

Abstract
The RNA binding protein Human antigen R (HuR) interacts with specific AU-rich domains in target mRNAs and is highly expressed in many cell types, including cardiomyocytes. However, the role of HuR in cardiac physiology is largely unknown. Our results show that HuR undergoes cytoplasmic translocation, indicative of its activation, in hypertrophic cardiac myocytes. Specifically, HuR cytoplasmic translocation is significantly increased in NRVMs (neonatal rat ventricular myocytes) following treatment with phenylephrine or angiotensin II, agonists of two independent Gαq-coupled GPCRs known to induce hypertrophy. This Gq-mediated HuR activation is dependent on p38 MAP kinase, but not canonical Gq-PKC signaling. Furthermore, we show that HuR activation is necessary for Gq-mediated hypertrophic growth of NRVMs as siRNA-mediated knockdown of HuR inhibits hypertrophy as measured by cell size and expression of ANF (atrial natriuretic factor). Additionally, HuR overexpression is sufficient to induce hypertrophic cell growth. To decipher the downstream mechanisms by which HuR translocation promotes cardiomyocyte hypertrophy, we assessed the role of HuR in the transcriptional activity of NFAT (nuclear factor of activated T cells), the activation of which is a hallmark of cardiac hypertrophy. Using an NFAT-luciferase reporter assay, we show an acute inhibition of NFAT transcriptional activity following pharmacological inhibition of HuR. In conclusion, our results identify HuR as a novel mediator of cardiac hypertrophy downstream of the Gq-p38 MAPK pathway, and suggest modulation of NFAT activity as a potential mechanism.
Keywords: HuR, Heart, Hypertrophy, RNA Binding Protein, p38 MAPK
1. Introduction
Human antigen R (HuR) is a widely expressed RNA binding protein that interacts with specific AU-rich domains in target mRNAs and exerts post-transcriptional regulation of target mRNA by a number of means including RNA stability, translation, splicing, polyadenylation, or microRNA targeting.[1-4] While relatively little is known about the role of HuR in the myocardium, RNA binding proteins such as HuR are becoming recognized as potentially central regulators of cardiac physiology and pathology.[5,6] Recent work by Krishnamurthy et al suggests that HuR is likely to play a central role in the cardiac response to stress.[7,8] They showed that HuR expression increased following ischemic injury and knockdown with shRNA delivered via intra-myocardial injection significantly reduced post- infarct remodeling and was accompanied by a decrease in transforming growth factor-β (TGF-β) expression. However, it is unclear from this work whether HuR plays a direct role in cardiac myocytes. Furthermore, this prior work focused on the role of HuR on fibrosis and ventricular remodeling following acute ischemic injury, but its role in the development of cardiac hypertrophy is completely unknown.
The initial development of hypertrophy is a beneficial and compensatory response to maintain cardiac output in the face of hemodynamic stress. However, cardiac hypertrophy in response to pathological etiologies such as hypertension or valvular dysfunction is a known driver of heart failure, and some reports have also questioned its necessity as a compensatory development and suggest a benefit to suppressing the initial development of hypertrophy.[9] Determination of the functional role of HuR in pathological cardiac hypertrophy would represent a significant advancement of our current knowledge of hypertrophic signaling pathways. Thus, the goal of this work is to determine the role that HuR activation in cardiomyocytes plays in hypertrophic signaling. Herein, we demonstrate the activation of HuR in hypertrophic cardiac myocytes via a Gαq-p38 MAPK-dependent signaling pathway. Importantly, this activation of HuR appears to be necessary for hypertrophic cell growth in NRVMs (neonatal rat ventricular myocytes), as siRNA-mediated knockdown or pharmacological inhibition of HuR prevents hypertrophic cell growth and activation of the pro-hypertrophic transcription factor NFAT (nuclear factor of activated T cells). In addition, HuR overexpression alone is sufficient to induce NRVM hypertrophy. Thus, these results demonstrate for the first time that HuR is necessary and sufficient to induce hypertrophic signaling in cardiac myocytes.
2. Methods
2.1 Neonatal Rat Ventricular Myocyte Isolation and Cell Culture
NRVMs were isolated using collagenase digestion and adhesion differential from fibroblasts as described.[10] Briefly, Sprague Dawley neonatal rats (1-2 days old) (Taconic) were decapitated and the hearts were isolated. Following removal of the atria, the ventricles were cut into small pieces and digested first in .05% trypsin/EDTA (Corning) overnight, then in collagenase II (Gibco) for 30 minutes. Cells were then spun at 100 × g followed by a 40 minute pre-plating process on non-treated plates to allow the fibroblasts to adhere. The non-adherent NRVMs were then transferred to cell culture-treated dishes in MEM alpha media (Gibco) with 10% FBS.
The study was performed under protocol #13-08-29-01, which has been approved by the University of Cincinnati Institutional Animal Care and Use Committee, and the animals received humane care in compliance with the National Research Council's criteria as outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Institutes of Health.
2.2 HuR siRNA-mediated gene silencing and overexpression
To achieve siRNA-mediated knockdown of HuR expression, NRVMs were seeded at ∼75% confluency and transfected with HuR or non-targeting control siRNA (80 nM) (Santa Cruz Biotechnology) 24 hours after plating using Lipofectamine 3000 (ThermoFisher Scientific) as per manufacturer's instructions. Cells were grown for 48 hours post-transfection prior to treatment with phenylephrine (PE). To achieve HuR overexpression, the full-length HuR coding region was cloned from mouse cDNA via PCR and inserted into a modified pGL4.1 expression vector driven by a constitutively active CMV promoter. NRVMs were seeded at ∼75% confluency and transfected with either HuR overexpression vector or equal amounts of a control vector (coding for overexpression of luciferase). Cells were grown for 24 hours post-transfection prior to treatment with PE. HuR knockdown (>80%) and overexpression (∼5-fold, Fig. S1) was confirmed via Western blotting.
2.3 RNA Isolation and qRT-PCR
RNA was isolated using a Macherey-Nagel NucleoSpin RNA kit and cDNA was synthesized using a BioScript All-in-One cDNA Synthesis SuperMix (Biotool). Samples were run on Stratagene Mx3005P (Agilent Technologies) using SYBR Green qPCR Master Mix (Biotool) to assess levels of GAPDH, ANF (atrial natriuretic factor), and RCAN1 (Regulator of Calcineurin 1). Results were analyzed using the ΔΔCt method.[11] Primers are as listed: GAPDH, F, 5′-ACCACAGTCCATGCCATCAC-3′, R, 5′-TCCACCACCCTGTTGCTGTA-3′; ANF, F, 5′-AGGAGAAGATGCCGGTAG-3′, R, 5′-GCTTTTCAAGAGGGCAGA-3′; RCAN, F, 5′-GGGCCAAATTTGAATCCCTCTTC-3′, R, 5′-GGAGCCAGGTGTGAACTTCC-3′.
2.4 Protein Isolation and Western Blotting
Total protein was isolated from in vitro cell cultures using a solution of 10 mM HEPES, (pH 9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.2 mM sodium-orthovanadate, and a protease inhibitor mixture tablet (Complete mini; Roche Applied Science). 25 ug of protein extract per lane was separated on a 10% polyacrylamide gel and transferred to a nitrocellulose membrane. Blocking was performed for 1 hour at room temperature using 5% dry milk in 0.1% Tween 20, tris-buffered saline (T-TBS). Primary antibodies for HuR or GAPDH (Santa Cruz Biotechnology) were incubated overnight at 4 °C, and secondary antibodies were incubated for 1–2 h at room temperature in T-TBS.
2.5 Immunohistochemistry, Wheat Germ Agglutinin Staining, and Microscopy
NRVMs were grown on coverslips (pre coated with 0.2% gelatin) in 12-well plates, and treated with PE (10 μM), angiotensin II (AngII, 100 nM), PMA (2 μM), SB203580 (10 μM), or chelerythrine (10 μM) for the indicated amount of time. Following treatment, cells were fixed with 4% paraformaldehyde for 15 minutes, followed by permeabilization with 100% methanol for 15 minutes, dehydration with 70% ethanol for 15 minutes and blocking with 6% bovine serum albumin (BSA) for 1 hour at RT. Cells were incubated in primary antibody for HuR (1:500) for 1 hour at RT followed by secondary antibody for Alexa Fluor 488 (1:2000) (ThermoFisher) for 1 hour at RT. All antibodies were made up in 0.6% BSA in PBS. For wheat germ agglutinin (WGA) staining, a Texas Red-X conjugate was used per manufacturer's instructions (ThermoFisher). All slides were imaged with the BioTek Cytation 3 image reader and quantitative assessment of HuR translocation was measured as the ratio of cytoplasmic to nuclear fluorescent intensity (adjusted for mean background fluorescence and integrated area) using ImageJ
2.6 NFAT-Luciferase Reporter Assays
NRVMs were transfected with 75 ng per well (in a 96-well plate) of NFAT-luciferase reporter plasmid acquired from AddGene (51941).[12] 48 hours after transfection, cells were treated with 10uM PE for 24 hours to induce NFAT reporter activity. To quantify luciferase reporter expression, cells were rinsed with sterile PBS and then lysed with 50 μL Cell Lysis Buffer (Promega) for 5 min at room temperature, followed by addition of 100 μL luciferase assay reagent (Promega); luminescence was read immediately on the BioTek Cytation 3 image reader. HuR inhibitors were previously identified by Wu et. al., and used here as described.[13]
2.7 Statistical Analysis
All results represent an N of at least 3 per group and are reported as the mean ± SEM. Results were analyzed with unpaired Student's t-tests and one-way ANOVA as appropriate. Statistical significance between groups was considered at P ≤ 0.05.
3. Results
3.1 HuR is activated in hypertrophic neonatal rat ventricular myocytes (NRVMs)
HuR activation is regulated by phosphorylation, and activation is commonly marked by translocation from the nucleus to cytoplasm.[2] Our results show a significant increase in HuR translocation to the cytoplasm in hypertrophic myocytes (Fig. 1).
Figure 1.
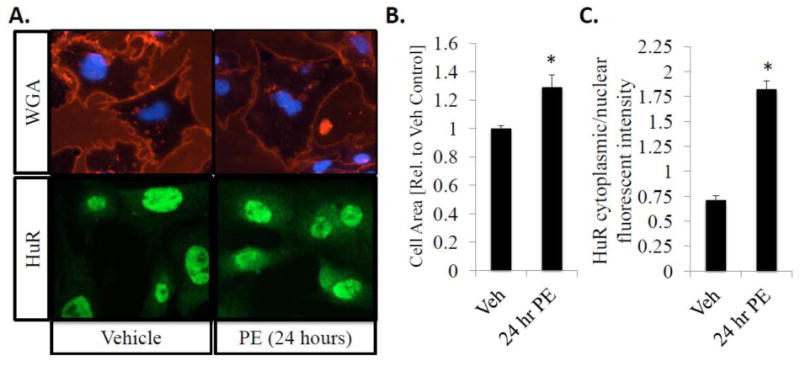
HuR nucleo-cytoplasmic shuttling correlates with an increase in cell size/hypertrophic growth in neonatal rat ventricular myocytes (NRVMs). Following a 24-hour exposure to PE (10 μM), NRVMs show a significant increase in cell area as indicated by WGA staining (A, top panel) and cytoplasmic translocation of HuR as determined by HuR IHC (A, bottom panel). (B) Cell surface area was quantitatively determined using NIH Image J, and is expressed as fold-increase in area compared to vehicle control treated cells. (C) HuR translocation was quantified as the ratio of cytoplasmic to nuclear fluorescent intensity. N ≥ 4 for each group (each N represents the average measurement of 10 cells per well). *P ≤ 0.05 vs. Veh control.
First, we demonstrate an increase in total cell area indicative of hypertrophic cell growth following 24 hours of treatment with 10 μM phenylephrine using wheat germ agglutinin (WGA) staining (1.29 ± .08-fold increase in cell area in 24 hr PE-treated cells compared to vehicle treated control cells; P<0.01) (Fig. 1A; top panel and 1B). Next, we show an increase in HuR translocation in hypertrophic NRVMs. HuR nuclear-cytoplasmic translocation was quantified following immunostaining by measuring the ratio of cytoplasmic to nuclear fluorescent intensity, and is significantly increased in hypertrophic cells (0.71 ± 0.04 in vehicle control vs. 1.82 ± 0.08 in 24 hr PE-treated cells; P<0.001) (Fig. 1A; lower panel and 1C).
3.2 HuR knockdown or pharmacological inhibition reduces cardiomyocyte hypertrophic growth
To determine if HuR is necessary for PE-mediated NRVM hypertrophy, expression of HuR protein was knocked down via transient transfection with HuR siRNA (Fig. 2A; top panel). Results show that while transfection of non-targeting control siRNA had no effect on hypertrophic cell growth, HuR knockdown significantly reduced the PE-mediated increase in cell area as determined via WGA staining (1.23 ± 0.07-fold increase in cell size in non-targeting control siRNA + PE treated cells vs. 0.87 ± 0.05-fold in HuR siRNA + PE-treated cells; P<0.01) (Fig. 2A; bottom panel and 2B). Furthermore, we show that the PE-induced increase in expression of ANF, a common gene marker of cardiac myocyte hypertrophic growth, is completely inhibited by HuR knockdown (1.95 ± 0.3 fold-induction in control siRNA + PE treated cells vs. 0.80 ± 0.35 fold-induction in HuR siRNA + PE-treated cells; P<0.05) (Fig. 2C).
Figure 2.
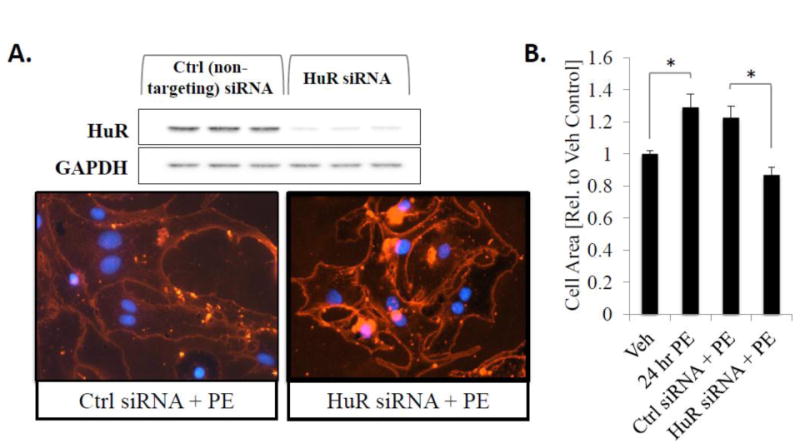
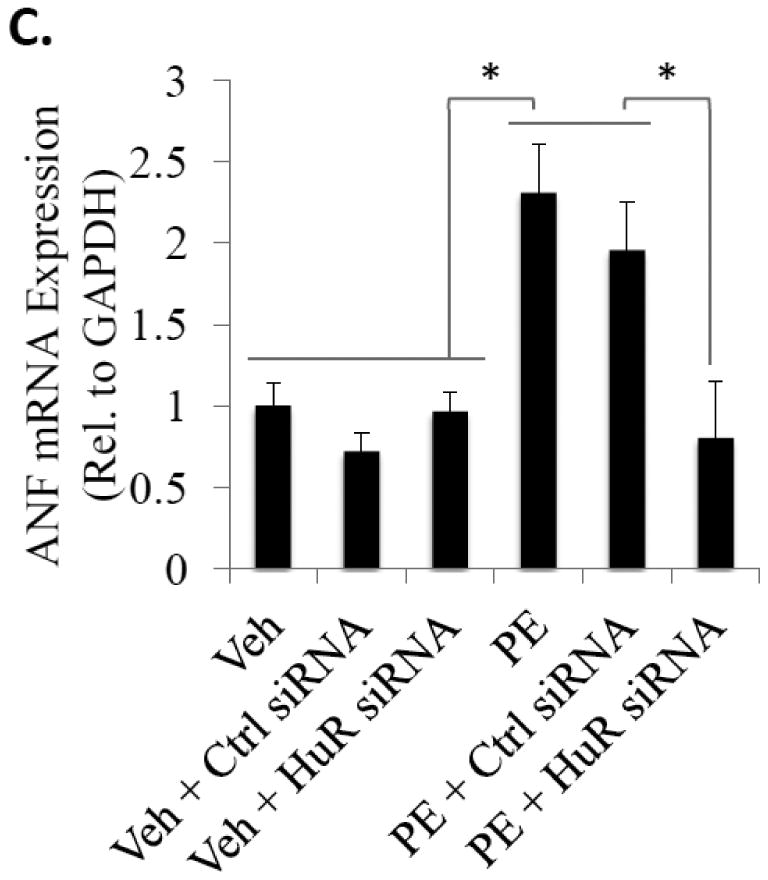
Knockdown of HuR expression inhibits hypertrophic cell growth. (A) NRVMs were transfected with non-targeting control or HuR siRNA 48 hours prior to treatment with PE for 24 hours to induce hypertrophic cell growth and stained with WGA to determine cell size. (B) Cell surface area was quantitatively determined using NIH Image J. N ≥ 3 for each group (each N represents the average measurement of 10 cells per well). *P ≤ 0.05. (C) In addition, RNA was isolated from a subset of cells after 24 hours of PE treatment and expression level of ANF, a hypertrophic marker gene, was assessed by qRT-PCR. N ≥ 6. *P ≤ 0.05.
We also show that a similar reduction in PE-induced hypertrophic growth can be achieved via pharmacological inhibition of HuR using newly developed small-molecule inhibitors of HuR that act through disruption of HuR binding to target mRNA.[13] Simultaneous treatment with the HuR inhibitors CMLD1 and CMLD2 (both at their respective IC50 values) significantly inhibits the increase in PE-induced cell size (1.39 ± 0.07-fold increase in cell size in vehicle + PE treated cells vs. 1.02 ± 0.03-fold in CMLD1/2 + PE-treated cells; P<0.01) (Fig. 3A; bottom panel and 3B). Similar to siRNA-mediated knockdown of HuR, we also show that pharmacological inhibition of HuR reduces PE-induced ANF expression (5.4 ± 1.4 fold-induction in vehicle + PE treated cells vs. 1.3 ± 0.1 fold-induction in CMLD1/2 + PE-treated cells; P<0.05) (Fig. 3C). These results indicate that HuR is necessary for PE-induced NRVM hypertrophy.
Figure 3. Pharmacological inhibition of HuR reduces hypertrophic cell growth and ANF expression.
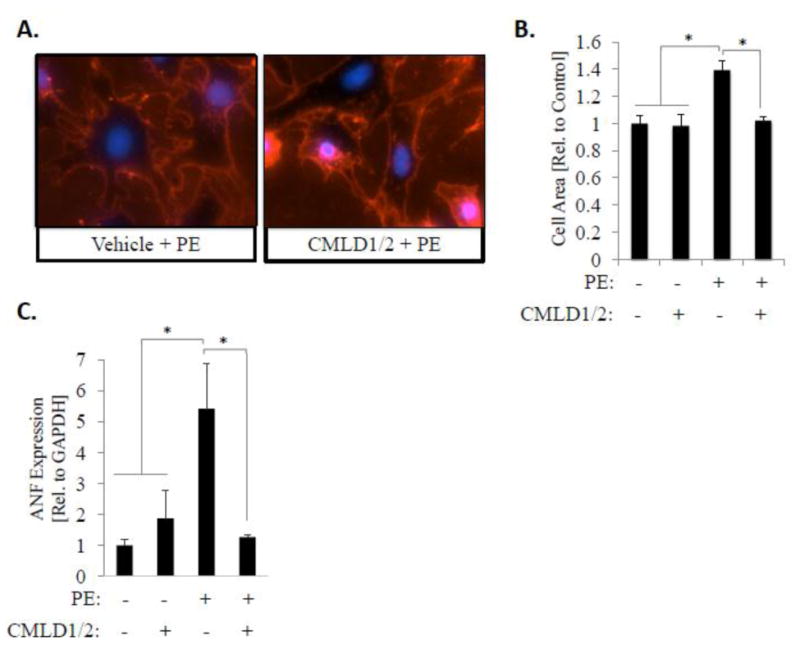
(A) NRVMs were treated simultaneously with PE and two novel small molecule inhibitors of HuR described by Wu et al.[13]. Hypertrophic growth was assessed 24 hours later via WGA staining. (B) Cell surface area was quantitatively determined using NIH Image J. N=6 for each group (each N represents the average measurement of 10 cells per well). *P ≤ 0.05. (C) Total RNA was also isolated from a subset of cells and expression level of ANF was assessed by qRT-PCR. *P ≤ 0.05.
3.3 HuR translocation downstream of Gαq-coupled receptors is dependent on p38 MAPK, but not canonical Gq-PKC signaling
PE induces myocyte hypertrophy downstream of Gαq-coupled α1-adrenergic receptors (α1-ARs), and the heterotrimeric Gq proteins have long been known to be necessary and sufficient for the development of pathological cardiac hypertrophy.[14] Thus, to determine if the signaling pathways by which HuR is activated in cardiac myocytes are downstream of Gαq, we treated NRVMs with agonists of two independent Gαq-coupled receptors: PE (an α1-AR agonist) and AngII (an angiotensin I receptor, AT1, agonist). Indeed, results show a significant cytoplasmic translocation of HuR within 15 minutes after treatment with either PE or AngII (0.71 ± 0.04 in vehicle control vs. 1.05 ± 0.06 in 15 hr PE-treated cells, P<0.001 vs. Ctrl and 1.06 ± 0.04 in 15 hr AngII-treated cells, P<0.001 vs. Ctrl) (Fig. 4).
Figure 4. Nucleo-cytoplasmic shuttling of HuR downstream of AngII and PE is dependent on p38 MAPK, but not PKC.
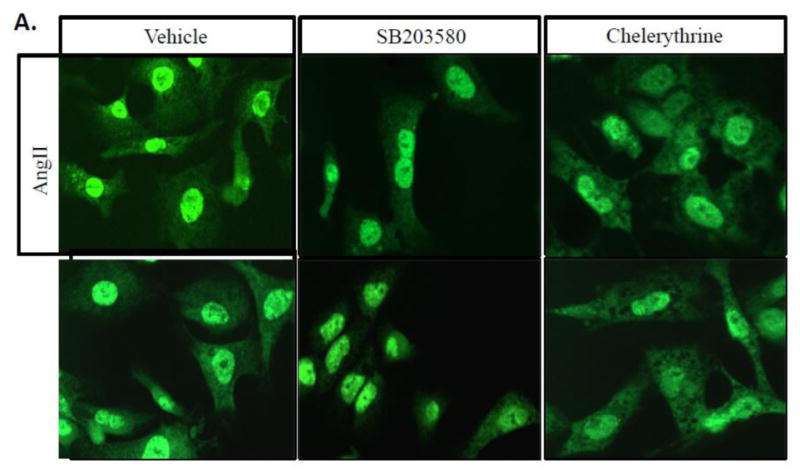
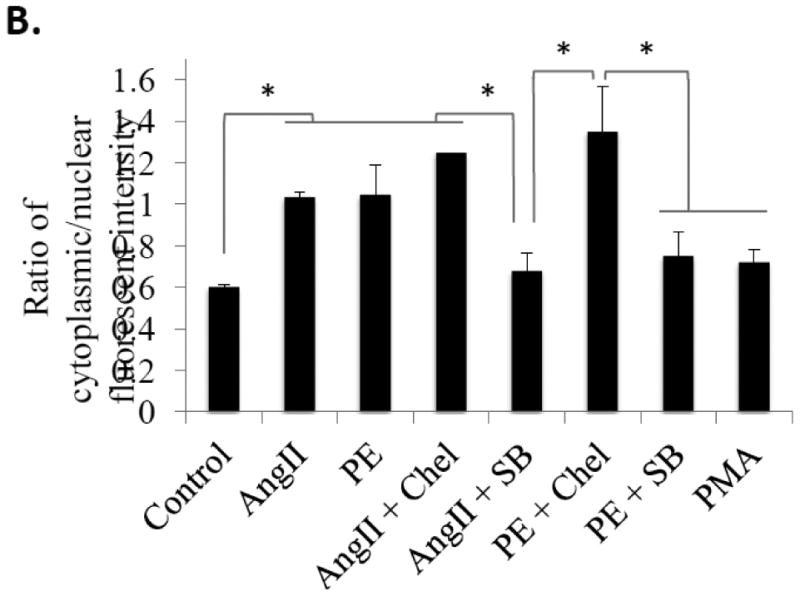
(A) NRVMs treated with AngII (100 nM) or PE (10 μM) show significant cytoplasmic translocation of HuR vs. control (vehicle) cells as measured by IHC 15 minutes after treatment. Treatment with the p38 inhibitor SB203580 shows that this translocation is dependent on p38 MAPK, but treatment with the PKC inhibitor chelerythrine does not affect HuR translocation in myocytes. (B) HuR translocation was quantified as the ratio of cytoplasmic to nuclear cell fluorescent intensity. N ≥ 5 for each group (each N represents the average measurement of 10 cells per well). *P ≤ 0.05.
It has previously been shown that HuR is activated downstream of p38 MAPK and PKC in other tissue types, and both p38 MAPK and PKC are known to signal downstream of Gq proteins and play a role in pathological hypertrophy.[15,16] Thus, we employed pharmacological inhibition of p38 MAPK and PKC (via SB203580 and chelerythrine, respectively) to determine the role of these kinases in PE/AngII-mediated translocation of HuR prior to myocyte hypertrophy. Our results demonstrate that HuR translocation downstream of both PE and AngII is dependent on p38 MAPK, but not PKC (Fig. 4). SB203580-mediated inhibition of p38 MAPK significantly reduced HuR translocation following treatment with either PE (0.68 ± 0.08, P<0.001) or AngII (0.76 ± 0.07, P<0.01) compared to PE alone (1.05 ± 0.06). Given that treatment with chelerythrine had no effect on HuR translocation, proper inhibition of PKC was confirmed by showing a reduction of PKC substrate phosphorylation (Fig. S2). A lack of HuR translocation following application of a direct activator of PKC (PMA, 2 μM) also confirms that PKC activation alone is insufficient to induce HuR translocation in cardiac myocytes (Fig. 4B). These results identify p38 MAPK as an upstream signaling mediator of HuR translocation in hypertrophic cardiac myocytes.
3.4 HuR mediates the activity of the pro-hypertrophic transcription factor NFAT
To begin to decipher the downstream mechanisms by which HuR translocation promotes cardiomyocyte hypertrophy, we assessed the effect of HuR knockdown and inhibition on the transcriptional activity of NFAT, the activation of which is a hallmark of cardiac hypertrophy.[12] First, we show that the PE-induced expression of the NFAT target gene RCAN1, a commonly used downstream gene marker of NFAT activation, is inhibited following siRNA-mediated knockdown of HuR (4.43 ± 0.77 fold-induction in PE treated cells vs. 1.04 ± 0.19 fold-induction in HuR siRNA + PE-treated cells; P<0.05) (Fig. 5A).
Figure 5. HuR knockdown or inhibition inhibits NFAT transcriptional activation and downstream gene expression.
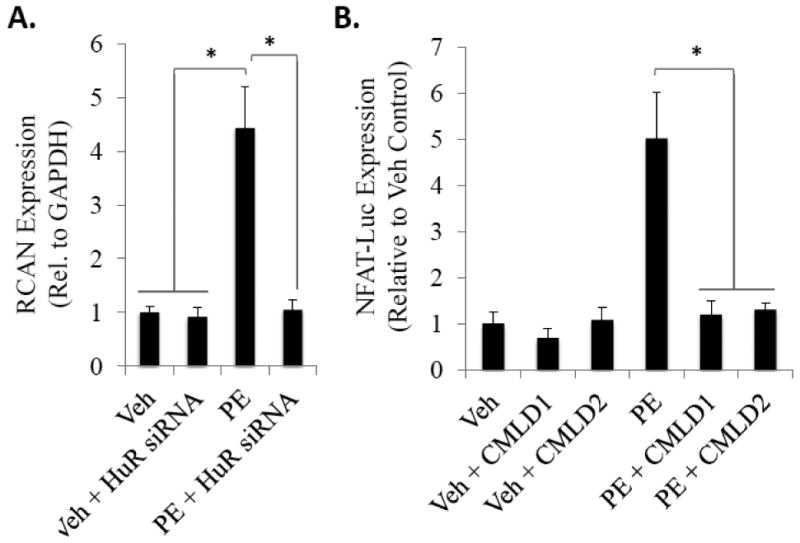
(A) Expression level of the NFAT-dependent gene RCAN was assessed via qRT-PCR in control or PE-treated NRVMs with and without siRNA-mediated HuR knockdown. N ≥ 3. *P ≤ 0.05. (B) NFAT transcriptional activation was directly assessed via transient transfection of NRVMs with an NFAT-luciferase reporter. Consistent with other assessed timepoints of NRVM hypertrophy, luciferase activity was measured 24 hours following treatment with PE and HuR pharmacological inhibition (via CMLD1 and CMLD2). N ≥ 8. *P ≤ 0.05.
Using a luciferase reporter assay to more directly probe NFAT transcriptional activity, we also show a significant inhibition of NFAT reporter activity after a 24-hour exposure to PE using pharmacological inhibition of HuR (Fig. 5B). Treatment with the HuR inhibitors CMLD1 or CMLD2, (each at their determined IC50 values for HuR inhibition of 4.0 μm, 2.4 μm, respectively [13]) has no significant effect on basal NFAT activity. However, when administered simultaneously with PE, CMLD1 and CMLD2 completely inhibited the increase in PE-mediated NFAT reporter activity (1.19 ± 0.30-fold vs. vehicle control in PE + CMLD1 treated and 1.30 ± 0.14 in PE + CMLD2 treated compared to 5.02 ± 0.98 in cells treated with PE alone, both are P<0.001 vs. PE alone) (Fig. 5B). Taken together with the reduction in NFAT-dependent RCAN expression, these results demonstrate that HuR inhibition prevents transcriptional activation of NFAT.
3.5 HuR overexpression is sufficient to drive hypertrophic cell growth
Finally, our results demonstrate that overexpression of HuR resulted in a significant increase in NRVM cell size (1.31 ± 0.03-fold larger than cells treated with a control plasmid, P<0.001) (Fig. 6A and B). In addition, overexpression of HuR was found to induce a 2.2-fold increase in NFAT reporter activity compared to control plasmid (P<0.001) (Fig. 6C). This suggests that not only is HuR necessary for the development of hypertrophic signaling in cardiac myocytes, but also the overexpression of HuR is sufficient to induce hypertrophic signaling.
Figure 6. HuR overexpression is sufficient to induce hypertrophic cell growth and NFAT transcriptional activation.
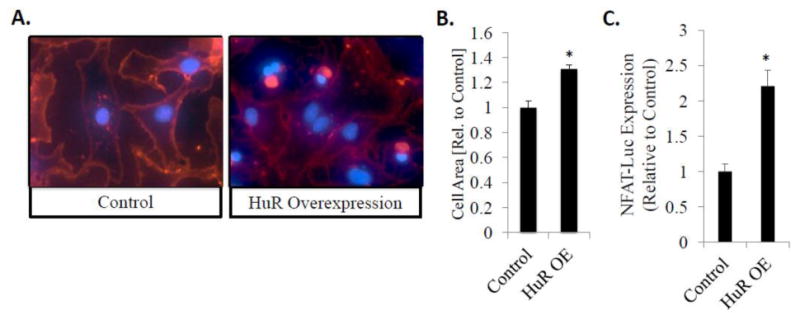
(A) Overexpression of HuR induces hypertrophic cell growth, as observed by an increase in total cell area, compared to treatment with a control plasmid. N ≥ 3. *P ≤ 0.05. (B) Overexpression of HUR is sufficient to induce NFAT-luciferase reporter expression compared to transfection with a control plasmid. N = 8. *P ≤ 0.05.
4. Discussion
This work is the first to show that HuR plays a direct role in cardiomyocyte hypertrophy. Not only does HuR knockdown or pharmacological inhibition prevent myocyte hypertrophy in an established model of phenylephrine-induced NRVM hypertrophy, but HuR overexpression is alone sufficient to induce hypertrophic signaling. To begin to elucidate the mechanisms by which HuR promotes hypertrophy, we also show that cytoplasmic translocation of HuR downstream of two separate Gαq-coupled GPCRs (AT1 and α1-AR) is dependent on p38 MAPK but not PKC. In addition, the downstream mechanisms of HuR in cardiomyocyte hypertrophy appear to be mediated through transcriptional activation of the pro-hypertrophic transcription factor NFAT.
Activation of heterotrimeric Gαq (Gq) proteins downstream of Gq-coupled GPCRs such as angiotensin receptors (AT1) or α1-ARs is a known mediator of pathological hypertrophy.[14,17] Furthermore, cardiac specific overexpression of Gq is sufficient to induce cardiac hypertrophy.[18] Activation of the Gq-protein is observed downstream of nearly every stimulus of pathological hypertrophy, but the specific mechanisms by which its activation results in cardiac hypertrophy have yet to be fully elucidated. Thus, identification of HuR as a key signaling node downstream of Gq activation in the hypertrophic myocyte would represent a significant enhancement in our understanding of Gq-mediated hypertrophy.
Prior work demonstrates that both p38 MAPK and PKC are known to signal downstream of Gq proteins and play a role in pathological cardiac hypertrophy.[15,16] HuR has also been shown to be targeted by both PKC (S158, S221, and S318) and p38 MAPK (T118), with phosphorylation at each of these sites shown to induce HuR translocation and RNA binding activity in other cell types.[19-23] However, these results are the first to show a functional link between Gq signaling pathways and HuR activation via p38 MAPK in cardiac myocytes. Surprisingly, our data suggests that PKC activity does not mediate acute HuR translocation in cardiac myocytes. This was an unexpected result given prior work showing that PKC modulates HuR translocation downstream of AngII in human mesangial cells.[19] Conversely, our results suggest that non-selective inhibition of all PKC isoforms using chelerythrine shows a trend to enhance HuR translocation in myocytes. While our results suggests that PKC does not mediate HuR translocation in cardiac myocytes, it is possible that PKC still plays a role in HuR binding to target mRNA. To this end, Schulz et al showed that the effect of PKC phosphorylation on HuR function is dependent on the specific site of phosphorylation.[24] Specifically, they used phosphor-mimetic mutants to show that only PKC phosphorylation of Ser221 mediates HuR nucleo-cytoplasmic shuttling, while phosphorylation at either Ser158 or Ser318 mediates RNA binding specificity.[24] Future work will be needed to identify the role that specific HuR phosphorylation sites play in myocyte hypertrophy.
Transcriptional activation of NFAT is recognized as a hallmark event in pathological cardiac hypertrophy.[12] Our data shows that HuR knockdown using siRNA inhibits expression of the NFAT-dependent gene RCAN, while pharmacological inhibition of HuR blocks expression of an NFAT-luciferase reporter following phenylephrine. This acute inhibition of NFAT suggests a possible mechanism for reduced hypertrophic signaling following HuR knockdown or inhibition, though a potential direct link between HuR and NFAT remains to be elucidated. Interestingly, HuR overexpression alone is also sufficient to induce NFAT transcriptional activity.
One significant aspect of this work is the application of novel pharmacological inhibitors of HuR recently described by Wu et al (Figures 3 and 5).[13] This is the first application of pharmacological inhibition of HuR in cardiac myocytes, and given the high recent interest in developing a pharmacological inhibitor of HuR for therapeutic use [13,25-27], represents a potential means for long-term translation of these basic science findings. Importantly, HuR knockdown or inhibition in basal/resting NRVMs appears to have very little effect. We confirmed this by performing RNA-sequencing on NRVMs following HuR-knockdown and found that only 24 total transcripts were changed between control and siRNA-treated cells (Supplemental Figure S3). This is significant in that it suggests that HuR plays only a very minor role in healthy, non-stressed cardiac myocytes and that HuR inhibition in these cells has little effect on cell function and/or gene expression. Thus, we believe that this work introduces HuR as a novel myocyte-centric target to manage hypertrophic growth of cardiac myocytes and these findings should be followed up using in vivo models.
5. Conclusions
In conclusion, this work identifies HuR activation downstream of p38 MAPK as a novel mediator of Gq-dependent cardiomyocyte hypertrophy, and suggests modulation of NFAT transcriptional activity as a potential mechanism for HuR-mediated hypertrophy. These results are the first to demonstrate that HuR is necessary and sufficient to induce hypertrophic signaling in cardiac myocytes.
Supplementary Material
Highlights.
Activation (cytoplasmic translocation) of the RNA binding protein HuR is necessary and sufficient for the development of hypertrophy in cardiac myocytes.
HuR translocation downstream of phenylephrine and angiotensin II, two pharmacological agonists of hypertrophy, is dependent on p38 MAPK.
HuR modulation of NFAT transcriptional activation is suggested as a likely mechanism for HuR-dependent hypertrophic signaling.
This work is the first to demonstrate a role for the RNA binding protein HuR in cardiac hypertrophy.
Acknowledgments
Grant Support: This study was supported in part by an American Heart Association Scientist Development Grant (16SDG27360004) to M.T. and National Institutes of Health grants (R01-HL132111, R01-CA178831 and R01-CA191785) to MT., L.X., and J.A., respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Izquierdo JM. Hu antigen R (HuR) functions as an alternative pre-mRNA splicing regulator of Fas apoptosis-promoting receptor on exon definition. J Biol Chem. 2008;283:19077–19084. doi: 10.1074/jbc.M800017200. [DOI] [PubMed] [Google Scholar]
- 2.Doller A, Pfeilschifter J, Eberhardt W. Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell Signal. 2008;20:2165–2173. doi: 10.1016/j.cellsig.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 4.Durie D, Lewis SM, Liwak U, Kisilewicz M, Gorospe M, Holcik M. RNA-binding protein HuR mediates cytoprotection through stimulation of XIAP translation. Oncogene. 2011;30:1460–1469. doi: 10.1038/onc.2010.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajasingh J. The many facets of RNA-binding protein HuR. Trends in Cardiovascular Medicine. 2015:1–3. doi: 10.1016/j.tcm.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Babu SS, Joladarashi D, Jeyabal P, Thandavarayan RA, Krishnamurthy P. RNA-stabilizing proteins as molecular targets in cardiovascular pathologies. Trends in Cardiovascular Medicine. 2015:1–8. doi: 10.1016/j.tcm.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R. IL-10 Inhibits Inflammation and Attenuates Left Ventricular Remodeling After Myocardial Infarction via Activation of STAT3 and Suppression of HuR. Circ Res. 2009;104:e9–e18. doi: 10.1161/CIRCRESAHA.108.188243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krishnamurthy P, Lambers E, Verma S, Thorne T, Qin G, Losordo DW, et al. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. The FASEB Journal. 2010;24:2484–2494. doi: 10.1096/fj.09-149815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 10.Ehler E, Moore-Morris T, Lange S. Isolation and culture of neonatal mouse cardiomyocytes. J Vis Exp. 2013:e50154–e50154. doi: 10.3791/50154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 12.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 13.Wu X, Lan L, Wilson DM, Marquez RT, Tsao WC, Gao P, et al. Identification and Validation of Novel Small Molecule Disruptors of HuR-mRNA Interaction. ACS Chem Biol. 2015:150317154550000–9. doi: 10.1021/cb500851u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams JW, Brown JH. G-proteins in growth and apoptosis: lessons from the heart. Oncogene. 2001;20:1626–1634. doi: 10.1038/sj.onc.1204275. [DOI] [PubMed] [Google Scholar]
- 15.Abdelmohsen K, Lal A, Kim HH, Gorospe M. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle. 2007;6:1288–1292. doi: 10.4161/cc.6.11.4299. [DOI] [PubMed] [Google Scholar]
- 16.Dorn GW, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mishra S, Ling H, Grimm M, Zhang T, Bers DM, Brown JH. Cardiac Hypertrophy and Heart Failure Development Through Gq and CaM Kinase II Signaling. Journal of Cardiovascular Pharmacology. 2010;56:598–603. doi: 10.1097/FJC.0b013e3181e1d263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, et al. Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S a. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doller A, Akool ES, Huwiler A, Müller R, Radeke HH, Pfeilschifter J, et al. Posttranslational modification of the AU-rich element binding protein HuR by protein kinase Cdelta elicits angiotensin II-induced stabilization and nuclear export of cyclooxygenase 2 mRNA. Mol Cell Biol. 2008;28:2608–2625. doi: 10.1128/MCB.01530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doller A, Winkler C, Azrilian I, Schulz S, Hartmann S, Pfeilschifter J, et al. High-constitutive HuR phosphorylation at Ser 318 by PKC{delta} propagates tumor relevant functions in colon carcinoma cells. Carcinogenesis. 2011;32:676–685. doi: 10.1093/carcin/bgr024. [DOI] [PubMed] [Google Scholar]
- 21.Doller A, Huwiler A, Müller R, Radeke HH, Pfeilschifter J, Eberhardt W. Protein kinase C alpha-dependent phosphorylation of the mRNA-stabilizing factor HuR: implications for posttranscriptional regulation of cyclooxygenase-2. Mol Biol Cell. 2007;18:2137–2148. doi: 10.1091/mbc.E06-09-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lafarga V, Cuadrado A, López de Silanes I, Bengoechea R, Fernandez-Capetillo O, Nebreda AR. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol Cell Biol. 2009;29:4341–4351. doi: 10.1128/MCB.00210-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdelmohsen K, Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip Rev RNA. 2010;1:214–229. doi: 10.1002/wrna.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schulz S, Doller A, Pendini NR, Wilce JA, Pfeilschifter J, Eberhardt W. Domain-specific phosphomimetic mutation allows dissection of different protein kinase C (PKC) isotype-triggered activities of the RNA binding protein HuR. Cell Signal. 2013;25:2485–2495. doi: 10.1016/j.cellsig.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 25.Wang Z, Bhattacharya A, Ivanov DN. Identification of Small-Molecule Inhibitors of the HuR/RNA Interaction Using a Fluorescence Polarization Screening Assay Followed by NMR Validation. PLoS ONE. 2015;10:e0138780. doi: 10.1371/journal.pone.0138780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D'Agostino VG, Adami V, Provenzani A. A novel high throughput biochemical assay to evaluate the HuR protein-RNA complex formation. PLoS ONE. 2013;8:e72426. doi: 10.1371/journal.pone.0072426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zucal C, D'Agostino V, Loffredo R, Mantelli B, NatthakanThongon, Lal P, et al. Targeting the multifaceted HuR protein, benefits and caveats. Curr Drug Targets. 2015;16:499–515. doi: 10.2174/1389450116666150223163632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.