et al., 2015, Guo et al., 2014, Speth et al., 2014). In addition, activation of HIF1α signaling in NSCs can trigger biological events not consistent with their intended use as therapeutic carriers. Therefore, given the established CXCR4/SDF-1 axis in tropism of NSCs toward glioma, it is of high interest to examine whether modification of NSCs via ectopic overexpression of CXCR4 results in phenotypic stability of NSCs. The analysis of markers of stemness and differentiation at mRNA and protein levels in CXCR4NSCs confirmed that overexpression of CXCR4 minimally affected the expression profiles of hNESTIN, SOX2, and βIII TUBULIN in CXCR4NSCs. Importantly, cytogenetic analysis revealed that CXCR4NSCs cells retain their normal karyotype after genetic modification via lentiviral transduction (Figure 4D).
XRT Increases SDF-1 Expression in GBM43 Glioma Xenografts
To confirm the potential significance of SDF-1 in contributing to migration of NSCs in vivo, we euthanized control and XRT-treated mice and compared brain tissues for SDF-1 expression. The analysis of H&E staining and ICC of brain tissue sections from control and irradiated mice demonstrates reduced tumor burden after XRT, and significantly increased SDF-1 levels at the tumor sites after XRT treatment (Figure 5A; n = 22 sections from 3 animals/group, ∗∗∗p < 0.001). Quantitative analysis for positive immunostaining demonstrated a nearly 4-fold increase in XRT-treated mice versus control animals (Figure 5B). These findings are in agreement with our previous observation of an increase in SDF-1 expression in irradiated U87MG xenografts and highlight the significance of utilizing the SDF-1/CXCR4 axis to advance NCSs as therapeutic carriers to glioblastoma in conjunction with XRT treatment (Balyasnikova et al., 2014).
Figure 5.

Irradiation Increases SDF-1 Expression in GBM43 Xenograft
The analysis of H&E staining and immunocytochemistry of brain tissue sections from control and irradiated mice demonstrates reduced tumor burden but increased SDF-1 levels at the tumor sites after XRT treatment.
(A) SDF-1 expression (green) is strongly localized at the tumor site (white boxes) after irradiation (2 Gy × 5 days).
(B) Densitometry quantification of the viewing fields demonstrates a 4-fold increase in SDF-1 expression in XRT-treated animals in comparison with control animals (n = 3 experiments; ∗∗∗p < 0.001, Student's t test, error bars with SEM).
Hypoxia-Treated NSCs Support the Loading and Replication of CRAd-S-pK7
Propagation of OVs in NSCs (HB1.F3.CD cells) utilized in this study have been extensively characterized by our group (Ahmed et al., 2011a, Ahmed et al., 2011b, Ahmed et al., 2013). We have previously demonstrated that CRAd-S-pK7 can replicate in NSCs as well as in glioblastoma multiforme (GBM) cells both in vitro and in vivo. To evaluate whether hypoxia-treated or genetically modified NSCs can effectively deliver the OVs to the tumor, we first evaluated the replication of OV in HNSCs or CXCR4NSCs in comparison with respective controls (NNSCs, VCNSCs). NSCs were infected with CRAd-S-pK7 at 50 viral particles per cell. Figures S3A and S3B show that treated NSCs (HNSCs and CXCR4NSCs) maintain their capacity for viral replication, albeit with mildly reduced progeny release compared with respective controls. These results confirm the ability of modified NSCs to serve as carriers of OVs for therapeutic studies.
CXCR4NSCs Efficiently Deliver OVs via Intranasal Route to Patient-Derived Glioma Xenografts and Improve Survival in a Mouse Model
Delivery of OVs to glioma in vivo through NSCs (NSC-OVs) depends on the efficient migration of NSCs toward the tumors when injected via the intranasal route. To evaluate whether HNSC-OV therapy provides survival benefits, we tested two different patient-derived xenograft (PDX) models (GBM43 and GBM6). Without XRT, there was no survival benefit of HNSC-OV intranasal treatments (Figure S4A). Following the standardized treatment protocol, we established that a fractionated dose of XRT significantly improves survival of mice with both GBM43 (extension of 9 days of median survival over PBS only with XRT; n = 8 animals/group, ∗p < 0.05, log-rank test) and GBM6 (extension of 8 days of median survival over PBS only with XRT; n = 5, ∗p < 0.05) intracranial xenografts (Figure 6A). Treatments of NNSCs alone, OVs alone, or NNSC-OVs did not further improve the survival of animals. However, mice treated with HNSCs-OVs showed prolonged survival (median survival benefit of 5 days) in both GBM43 (n = 8, ∗p < 0.05; Figure 6B) and GBM6 (n = 5, ∗p < 0.05) models (Figure S4B). These data confirm our hypothesis that hypoxic preconditioning of intranasally delivered NSCs not only enhances tumor tropism and OV delivery but also results in therapeutic benefits in aggressive PDX models of GBMs.
Figure 6.

Hypoxia Pretreatment and CXCR4 Overexpression in NSCs Are Necessary for Effective Intranasal Delivery of OV-Loaded NSCs in Irradiated Animals
(A) When given only PBS intranasally, radiation therapy significantly enhanced survival of mice bearing aggressive patient-derived GBM43 glioma xenografts (median survival increased by 9 days; n = 8 animals/group; ∗∗∗p < 0.001, log-rank test).
(B) However, there was no further improvement in survival when mice were treated with intranasal OV, NNSCs, or NNSCs + OV following XRT treatment. Treatment of mice with OV-loaded HNSCs resulted in significant improvement in the survival of mice (5 days; n = 8; ∗p < 0.05, log-rank test) after irradiation.
(C) Significant improvement in survival of mice after XRT was also achieved with OV-loaded genetically enhanced CXCR4NSCs (9 days of median survival benefits; n = 8; ∗∗p = 0.0013, log-rank test).
The ability of CXCR4NSCs to deliver OV payload to tumor burden in a GBM43 PDX model was also evaluated. Control VCNSCs or CXCR4NSCs were loaded with OVs and intranasally delivered in irradiated mice according to the standardized regimen. Figure 6C shows that CXCR4NSC-OV therapy significantly improves the survival of animals in comparison with control VCNSC-OV. These results demonstrate that CXCR4NSCs migrate toward glioma and successfully deliver OVs, which translates into improved survival in the PDX glioma model.
To further confirm that the observed therapeutic effect is due to enhanced migration of HNSCs or CXCR4NSCs toward tumor burden, we implanted GBM43 glioma cells in the brain and the mice were randomized to designated treatment groups. On the eighth day after tumor injection, mice were subjected to XRT (2 Gy for 5 days), which was followed by intranasal delivery of NNSC-OVs/HNSC–OVs or VCNSCs/CXCR4NSCs. To evaluate the delivery efficiency of OVs by NSCs to the tumor site, we stained brain tissue sections for hexon proteins of viral capsid. As shown in Figure 7A, NNSCs delivered the OVs to glioma (arrows); moreover, hexon immunostaining was nearly twice as high in the HNSCs group, as determined by the hexon staining measured as the mean fluorescence intensity in analyzed tissue sections (n = 10 from 3 animals/group, ∗∗∗p < 0.001). Furthermore, staining of tissue sections for hexon after CXCR4NSC-OV therapy also confirmed that the ability of CXCR4NSC to deliver OVs to the tumor burden is substantially improved in comparison with OV therapy delivered by control VCNSCs (Figure 7B).
Figure 7.

Analysis of Staining for Expression of Viral Capsid Hexon Proteins in GBM43 Xenograft Tissue from Mice Intranasally Treated with OV-Loaded NSCs
Analysis of hexon immunofluorescence staining (green spots, yellow arrows) indicates significantly improved OV delivery to the tumor site (white dashed outline) in mice treated with modified NSCs.
(A) Hexon signal (green) is significantly higher at the tumor site (staining for hNESTIN, red) in tissue sections from mice treated with CRAd-pk7-loaded HNSCs in comparison with NNSCs (n = 10 sections from 3 animals/group; ∗∗∗p < 0.001, Student's t test).
(B) Using GBM43 cells expressing blue fluorescent protein, we also demonstrate much improved OV delivery to the tumor site using CXCR4NSCs when compared with VCNSCs (n = 10 sections from 3 animals/group; ∗∗∗p < 0.001, Student's t test, error bars with SEM).
Discussion
In this study we explored the strategy of enhancing the migratory phenotype of NSCs for intranasal delivery of therapeutic cell carriers for the treatment of MG. This approach is thought to overcome several hurdles that stand in the way of an effective glioma-targeting treatment regimen. We delivered multiple doses of hypoxia-preconditioned NSCs loaded with OVs directly to the nasal cavity of mice bearing intracranial tumors. Mice treated with this modality had significant survival advantage over other control groups. Furthermore, selective genetic enhancement in NSC surface expression of CXCR4 receptor mimics the beneficial impact of hypoxia on NSC migration without the hypoxia-associated phenotypic changes in NSCs. Thus, stable genetic manipulation of NSCs can selectively augment their therapeutic functions against GBM, while maintaining intracellular OV replication.
Intranasal delivery effectively addresses the issue of circumventing the BBB and allows for non-invasive repeated dosing. Although this modality has been successfully utilized for delivering a variety of growth factors, hormones, and neuropeptides (Chapman et al., 2013) to the CNS, the efficacy of this route to deliver cell-based carriers is still in its early phase of exploration. As the tropism of NSCs to intracranial glioma and other malignancies is well validated (Aboody et al., 2000, Gutova et al., 2015, Kanojia et al., 2015), the intranasal delivery imposes strict requirements for fast migration of therapeutic cell carriers from the nasal cavity to prevent rapid clearance from the nasal cavity. In this respect, MSCs genetically modified to overexpress CXCR4 have been shown to possess higher migratory properties and therapeutic potential in an acute lung injury model (Yang et al., 2015), better homing capacity to bone marrow in irradiated mice (Chen et al., 2013), and mobilization and enhanced neuroprotection in a rat cerebral ischemia model (Yu et al., 2012). However, genetic modification of NSCs to enhance their migration toward brain malignancy has never previously been explored.
Non-immunogenic glioma-tropic NSCs have been known to be an effective carrier of OVs (Ahmed et al., 2011a) in the mouse model of glioma. A study by Zhao et al. (2008) showed that hypoxia is a key factor in determining NSC tropism to glioma and that multiple signaling pathways mediate this phenomenon. Several other studies delineate a critical role of hypoxia in promoting survival and increasing the motility and regenerative capacity of stem cells (Rosova et al., 2008, Wang et al., 2013). In line with the current literature, our results also show that hypoxic preconditioning of NSCs results in an increased migratory phenotype of the NSCs, in part due to enhanced expression of CXCR4 on the cell surface, and allows for efficient intracellular viral replication. However, hypoxic preconditioning is known to initiate other changes in signaling, leading to upregulation of numerous chemokine receptors and proteins involved in migration of NSCs (Zhao et al., 2008). In our study, hypoxia preconditioning affected mRNA expression of several stemness and differentiation markers in NSCs. To overcome this limitation, we generated genetically modified NSCs overexpressing CXCR4, thus providing certainty to the fate of therapeutic stem cells. Genetic modification of NSCs had no effect on the NSC phenotype at either RNA or protein level and allowed for effective viral replication. Moreover, CXCR4-overexpressing NSCs demonstrated genetic stability as assessed by cytogenetic analysis. CXCR4/SDF-1 interaction is necessary for the migration of neural progenitors in the developing brain (Li and Ransohoff, 2008). The binding of SDF-1 to CXCR4 can lead to the activation of multiple signaling pathways in the CNS, such as phosphatidylinositol 3-kinase-AKT, mitogen-activated protein kinase, and nuclear factor κB (Li and Ransohoff, 2008). Migration is a complex process involving cooperative signaling events. SDF-1/CXCR4 has been shown to play a critical role in NSC migration through activation of p38 MAPK, PAXILLIN, and cJUN (Imitola et al., 2004). These reports corroborate our data demonstrating that overexpression of CXCR4 endows increased migration ability to the HB1F3.CD cells.
MGs are known to secrete SDF-1, and radiation therapy further increases SDF-1 levels (Bajetto et al., 2006, Balyasnikova et al., 2014, Wang et al., 2013). Thus, the CXCR4/SDF-1 axis may be essential for increased NSC motility toward GBM. Indeed, after radiation animals treated with either HNSCs-OVs or CXCR4NSC-OVs demonstrated higher presence of viral protein in the tumor vicinity and survived significantly longer than animals treated with NNSCs-OVs or VCNSCs-OVs. As radiation therapy is an integral part of the current treatment regimen for MG, the strategy of enhancing the chemotactic properties of NSCs via modulation of CXCR4 expression should be further explored in various therapeutic settings. We envision that an ideal clinical timing for intranasal oncolytic therapy via CXCR4NSCs would be following standard radiotherapy. The non-invasive nature of intranasal delivery would allow for repeated treatments. CXCR4NSCs can be prepared in large quantities for clinical use, circumventing the logistically challenging hypoxia pretreatment.
Given the high translational value of this approach, we urge caution regarding the limitations of preclinical rodent models. It is well known that normal and cancerous mouse tissues are poorly permissive for OVs, and CRAds are rapidly cleared in immunocompetent hosts, rendering the immunocompetent mouse unsuitable for CRAd studies. Although both cotton rat and Syrian hamster are CRAd-permissive animal models with intact immune systems, no glioma models are currently available for studies in these animals (Ahmed et al., 2010). Our previous studies in the cotton rat model have demonstrated the improved dissemination and persistence of OV by autologous stem cells, which efficiently shielded virus and suppressed anti-CRAd immune response, allowing us to project efficient entry and propagation of CRAds in glioma cells in immunocompetent hosts (Ahmed et al., 2010). Furthermore, NSCs can successfully disseminate OV in the tumor environment (Morshed et al., 2015), which results in superior therapeutic outcome in comparison with CRAd virotherapy alone (Ahmed et al., 2013). Lastly, the immune response against OV in immunocompetent hosts (Jiang et al., 2014) may assist in the bystander anti-glioma activity of the proposed therapy.
Finally, we recognize that rodent models for intranasal delivery of NSCs have intrinsic limitations, such as the relative size and anatomical differences in the nasal and olfactory systems between humans and mice. Despite differences in the anatomy of the nasal cavity, larynx, and pharyngeal details (Harkema et al., 2006), the fundamental structures and layouts of the olfactory systems are well preserved across vertebrate species (Ache, 2010), offering a compelling basis for extrapolating basic cellular and molecular phenomena from rodent models to human scenarios. Unlike rodents, humans do not rely on olfactory sensation for survival and have smaller olfactory systems (3% of the nasal epithelium is olfactory) (Harkema et al., 2006) compared with rodents (∼50%) (Gross et al., 1982), which may imply that intranasal delivery would be a less effective route of NSC entry in humans. However, the rodent olfactory nasal epithelium surface area ranges between 125.5 ± 4.0 and 136.9 ± 7.3 mm2 in adult mice (Gross et al., 1982), whereas humans have about 500 mm2 on average (Harkema et al., 2006), which might offer more entry access for NSCs. In addition, trigeminal nerves offer additional entry routes for NSCs into the brain (Dhuria et al., 2010). Furthermore, we will be able to achieve more precise delivery of NSCs to the cribriform plate using catheter-based delivery. However, to truly understand the effectiveness of stem cell penetration to the brain from the nasal cavity, studies in large animal models such as monkey should be conducted.
Our study demonstrates that for intranasal delivery of NSCs-OV to be efficacious, hypoxia preconditioning or genetically engineering NSCs to overexpress CXCR4 is required to further enhance the penetration of NSCs into the brain. This corroborates our previous observation that direct intratumoral delivery of NSCs loaded with OVs generates a more potent response than a distant site of delivery (Ahmed et al., 2013) due to the lower number of NCSs reaching the tumor from a distant site. Although both intracavitary and intrathecal delivery of NSCs in the context of brain tumor are clinically relevant, they are invasive and do not lend themselves to multiple dosing. The restrictions of systemic delivery of therapeutic agents for brain tumor have been well documented in the literature, with a major limitation being the phenomenon of first-pass clearance (Lesniak and Brem, 2004). For similar reasons, we did not consider the systemic route of delivery as viable for NSC-based OV therapy due to the fast elimination of NSCs from systemic circulation (Aboody et al., 2006), high uptake of CRAd by liver tissue (Waddington et al., 2008), and potential hepatotoxicity. Intranasal delivery is an attractive alternative due to its non-invasive and repeatable nature and its potential for targeting inoperable brain malignancies, such as multiple brain metastases or invasive tumors. We hypothesize that CXCR4 enhancement would be beneficial to all applicable routes of delivery, given that the SDF-1 chemotaxis gradient derived from tumor growth and heightened by radiotherapy may be established across the CNS. Therefore, the development of strategies enhancing the number of therapeutic NSCs arriving at the brain tumor using the intranasal route should be further pursued.
In summary, the current report builds on our previous validation of the intranasal delivery of stem cells as a feasible route for anti-glioma therapy. Here we show that NSCs can be repeatedly delivered intranasally to extend the survival benefits of oncolytic virotherapy against glioma in animal models. We identified that hypoxia pretreatment of NSCs enhances the migratory potential of NSCs via the SDF-1/CXCR4 signaling axis. Furthermore, NSCs genetically modified to overexpress CXCR4 provide efficient delivery of the OV payload to glioma, resulting in significant survival benefits in conjunction with radiotherapy. The strategy of intranasal delivery of various therapeutics by potent stem cell carriers should be further evaluated in the context of brain malignancies for future transition to the clinic.
Experimental Procedures
See also Supplemental Experimental Procedures.
Cell Culture and Reagents
U87MG cells and human NSCs (HB1.F3.CD) were maintained in DMEM (Corning) with 10% fetal bovine serum (FBS; Hyclone). Patient-derived GBM43 and GBM6 glioma cells were routinely maintained as mouse flank tumors, and were subsequently isolated and maintained in 10% FBS (Hyclone) DMEM (Sarkaria et al., 2006) or in serum-free neurobasal medium supplemented with B27, N2, basic fibroblast growth factor (20 ng/mL), and epidermal growth factor (20 ng/mL; all reagents from Life Technologies). CXCR4-overexpressing NSCs (CXCR4NSCs) were generated by transduction of HB1.F3.CD cells with lentivirus encoding for CXCR4 cDNA (GenTarget), along with vector-only control NSCs (VCNSCs) from the same passage of NSCs.
Hypoxic Culture
NSCs were cultured in a humidified hypoxic gas flow chamber at 37°C (Coy Laboratory Products) with sustained 1% O2 and 5% CO2 for 24 hr (HNSCs). Control NSCs were maintained under the same culture condition but at atmospheric O2 levels (NNSCs).
Loading of NSCs with Oncolytic Virus
The generation of replication competent adenoviral vector CRAd-S-pk7, which contains the wild-type adenovirus replication protein E1A under the control of human survivin promoter, has been previously described (Ulasov et al., 2007). For analysis of viral replication and progeny release, NNSCs, HNSCs, VCNSCs, and CXCR4NSCs were infected with 50 CRAd-S-pk7 particles/cell. After 1 hr of incubation, cells were washed three times to remove unbound viral particles and grown in fresh complete growth medium. After 72 hr, cells and medium were collected and freeze-thawed three times to release infectious progeny. Lysates were cleared by centrifugation and titrated over adherent HEK293 cells with 10-fold dilution. After 48 hr, cells were fixed and stained for viral hexon protein detection using an Adeno-X Rapid Titer Kit (Clontech). The titration (i.e., infectious) units were calculated to determine the viral progeny release.
Patient-Derived Xenograft Mouse Model of GBM
All protocols were approved by the Institutional Committee on Animal Use at The University of Chicago. The surgical procedures were conducted according to the NIH guidelines on the care and use of laboratory animals. Six- to eight-week-old athymic nude male mice were obtained from Harlan Laboratories and maintained in a pathogen-free facility. Mice were anesthetized with ketamine-HCl (25 mg/mL)/xylazine (2.5 mg/mL) solution, and GBM43 or GBM6 cells were injected in the mouse brain at 2 × 104 or 1 × 105 cells in 2.5 μL of sterile saline, respectively, using the established procedure (Ahmed et al., 2013). Standard post-surgery care was given following a protocol approved by the Institutional Animal Care and Use Committee.
Radiation Therapy and Intranasal Stem Cell Therapy
Select groups of glioma xenograft mouse models underwent XRT as detailed in the Results section. Specifically, after ketamine and xylazine anesthesia, the entire body of animals excluding the head was shielded with lead and was exposed to a 2-Gy daily dose of radiation for five consecutive days starting on the eighth day after tumor cell xenograft (days 8–12). Seven days following the first XRT treatment and weekly thereafter (days 14, 21, and 28), animals were treated either with sterile saline or NSCs loaded with OV (see below). In brief, after anesthesia the mice were inoculated with 5 × 105 NSCs in 12 μL of sterile saline (given as three fractions at 5-min intervals) via the nasal cavity as previously described with small modifications (Balyasnikova et al., 2014). All mice were followed to assess their survival. Upon appearance of signs of morbidity, mice were euthanized and whole brain tissues were excised immediately or after intracardiac perfusion with PBS or 4% paraformaldehyde, and embedded in OCT compound (Sakura Finetek). Sections (10–20 μm) were cut, air dried, and stained with H&E or processed for SDF-1 or hexon fluorescence staining as detailed under Immunocytochemistry in Supplemental Experimental Procedures.
Statistical Analysis
All statistical analyses were performed with Student's t test using GraphPad Prism 4 (GraphPad Software) unless specified otherwise. The sample size for each group was ≥3, where n represents biological replicates unless otherwise indicated. All numerical data are reported as means ± SEM. A Kaplan-Meier survival curve was generated and a log-rank test was applied to compare survival distributions. For all survival experiments, n represents the number of animals per group. All reported p values were two-sided and were considered to be statistically significant at ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Author Contributions
M.D. and D.Y.: conception and design, collection and/or assembly of data, data analysis and interpretation, and manuscript writing. D.K.: collection and/or assembly of data, and data analysis and interpretation. G.L., M.S., D.A.S., K.C.P., and Y.H.: collection and/or assembly of data. L.Z.: data analysis and interpretation. A.U.A.: data analysis and interpretation and manuscript writing. K.S.A.: provision of study material. M.S.L.: conception and design, financial support, and data analysis and interpretation. I.V.B.: conception and design, financial support, data analysis and interpretation, manuscript writing, and final approval of manuscript.
Acknowledgments
This work was supported by NIH R01NS087990 (M.S.L., I.V.B.) and U01NS069997 (M.S.L.), and NIH/NRCDP K-12 award (M.D.). We thank Dr. Meijing Wu for her contributions to data analysis. We are grateful to Dr. C. David James for providing patient-derived GBM6 and GBM43 cell lines. K.S.A. is an uncompensated Board Member, Chief Scientific Officer, and shareholder of TheraBiologics.
Published: September 1, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.07.024.
Supplemental Information
References
- Aboody K.S., Brown A., Rainov N.G., Bower K.A., Liu S., Yang W., Small J.E., Herrlinger U., Ourednik V., Black P.M. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA. 2000;97:12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboody K.S., Bush R.A., Garcia E., Metz M.Z., Najbauer J., Justus K.A., Phelps D.A., Remack J.S., Yoon K.J., Gillespie S. Development of a tumor-selective approach to treat metastatic cancer. PLoS One. 2006;1:e23. doi: 10.1371/journal.pone.0000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ache B.W. Odorant-specific modes of signaling in mammalian olfaction. Chem. Senses. 2010;35:533–539. doi: 10.1093/chemse/bjq045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed A.U., Rolle C.E., Tyler M.A., Han Y., Sengupta S., Wainwright D.A., Balyasnikova I.V., Ulasov I.V., Lesniak M.S. Bone marrow mesenchymal stem cells loaded with an oncolytic adenovirus suppress the anti-adenoviral immune response in the cotton rat model. Mol. Ther. 2010;18:1846–1856. doi: 10.1038/mt.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed A.U., Thaci B., Alexiades N.G., Han Y., Qian S., Liu F., Balyasnikova I.V., Ulasov I.Y., Aboody K.S., Lesniak M.S. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol. Ther. 2011;19:1714–1726. doi: 10.1038/mt.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed A.U., Tyler M.A., Thaci B., Alexiades N.G., Han Y., Ulasov I.V., Lesniak M.S. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol. Pharm. 2011;8:1559–1572. doi: 10.1021/mp200161f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed A.U., Thaci B., Tobias A.L., Auffinger B., Zhang L., Cheng Y., Kim C.K., Yunis C., Han Y., Alexiades N.G. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J. Natl. Cancer Inst. 2013;105:968–977. doi: 10.1093/jnci/djt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade C. Intranasal drug delivery in neuropsychiatry: focus on intranasal ketamine for refractory depression. J. Clin. Psychiatry. 2015;76:e628–e631. doi: 10.4088/JCP.15f10026. [DOI] [PubMed] [Google Scholar]
- Auffinger B., Tobias A.L., Han Y., Lee G., Guo D., Dey M., Lesniak M.S., Ahmed A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014;21:1119–1131. doi: 10.1038/cdd.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajetto A., Barbieri F., Dorcaratto A., Barbero S., Daga A., Porcile C., Ravetti J.L., Zona G., Spaziante R., Corte G. Expression of CXC chemokine receptors 1-5 and their ligands in human glioma tissues: role of CXCR4 and SDF1 in glioma cell proliferation and migration. Neurochem. Int. 2006;49:423–432. doi: 10.1016/j.neuint.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Balyasnikova I.V., Prasol M.S., Ferguson S.D., Han Y., Ahmed A.U., Gutova M., Tobias A.L., Mustafi D., Rincon E., Zhang L. Intranasal delivery of mesenchymal stem cells significantly extends survival of irradiated mice with experimental brain tumors. Mol. Ther. 2014;22:140–148. doi: 10.1038/mt.2013.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriga E.H., Maxwell P.H., Reyes A.E., Mayor R. The hypoxia factor Hif-1alpha controls neural crest chemotaxis and epithelial to mesenchymal transition. J. Cell Biol. 2013;201:759–776. doi: 10.1083/jcb.201212100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman C.D., Frey W.H., 2nd, Craft S., Danielyan L., Hallschmid M., Schioth H.B., Benedict C. Intranasal treatment of central nervous system dysfunction in humans. Pharm. Res. 2013;30:2475–2484. doi: 10.1007/s11095-012-0915-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Li M., Cheng H., Yan Z., Cao J., Pan B., Sang W., Wu Q., Zeng L., Li Z. Overexpression of the mesenchymal stem cell Cxcr4 gene in irradiated mice increases the homing capacity of these cells. Cell Biochem. Biophys. 2013;67:1181–1191. doi: 10.1007/s12013-013-9632-6. [DOI] [PubMed] [Google Scholar]
- Dahan P., Martinez Gala J., Delmas C., Monferran S., Malric L., Zentkowski D., Lubrano V., Toulas C., Cohen-Jonathan Moyal E., Lemarie A. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: possible involvement in radioresistance. Cell Death Dis. 2014;5:e1543. doi: 10.1038/cddis.2014.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielyan L., Schafer R., von Ameln-Mayerhofer A., Bernhard F., Verleysdonk S., Buadze M., Lourhmati A., Klopfer T., Schaumann F., Schmid B. Therapeutic efficacy of intranasally delivered mesenchymal stem cells in a rat model of Parkinson disease. Rejuvenation Res. 2011;14:3–16. doi: 10.1089/rej.2010.1130. [DOI] [PubMed] [Google Scholar]
- Decleves X., Amiel A., Delattre J.Y., Scherrmann J.M. Role of ABC transporters in the chemoresistance of human gliomas. Curr. Cancer Drug Targets. 2006;6:433–445. doi: 10.2174/156800906777723930. [DOI] [PubMed] [Google Scholar]
- Dey M., Chang A.L., Wainwright D.A., Ahmed A.U., Han Y., Balyasnikova I.V., Lesniak M.S. Heme oxygenase-1 protects regulatory T cells from hypoxia-induced cellular stress in an experimental mouse brain tumor model. J. Neuroimmunol. 2014;266:33–42. doi: 10.1016/j.jneuroim.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhuria S.V., Hanson L.R., Frey W.H., 2nd Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J. Pharm. Sci. 2010;99:1654–1673. doi: 10.1002/jps.21924. [DOI] [PubMed] [Google Scholar]
- Elnaggar Y.S., Etman S.M., Abdelmonsif D.A., Abdallah O.Y. Intranasal piperine-loaded chitosan nanoparticles as brain-targeted therapy in Alzheimer's disease: optimization, biological efficacy, and potential toxicity. J. Pharm. Sci. 2015;104:3544–3556. doi: 10.1002/jps.24557. [DOI] [PubMed] [Google Scholar]
- Feng C., Zhang C., Shao X., Liu Q., Qian Y., Feng L., Chen J., Zha Y., Zhang Q., Jiang X. Enhancement of nose-to-brain delivery of basic fibroblast growth factor for improving rat memory impairments induced by co-injection of beta-amyloid and ibotenic acid into the bilateral hippocampus. Int. J. Pharm. 2012;423:226–234. doi: 10.1016/j.ijpharm.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Gross E.A., Swenberg J.A., Fields S., Popp J.A. Comparative morphometry of the nasal cavity in rats and mice. J. Anat. 1982;135:83–88. [PMC free article] [PubMed] [Google Scholar]
- Guan G., Zhang Y., Lu Y., Liu L., Shi D., Wen Y., Yang L., Ma Q., Liu T., Zhu X. The HIF-1alpha/CXCR4 pathway supports hypoxia-induced metastasis of human osteosarcoma cells. Cancer Lett. 2015;357:254–264. doi: 10.1016/j.canlet.2014.11.034. [DOI] [PubMed] [Google Scholar]
- Guo M., Cai C., Zhao G., Qiu X., Zhao H., Ma Q., Tian L., Li X., Hu Y., Liao B. Hypoxia promotes migration and induces CXCR4 expression via HIF-1alpha activation in human osteosarcoma. PLoS One. 2014;9:e90518. doi: 10.1371/journal.pone.0090518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutova M., Shahmanyan D., Oganesyan D., Abramyants Y., Danielyan L., Frey W.H., II, Khankaldyyan V., Najbauer J., Balyasnikova I.V., Moats R.A. Intranasal delivery of therapeutic neural stem cells to target intracerebral glioma. Enliven J. Stem Cell Res. Regen. Med. 2015;1 007. [Google Scholar]
- Harkema J.R., Carey S.A., Wagner J.G. The nose revisited: a brief review of the comparative structure, function, and toxicologic pathology of the nasal epithelium. Toxicol. Pathol. 2006;34:252–269. doi: 10.1080/01926230600713475. [DOI] [PubMed] [Google Scholar]
- Imitola J., Raddassi K., Park K.I., Mueller F.J., Nieto M., Teng Y.D., Frenkel D., Li J., Sidman R.L., Walsh C.A. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc. Natl. Acad. Sci. USA. 2004;101:18117–18122. doi: 10.1073/pnas.0408258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Clise-Dwyer K., Ruisaard K.E., Fan X., Tian W., Gumin J., Lamfers M.L., Kleijn A., Lang F.F., Yung W.K. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS One. 2014;9:e97407. doi: 10.1371/journal.pone.0097407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Li Y., Liu X. Intranasal delivery: circumventing the iron curtain to treat neurological disorders. Expert Opin. Drug Deliv. 2015;12:1717–1725. doi: 10.1517/17425247.2015.1065812. [DOI] [PubMed] [Google Scholar]
- Kanojia D., Balyasnikova I.V., Morshed R.A., Frank R.T., Yu D., Zhang L., Spencer D.A., Kim J.W., Han Y., Yu D. Neural stem cells secreting anti-HER2 antibody improve survival in a preclinical model of HER2 overexpressing breast cancer brain metastases. Stem Cells. 2015;33:2985–2994. doi: 10.1002/stem.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keunen O., Johansson M., Oudin A., Sanzey M., Rahim S.A., Fack F., Thorsen F., Taxt T., Bartos M., Jirik R. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA. 2011;108:3749–3754. doi: 10.1073/pnas.1014480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I.D., Shin J.H., Kim S.W., Choi S., Ahn J., Han P.L., Park J.S., Lee J.K. Intranasal delivery of HMGB1 siRNA confers target gene knockdown and robust neuroprotection in the postischemic brain. Mol. Ther. 2012;20:829–839. doi: 10.1038/mt.2011.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesniak M.S., Brem H. Targeted therapy for brain tumours. Nat. Rev. Drug Discov. 2004;3:499–508. doi: 10.1038/nrd1414. [DOI] [PubMed] [Google Scholar]
- Lesniak M.S., Langer R., Brem H. Drug delivery to tumors of the central nervous system. Curr. Neurol. Neurosci. Rep. 2001;1:210–216. doi: 10.1007/s11910-001-0020-z. [DOI] [PubMed] [Google Scholar]
- Li M., Ransohoff R.M. Multiple roles of chemokine CXCL12 in the central nervous system: a migration from immunology to neurobiology. Prog. Neurobiol. 2008;84:116–131. doi: 10.1016/j.pneurobio.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller B.J., Cao Y., Li C.Y., Dewhirst M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5:429–441. doi: 10.1016/s1535-6108(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Morshed R.A., Gutova M., Juliano J., Barish M.E., Hawkins-Daarud A., Oganesyan D., Vazgen K., Yang T., Annala A., Ahmed A.U. Analysis of glioblastoma tumor coverage by oncolytic virus-loaded neural stem cells using MRI-based tracking and histological reconstruction. Cancer Gene Ther. 2015;22:55–61. doi: 10.1038/cgt.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistollato F., Abbadi S., Rampazzo E., Persano L., Della Puppa A., Frasson C., Sarto E., Scienza R., D'Avella D., Basso G. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells. 2010;28:851–862. doi: 10.1002/stem.415. [DOI] [PubMed] [Google Scholar]
- Reitz M., Demestre M., Sedlacik J., Meissner H., Fiehler J., Kim S.U., Westphal M., Schmidt N.O. Intranasal delivery of neural stem/progenitor cells: a noninvasive passage to target intracerebral glioma. Stem Cells Transl. Med. 2012;1:866–873. doi: 10.5966/sctm.2012-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosova I., Dao M., Capoccia B., Link D., Nolta J.A. Hypoxic preconditioning results in increased motility and improved therapeutic potential of human mesenchymal stem cells. Stem Cells. 2008;26:2173–2182. doi: 10.1634/stemcells.2007-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safa A.R., Saadatzadeh M.R., Cohen-Gadol A.A., Pollok K.E., Bijangi-Vishehsaraei K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015;2:152–163. doi: 10.1016/j.gendis.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkaria J.N., Carlson B.L., Schroeder M.A., Grogan P., Brown P.D., Giannini C., Ballman K.V., Kitange G.J., Guha A., Pandita A. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin. Cancer Res. 2006;12:2264–2271. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- Speth J.M., Hoggatt J., Singh P., Pelus L.M. Pharmacologic increase in HIF1alpha enhances hematopoietic stem and progenitor homing and engraftment. Blood. 2014;123:203–207. doi: 10.1182/blood-2013-07-516336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J., Belanger K., Brandes A.A., Marosi C., Bogdahn U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Tian T., Li A., Lu H., Luo R., Zhang M., Li Z. TAZ promotes temozolomide resistance by upregulating MCL-1 in human glioma cells. Biochem. Biophysical Res. Commun. 2015;463:638–643. doi: 10.1016/j.bbrc.2015.05.115. [DOI] [PubMed] [Google Scholar]
- Ulasov I.V., Zhu Z.B., Tyler M.A., Han Y., Rivera A.A., Khramtsov A., Curiel D.T., Lesniak M.S. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum. Gene Ther. 2007;18:589–602. doi: 10.1089/hum.2007.002. [DOI] [PubMed] [Google Scholar]
- van Velthoven C.T., Kavelaars A., van Bel F., Heijnen C.J. Nasal administration of stem cells: a promising novel route to treat neonatal ischemic brain damage. Pediatr. Res. 2010;68:419–422. doi: 10.1203/PDR.0b013e3181f1c289. [DOI] [PubMed] [Google Scholar]
- Vordermark D., Katzer A., Baier K., Kraft P., Flentje M. Cell type-specific association of hypoxia-inducible factor-1 alpha (HIF-1 alpha) protein accumulation and radiobiologic tumor hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2004;58:1242–1250. doi: 10.1016/j.ijrobp.2003.11.030. [DOI] [PubMed] [Google Scholar]
- Waddington S.N., McVey J.H., Bhella D., Parker A.L., Barker K., Atoda H., Pink R., Buckley S.M., Greig J.A., Denby L. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. 2008;132:397–409. doi: 10.1016/j.cell.2008.01.016. [DOI] [PubMed] [Google Scholar]
- Wang S.C., Yu C.F., Hong J.H., Tsai C.S., Chiang C.S. Radiation therapy-induced tumor invasiveness is associated with SDF-1-regulated macrophage mobilization and vasculogenesis. PLoS One. 2013;8:e69182. doi: 10.1371/journal.pone.0069182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womeldorff M., Gillespie D., Jensen R.L. Hypoxia-inducible factor-1 and associated upstream and downstream proteins in the pathophysiology and management of glioblastoma. Neurosurg. Focus. 2014;37:E8. doi: 10.3171/2014.9.focus14496. [DOI] [PubMed] [Google Scholar]
- Yang J.X., Zhang N., Wang H.W., Gao P., Yang Q.P., Wen Q.P. CXCR4 receptor overexpression in mesenchymal stem cells facilitates treatment of acute lung injury in rats. J. Biol. Chem. 2015;290:1994–2006. doi: 10.1074/jbc.M114.605063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Chen D., Zhang Y., Wu X., Huang Z., Zhou H., Zhang Y., Zhang Z. Overexpression of CXCR4 in mesenchymal stem cells promotes migration, neuroprotection and angiogenesis in a rat model of stroke. J. Neurol. Sci. 2012;316:141–149. doi: 10.1016/j.jns.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Zhao D., Najbauer J., Garcia E., Metz M.Z., Gutova M., Glackin C.A., Kim S.U., Aboody K.S. Neural stem cell tropism to glioma: critical role of tumor hypoxia. Mol. Cancer Res. 2008;6:1819–1829. doi: 10.1158/1541-7786.MCR-08-0146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.