

Abstract
Objective:
To evaluate the influence of oral laquinimod, a candidate multiple sclerosis (MS) treatment, on induction of T follicular helper cells, development of meningeal B cell aggregates, and clinical disease in a spontaneous B cell–dependent MS model.
Methods:
Experimental autoimmune encephalomyelitis (EAE) was induced in C57BL/6 mice by immunization with recombinant myelin oligodendrocyte glycoprotein (rMOG) protein. Spontaneous EAE was evaluated in C57BL/6 MOG p35-55–specific T cell receptor transgenic (2D2) × MOG-specific immunoglobulin (Ig)H-chain knock-in (IgHMOG-ki [Th]) mice. Laquinimod was administered orally. T cell and B cell populations were examined by flow cytometry and immunohistochemistry.
Results:
Oral laquinimod treatment (1) reduced CD11c+CD4+ dendritic cells, (2) inhibited expansion of PD-1+CXCR5+BCL6+ T follicular helper and interleukin (IL)-21–producing activated CD4+CD44+ T cells, (3) suppressed B cell CD40 expression, (4) diminished formation of Fas+GL7+ germinal center B cells, and (5) inhibited development of MOG-specific IgG. Laquinimod treatment not only prevented rMOG-induced EAE, but also inhibited development of spontaneous EAE and the formation of meningeal B cell aggregates. Disability progression was prevented when laquinimod treatment was initiated after mice developed paralysis. Treatment of spontaneous EAE with laquinimod was also associated with increases in CD4+CD25hiFoxp3+ and CD4+CD25+IL-10+ regulatory T cells.
Conclusions:
Our observations that laquinimod modulates myelin antigen–specific B cell immune responses and suppresses both development of meningeal B cell aggregates and disability progression in spontaneous EAE should provide insight regarding the potential application of laquinimod to MS treatment. Results of this investigation demonstrate how the 2D2 × Th spontaneous EAE model can be used successfully for preclinical evaluation of a candidate MS treatment.
Laquinimod, a quinoline-3-carboxamide, is a novel oral agent with immunomodulatory properties that is being developed for the treatment of multiple sclerosis (MS).1 In 2 phase III placebo-controlled relapsing-remitting MS trials, laquinimod demonstrated more pronounced beneficial effects on disease progression and brain atrophy than on clinical or imaging markers of CNS inflammation,2–4 suggesting that it may also be beneficial in progressive MS. However, the mechanism(s) responsible for laquinimod's effects in MS is not completely understood. In studies of experimental autoimmune encephalomyelitis (EAE), laquinimod induced both innate and adaptive immune modulation.5–10 In this regard, laquinimod treatment promotes development of type II (M2) myeloid antigen-presenting cells (APCs) that inhibit development of proinflammatory Th1 and Th17 cells.5 Besides its established effects on myeloid cells and T cells, it is possible that laquinimod exerts activity on B cells, which could also contribute to its potential benefit in patients with MS.
Favorable responses to CD20-mediated B cell depletion in both relapsing-remitting MS and progressive MS have underscored the importance of B cells in MS pathogenesis.11–13 B cells may participate in MS pathogenesis by functioning as APCs, through cytokine secretion, and by serving as a source of antibody-secreting plasma cells.14,15 Ectopic meningeal B cell follicles have been identified in brain tissue from patients with secondary progressive MS, suggesting that B cells could also contribute to disease progression.16 Currently, information regarding the potential influence of laquinimod on B cells is limited. One investigation found that in vitro laquinimod treatment of peripheral blood mononuclear cells altered B cell expression of markers associated with regulation, suggesting that in vivo laquinimod treatment may similarly affect B cells.17 Previously, we demonstrated that in vivo laquinimod treatment causes a disproportionate reduction in the numbers of the CD11c+CD4+CD8α− (referred to as CD4+) dendritic cells (DCs).5 The CD4+ DC subpopulation is instrumental in promoting differentiation of T follicular helper (Tfh) cells,18–20 the CD4+ T cell subset that directs B cell differentiation, germinal center (GC) formation, and immunoglobulin (Ig) class switching.21 Therefore, we hypothesized that laquinimod could affect several B cell activities that contribute to CNS autoimmunity. In this study, we evaluated laquinimod treatment in acute inflammatory EAE and in a model of spontaneous EAE that requires cooperation between T cells and B cells and is associated with the development of ectopic meningeal B cell aggregates.
METHODS
Mice.
Female C57BL/6 mice, 7 to 8 weeks old, were purchased from Jackson Laboratories (Bar Harbor, ME). Myelin oligodendrocyte glycoprotein (MOG) peptide (p)35-55–specific T cell receptor transgenic 2D2 mice were provided by V.K. Kuchroo (Harvard Medical School, Boston, MA).22 C57BL/6J MOG-BCR knock-in (IgHMOG-ki, also referred to as Th) mice were provided by H. Wekerle (Max Planck Institute of Neurobiology, Martinsried, Germany).23 The University of California San Francisco Institutional Animal Care and Use Committee approved the experimental protocol (approval AN081032), in accordance with guidelines for animal use in research established by the NIH.
Antigens.
Mouse MOG p35-55 (MEVGWYRSPFSRVVHLYRNGK) was synthesized by Auspep (Melbourne, Australia). Recombinant (r) mouse rMOG protein was synthesized, purified, and refolded as previously reported.24
EAE induction and clinical assessment.
Female, 7- to 10-week-old C57BL/6 mice were injected subcutaneously with 100 μg rMOG in complete Freund's adjuvant (Difco Laboratories, Detroit, MI). Mice received intraperitoneal injections of 200 ng pertussis toxin on the day of immunization and 2 days later. Animals were examined daily, and clinical scores were assessed as follows: 0, no signs; 1, decreased tail tone; 2, mild monoparesis or paraparesis; 3, severe paraparesis; 4, paraplegia and/or quadriparesis; and 5, moribund or death. In all EAE experiments, mice were scored daily by an examiner who was blinded to the treatment assignment.
Laquinimod treatment.
Laquinimod (Teva Pharmaceutical Industries, Ltd., Petah Tikva, Israel) was dissolved in purified water. Mice received a daily dose of either laquinimod (25 mg/kg) or vehicle (water) by oral gavage. The dosing regimen was selected based on a previous report.25 Treatment started on the day of EAE induction. For prevention of spontaneous EAE, 2D2 × Th mice were treated with laquinimod (25 mg/kg) or vehicle starting at 19 days of age. In reversal of spontaneous EAE, mice were randomized to treatment with laquinimod or vehicle (water) when they developed an EAE score of ≥1. For in vitro APC-T cell assays, cells of interest were harvested following a 10-day in vivo treatment period with laquinimod or vehicle.
Adoptive transfer of B cells.
Splenic B cells were isolated from Th mice treated with laquinimod or vehicle for 10 days. Enriched B cell fractions obtained by magnetic cell sorting using B220+ magnetic beads (STEMCELL Technologies, Vancouver, Canada) exhibited a purity of at least 95%. Recipient 2D2 × JHT mice received 10 × 106 laquinimod- or vehicle-treated B cells and were immunized with rMOG the following day.
Generation of Tfh cells.
Naive T cells (CD4+CD62L+CD44+) were magnetically sorted from T cell receptor transgenic 2D2 mice (purity greater than 96%) using magnetic beads (Miltenyi Biotec, Auburn, CA) and stimulated with MOG p35-55 (20 μg/mL) in the presence of APCs (CD90− cells) at a T cell/APC ratio of 1:5 as previously described.5 Tfh differentiation was induced by addition of interleukin (IL)-6, IL-21, anti–IL-4, anti–IL-12, anti–interferon γ, and anti–transforming growth factor β antibodies. Cells were cultured for 3 to 4 days before intracellular evaluation of cytokine production using a FACSCanto flow cytometer (BD Biosciences, San Jose, CA).
Detection of anti-MOG antibodies.
Total serum MOG-specific IgG was quantified using a custom anti-rMOG IgG quantitative ELISA. MaxiSorp plates (96-well; Corning Inc., Corning, NY) were precoated with rMOG protein (10 μg/mL in phosphate-buffered saline), blocked with blocking buffer, and incubated overnight with sera at 1:100 dilution. After washing, MOG-specific IgG was detected with horseradish peroxidase–conjugated anti-mouse IgG. Results are expressed as optical density. A SpectraMax spectrophotometer (450-nm wavelength) and SoftMax software (Molecular Devices, Sunnyvale, CA) were used for data analysis.
T cell proliferation.
Purified B cells from laquinimod- or vehicle-treated Th mice were cocultured with CFSE-labeled naive CD4+ cells isolated from laquinimod- or vehicle-treated 2D2 mice in the presence of rMOG. Cells were cultured for 48 hours, and CFSE dilution was evaluated by flow cytometry.
Histology and immunohistochemistry.
The brains, spinal cords, and optic nerves were removed from mice and fixed in 10% neutral-buffered formalin, paraffin-embedded, and sectioned as described previously.5,15 Representative sections were stained with Luxol fast blue–hematoxylin & eosin and reticulin preparation (for connective tissue). Stained tissue specimens were examined by light microscopy. A blinded observer (R.A. Sobel) counted both meningeal and parenchymal inflammatory foci (>10 clustered inflammatory cells). Avidin-biotin immunohistochemical staining was performed on the sections with rabbit anti-mouse CD3 (Abcam, Cambridge, UK) and rat anti-mouse CD45R (B220; BD Biosciences) using reagents from Vector Laboratories (Burlingame, CA). As described previously,5,15 normal mouse spleen tissue served as positive staining controls. Negative controls included omission of the primary antibody and analysis of mouse CNS tissues from unimmunized mice.
Flow cytometry.
Single-cell suspensions were incubated with anti-CD16/CD32 (1:100) to prevent nonspecific antibody binding, then stained with anti-CD4, -CD44, -PD-1, -CXCR5, CD11b, -CD11c, -B220, -IgD, -GL7, -Fas, -MHCII, -CD40, -CD80, and -CD86 (all 1:100) (eBioscience, San Diego, CA) as described previously.5,15 Intracellular cytokine production by CD4+ T cells was analyzed by monitoring the expression of interferon γ, IL-17, IL-10 (eBioscience), and IL-21 (R&D Systems, Minneapolis, MN) as described previously.5,15,26 B cell lymphoma 6 (BCL6) and Foxp3 staining were performed according to the manufacturer's protocol (eBioscience). Intracellular cytokine staining was performed on T cells after stimulation with phorbol 12-myristate 13-acetate (50 ng/mL) and ionomycin (500 ng/mL) in the presence of monensin (BD Biosciences GolgiStop, 3 μM).5,15,26 Cells were analyzed by flow cytometry on a FACSCanto (BD Biosciences) as described previously.5,15,26
Statistical analysis.
Data are shown as mean ± SEM or SD. Statistical significance between groups was analyzed using Student t test or Mann–Whitney U test. Repeated-measurement mixed-effect models were used to compare clinical scores between treatment groups. Survival analysis was used to compare the occurrence of spontaneous EAE between treatment groups; hazard ratio was determined using Cox regression with the Breslow method for ties. A value of p ≤ 0.05 was considered significant.
RESULTS
Laquinimod alters the frequency and differentiation of Tfh cells.
Previous studies have demonstrated that laquinimod is effective in the treatment of EAE induced by MOG p35-55,5,6,25 an encephalitogenic peptide that does not efficiently activate B cells or lead to production of MOG-specific antibodies.15,27 To evaluate the effect of laquinimod on Tfh cell development and differentiation, we examined rMOG-immunized C57BL/6 mice, an EAE model that leads to activation of MOG-specific B cells and production of MOG-specific antibodies.15,27 Daily oral laquinimod treatment prevented rMOG-induced EAE (figure 1A) and was associated with a reduced frequency of CD4+ DCs, which are known to participate in the development of Tfh cells (figure 1B).
Figure 1. Laquinimod treatment reduces the frequency of Tfh cells and IL-21–producing T cells in rMOG-induced EAE.

(A–F) EAE was induced in C57BL/6 mice by immunization with rMOG. Mice were treated daily with laquinimod or vehicle from day of immunization. Lymph nodes and spleens were isolated 10 days later. (A) Laquinimod prevented the development of EAE. (B) The frequency of CD4+ dendritic cells was examined in the spleen and lymph nodes from laquinimod- and vehicle-treated mice. (C) Tfh cells were defined as PD-1+CXCR5+ cells among CD4+CD44hi T cells. Representative flow cytometry analysis and Tfh cell frequency are shown. (D) The expression of BCL6, a transcriptional repressor that directs Tfh cell differentiation, was examined within Tfh cells. The production of IL-21, a Tfh cell cytokine that contributes to the formation and function of germinal centers, was examined among activated T cells (E) and Tfh cells (F). *p < 0.05, **p < 0.001, Student t test. BCL6 = B cell lymphoma 6; EAE = experimental autoimmune encephalomyelitis; IL = interleukin; LAQ = laquinimod; LN = lymph nodes; PD-1 = programmed cell death protein 1; rMOG = recombinant myelin oligodendrocyte glycoprotein; Tfh = T follicular helper; Veh = vehicle.
Tfh cells are characterized by expression of various cell surface and intracellular markers, including CXCR5, inducible T cell costimulator, programmed cell death protein 1 (PD-1), the transcriptional repressor BCL6, which directs differentiation of Tfh, and IL-21, which participates in GC formation.28 Daily laquinimod treatment was associated with reduction of PD-1+CXCR5+ (figure 1C) and PD-1+CXCR5+BCL6+ Tfh (figure 1D) in lymph nodes. IL-21 participates in GC formation.29 Of note, laquinimod treatment reduced the frequency of IL-21–producing activated CD4+CD44hi T cells (figure 1E) and Tfh cells (figure 1F) in lymph nodes and spleen, suggesting that inhibition of T cell IL-21 secretion may be common to activated T cells.
Previously, we demonstrated that laquinimod exerts its immunomodulatory activity on effector T cells through alteration of APC function, but not on T cells directly.5 Thus, we wished to determine whether laquinimod similarly affects Tfh cells. APCs from laquinimod- or vehicle-treated mice were cocultured with vehicle-treated naive MOG p35-55–specific (2D2) T cells. In a reciprocal manner, naive T cells isolated from laquinimod- or vehicle-treated 2D2 mice were cultured with purified vehicle-treated APCs. We observed that APCs from laquinimod-treated mice, but not from untreated mice, suppressed differentiation of Tfh cells, regardless of whether naive 2D2 T cells were obtained from laquinimod- or vehicle-treated mice (figure 2). This observation is consistent with our earlier demonstration that laquinimod exerts its activity directly on APCs that are in turn responsible for adaptive T cell immune modulation.5
Figure 2. APCs from laquinimod-treated mice inhibit differentiation of Tfh cells.
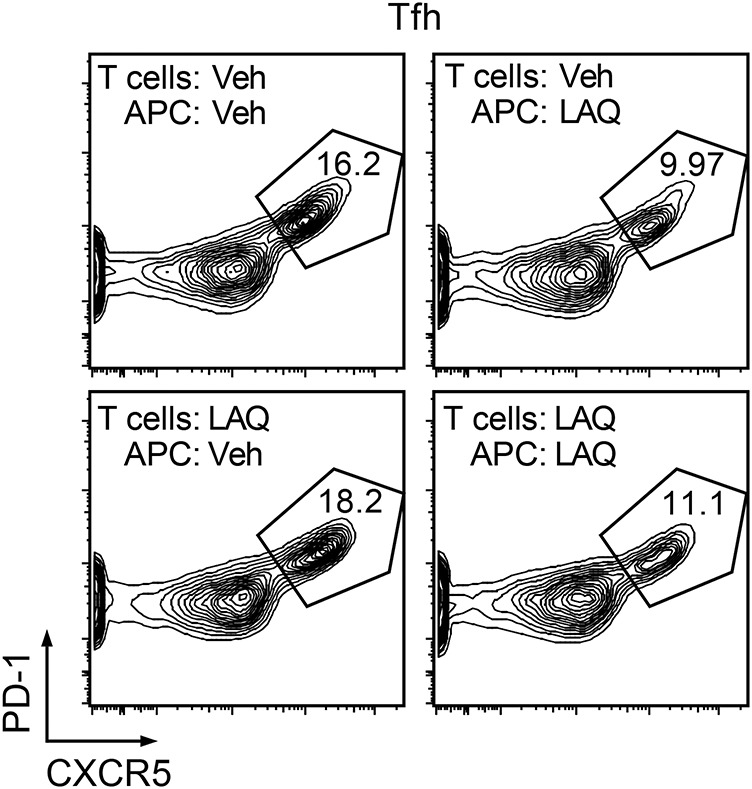
Splenic CD90.2− cells from mice treated in vivo with laquinimod or vehicle were used as APCs in a reciprocal manner to stimulate naive (CD4+CD44−CD62L+) T cells from MOG p35-55 T cell receptor transgenic mice (2D2) treated with laquinimod or vehicle. APCs and T cells were cultured with MOG p35-55 in Tfh-polarizing conditions (IL-6, IL-21, anti-IL-4, -IL-12, -IFN-γ and -TGF-β antibodies) from the start of culture. Flow cytometry analysis for expression of Tfh cell markers was performed after 3 days of culture. APC = antigen-presenting cell; IFN-γ = interferon γ; IL = interleukin; LAQ = laquinimod; MOG = myelin oligodendrocyte glycoprotein; PD-1 = programmed cell death protein 1; Tfh = T follicular helper; TGF-β = transforming growth factor β; Veh = vehicle.
Laquinimod reduces the frequency of GC B cells and formation of MOG-specific IgG.
Tfh cells participate in maturation and differentiation of GC B cells, antibody class switch recombination, and production of IgG antibodies.18,20,21 Because we observed that laquinimod reduced the frequency of Tfh, we questioned whether laquinimod treatment might decrease the frequency of GC B cells and suppress IgG formation. Oral laquinimod treatment reduced the frequency of GC B cells within the draining lymph nodes (figure 3A) and was associated with a marked decrease in anti-MOG IgG antibody titers (figure 3B). While CD4+ DCs (CD4+CD8α− follicular DCs [FDCs]) have a key role in Tfh cell development, B cells also contribute to Tfh cell homeostasis.30 We therefore examined whether the influence of in vivo laquinimod treatment on B cells could also contribute to reduction of Tfh. B cells were isolated from MOG-specific BCR transgenic (Th) mice after 10 days of treatment with laquinimod or vehicle and then transferred into B cell–deficient (JHT) 2D2 recipient mice. Recipient mice were then immunized with rMOG. Examination of draining lymph nodes revealed a reduced frequency of Tfh cells in laquinimod-treated recipient mice (figure 3C). These findings indicate that laquinimod can act directly on B cells, which may also contribute to the observed reduction of Tfh cells during laquinimod treatment.
Figure 3. Laquinimod treatment reduces the frequency of GC B cells and IgG antibody production in rMOG-immunized mice.

Lymph node cells were isolated 10 days after immunization with rMOG. (A) GC B cells were defined as Fas+GL7+ cells among B220+IgDlow B cells. Representative flow cytometry analysis (left panel) and GC B cell frequency from all mice (right panel) are shown. (B) Serum anti-rMOG IgG levels were determined by ELISA 10 days after immunization. (C) B cells isolated from mice treated or not treated with laquinimod were transferred to JHT (B cell–deficient) mice. Lymph node cells were isolated 10 days after immunization with rMOG. The frequency of Tfh cells is shown. (D) CD4+ T cells from 2D2 mice were cocultured with B cells treated or not treated with laquinimod in the presence of rMOG. Proliferation of CD4+ T cells was evaluated after 72 hours. (E) Lymph node and spleen cells were isolated 10 days later after immunization with rMOG. B cell CD40 expression was examined by flow cytometry. *p < 0.05, ***p < 0.001, Student t test. CDI = cell division index; GC = germinal center; Ig = immunoglobulin; LAQ = laquinimod; LN = lymph nodes; MOG = myelin oligodendrocyte glycoprotein; OD = optical density; PD-1 = programmed cell death protein 1; rMOG = recombinant myelin oligodendrocyte glycoprotein; Tfh = T follicular helper; Veh = vehicle.
We also examined whether in vivo laquinimod treatment influences B cell APC function. B cells from laquinimod-treated mice stimulated proliferation of 2D2 T cells as well as B cells from vehicle-treated mice, suggesting that B cell APC function, unlike APC function of myeloid cells,5 was not affected by laquinimod treatment (figure 3D). Of note, laquinimod treatment did not alter B cell expression of major histocompatibility complex (MHC) II, CD80, or CD86 costimulatory molecules (figure e-1 at Neurology.org/nn), although it reduced cell surface expression of CD40 (figure 3E), the costimulatory molecule that is required for the maintenance of Tfh cells.30
Laquinimod inhibits disease development and meningeal follicle-like structure formation in spontaneous EAE.
Laquinimod treatment was tested in the B cell–dependent model of spontaneous EAE that develops when MOG-specific T cell receptor transgenic (2D2) mice are crossed with MOG-specific B cell receptor transgenic (Th) mice.15,31,32 Laquinimod administration before the development of first clinical signs resulted in delayed onset and reduced disease incidence when compared to vehicle-treated mice (figure 4A). Among mice that developed clinical signs of EAE, those that received laquinimod had a significantly lower maximal clinical score (figure 4B). Meningeal B cell aggregates, which are sometimes detected in secondary progressive MS,16 are associated with clinical disease in 2D2 × Th mice.15,31,32 B cell aggregates were located in the leptomeninges and subpial parenchyma (figure 5A). There was a reduction in size and markedly significant decrease in number of the B cell aggregates in laquinimod-treated mice (figure 5B).
Figure 4. Laquinimod treatment suppresses development of spontaneous EAE.

(A) 2D2 × Th mice were treated with laquinimod (25 mg/kg) or vehicle (water) daily by oral gavage starting 19 days after birth (see arrow). Mice were examined once a day for onset of clinical EAE. The first quartile of mice that developed EAE is indicated since only 29% of laquinimod-treated mice developed disease throughout the observation period. Hazard ratio was determined using Cox regression with the Breslow method for ties. (B) Daily oral laquinimod treatment decreases the severity of spontaneous EAE in 2D2 × Th mice. Data represent the mean maximal scores of those mice that developed EAE. *p < 0.05, Mann-Whitney U test. EAE = experimental autoimmune encephalomyelitis; LAQ = laquinimod; Veh = vehicle.
Figure 5. Laquinimod treatment prevents development of meningeal follicles in spontaneous EAE.

(A) Laquinimod influences CNS inflammation and demyelination in spontaneous EAE (LH&E). The apparent reduction in typical EAE CNS inflammatory foci was not statistically significant. (B) In vehicle-treated mice, extensive mononuclear cell infiltrates in leptomeninges and parenchyma with organized lymphoid follicle-like aggregates containing T cells and B cells are observed (a–d). Laquinimod treatment is associated with reduced size and number of meningeal follicle-like structures in the leptomeninges and subpial parenchyma (e–h). Boxes in a and e correspond to fields in b–d and f–h, respectively. (a, b, e, and f) LH&E. (c and g) anti-CD3. (d and h) anti-CD45R (B220). Scale bars: e, 200 μm (applies to a and e); f, 50 μm, applies to b–d, f–h. *p < 0.01, Mann–Whitney U test. EAE = experimental autoimmune encephalomyelitis; LAQ = laquinimod; LH&E = Luxol fast blue–hematoxylin & eosin; Veh = vehicle.
Laquinimod prevents clinical progression of spontaneous EAE.
Laquinimod was tested in mice after onset of spontaneous EAE for its ability to prevent disability progression. Mice were randomized to laquinimod or vehicle after onset of clinical EAE (mean score 2.25). There was a statistical trend for overall lower mean clinical scores with laquinimod (p = 0.077, repeated-measures mixed-effect linear regression). There was a clear interaction of laquinimod treatment with time (p = 0.0002) with the difference between groups becoming significant after 36 days (figure 6A). Laquinimod treatment of 2D2 × Th mice was associated with a significant reduction in the frequency of splenic Tfh cells as well as IL-21–secreting Tfh cells (figure 6, B and C). In contrast to laquinimod treatment in acute rMOG-induced EAE (figure 1C), we did not detect a similar decrease of Tfh in lymph nodes. For comparison to treatment of 2D2 × Th mice, we treated naive (unimmunized) C57BL/6 mice with laquinimod and similarly observed a reduction of splenic, but not lymph node, Tfh cells (figure 6D). In contrast to laquinimod treatment of acute EAE, we did not observe a reduction of Fas+GL7+ GC B cells (figure 6E), a finding that may not be surprising, as immunization of wild-type mice with protein (e.g., rMOG) promotes activation of GC B cells,27 and 2D2 × Th mice were not given an immunization.
Figure 6. Laquinimod treatment of spontaneous EAE prevents progression and reduces the frequency of Tfh cells.

(A) 2D2 × Th mice that had developed clinical signs of EAE (score ≥1) were randomly assigned to treatment with laquinimod (25 mg/kg) (n = 11) or vehicle (n = 11). Treatments were administered by oral gavage daily for 48 days. The frequency of Tfh cells (B), IL-21–producing Tfh cells (C), and germinal center B cells (E) were examined after 48 days of treatment. (D) Frequency of Tfh cells observed in lymph node and spleen cells isolated from naive mice treated with laquinimod or vehicle for 10 days. (F–G) Frequency of Treg cells (F) and IL-10–producing CD4+CD25+ T cells (G) detected in lymph node and spleen cells isolated from 2D2 × Th mice that had received laquinimod or vehicle treatment for 48 days. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, Student t test. EAE = experimental autoimmune encephalomyelitis; Ig = immunoglobulin; IL = interleukin; LAQ = laquinimod; LN = lymph nodes; PD-1 = programmed cell death protein 1; Tfh = T follicular helper; Treg = T regulatory; Veh = vehicle.
Previously, we observed that laquinimod treatment of wild-type mice was associated with an increase in regulatory T cells.5 Thus, we examined the frequency of regulatory T cells in laquinimod-treated and vehicle-treated 2D2 × Th mice. Reduction in disease progression by treatment with laquinimod was associated with increased frequencies of CD4+CD25hiFoxp3+ (figure 6F) and CD4+CD25+IL-10+ regulatory T cells (figure 6G).
DISCUSSION
In this report, we evaluated oral laquinimod in 2 EAE models that require B-T cell cooperation. Laquinimod treatment of rMOG-induced EAE interfered with development of Tfh, B cell activation, secretion of MOG-specific antibodies, and EAE. Similarly, laquinimod treatment of spontaneous EAE reduced expansion of Tfh cells but also prevented accumulation of meningeal B cell aggregates, lymphoid structures that have been observed in tissues of patients with progressive MS, and impeded disability progression when treatment was initiated after mice developed paralysis. Collectively, these findings may be relevant to the potential application of laquinimod to treatment of patients with progressive MS. Furthermore, our observation that laquinimod inhibits MOG-induced antibodies suggests that laquinimod could interfere with formation of pathogenic antigen-specific antibodies in humoral autoimmune diseases, including neuromyelitis optica.33
CD4+ DCs have a key role in the development of Tfh cells and humoral immune responses.18–20 Thus, our observation that laquinimod treatment reduces CD4+ DCs in rMOG-induced EAE, which confirms similar observations in MOG p35-55–induced EAE,5,6 suggests that the disproportionate decrease of this DC subset may contribute to the reduction of Tfh and anti-MOG antibodies. FDCs, which express CD21, CD32, and CD35, have an important role in the development of GCs and are also a hallmark cell type of meningeal B cell aggregates in secondary progressive MS. Thus, in further studies, one may wish to evaluate how laquinimod treatment influences FDCs. It is also of interest that while laquinimod treatment in spontaneous EAE was associated with a marked decrease in meningeal B cell aggregates, we did not observe a statistically significant reduction of typical meningeal and parenchymal lymphocytic infiltrates (figure 5A). It is possible that by examining a larger number of mice, the apparent decrease in lymphocytic infiltrates would have been statistically significant. Alternatively, and consistent with the observations that laquinimod treatment selectively reduces CD4+ DCs, laquinimod may have a more pronounced effect on B cell differentiation and development of meningeal B cell aggregates in spontaneous EAE. It is also recognized that laquinimod may enter the CNS, especially during inflammation.34 Thus, reduction of ectopic meningeal B cell aggregates may not be attributable solely to laquinimod's influence on peripheral B cell differentiation, but could conceivably also reflect direct inhibitory effects in situ.
Previous investigations indicate that laquinimod may promote neuroprotection.35–37 Laquinimod protects against cuprizone-induced demyelination,35,37 a model that leads to activation of glial cells, but is unaccompanied by significant CNS lymphocytic infiltration.38 Such findings suggest that, in addition to its known anti-inflammatory properties, laquinimod can also exert neuroprotective effects directly within the CNS. Prevention of disability progression and brain atrophy are clinical and imaging measures that are associated with neuroprotection.39,40 Of note, we observed that the reduction in disability was statistically significant, but only after 1 month of laquinimod treatment. One may therefore ask whether laquinimod treatment in this model was associated with neuroprotection, a possibility that we did not formally address. Because CNS damage in spontaneous EAE in 2D2 × Th mice occurs as a consequence of inflammation, the influence of laquinimod treatment on disability progression reported here could be a reflection of secondary, but not direct, CNS neuroprotection.
While laquinimod treatment was effective in both rMOG-induced EAE and in spontaneous EAE, there were notable differences in its influence on immune cell activation. For example, laquinimod treatment was associated with a marked reduction of activated GC B cells in rMOG-immunized mice (figure 3A), but not in treatment of spontaneous EAE (figure 6E). However, this observation is not necessarily surprising because protein immunization in complete Freund’s adjuvant is a potent stimulus that promotes activation of GC B cells.27 In addition, the influence of laquinimod treatment on certain immune cells is more easily detected when they have been activated. In myelin protein or peptide-induced EAE, peripheral T cell activation is most commonly assessed in draining lymph nodes or the spleen. It is important to recognize that the kinetics for T cell activation in these secondary lymphoid organs are not identical, and the identification of activated T cells in draining lymph nodes of antigen-primed mice often precedes detection in the spleen.41 In this regard, we observed a significant reduction of Tfh cells in lymph nodes, but not in the spleen, 10 days after rMOG immunization of laquinimod-treated mice (figure 1, C and D). Conceivably, evaluation of mice at later time points may have enabled us to detect activated GC B cells in the spleen. Since it is not necessarily feasible to evaluate the influence of a therapy on immune cells at successive times, it can be advantageous to evaluate those cells in multiple secondary lymphoid tissues simultaneously.
Recently, we reported that oral dimethyl fumarate (Tecfidera; Biogen, Cambridge, MA) prevented development of spontaneous EAE.42 In the present study, we found that laquinimod treatment not only prevented spontaneous EAE but also was successful when treatment was initiated after mice developed paralysis. In contrast, other investigators did not detect a therapeutic benefit of glatiramer acetate (Copolymer-1, Copaxone; Teva Pharmaceutical Industries) in 2D2 × Th mice and questioned whether the 2D2 × Th EAE model is appropriate for evaluating therapeutic intervention.43 However, it is important to recognize that just as there is heterogeneity in MS, EAE models also differ. Certain features of individual EAE models may recapitulate unique aspects of human CNS autoimmune disease, and therefore each model may not be optimal for evaluating all potential therapeutic agents. Although artificial, as T cells and B cells both target MOG,15,31,32 one strength of the 2D2 × Th model is that it permits evaluation of myelin-specific B cell activation and B-T cell cooperation in an EAE model that shares certain features with progressive MS. Use of this model has permitted us to evaluate and demonstrate how laquinimod treatment may influence B-T cell interaction and B cell participation in MS. Our results clearly demonstrate that the 2D2 × Th spontaneous EAE model can be used successfully to study therapeutic immune intervention.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Liat Hayardeny for providing laquinimod and Dr. Dirk Baumjohann for helpful discussion.
GLOSSARY
- APC
antigen-presenting cell
- BCL6
B cell lymphoma 6
- DC
dendritic cell
- EAE
experimental autoimmune encephalomyelitis
- FDC
follicular dendritic cell
- GC
germinal center
- Ig
immunoglobulin
- IL
interleukin
- MHC
major histocompatibility complex
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- p
peptide
- PD-1
programmed cell death protein 1
- rMOG
recombinant myelin oligodendrocyte glycoprotein
- Tfh
T follicular helper
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
M. Varrin-Doyer designed research, acquired and analyzed data, performed statistical analysis, and wrote the manuscript. K.L. Pekarek acquired and analyzed data, and discussed the results at all stages. C.M. Spencer analyzed data and contributed to critical revisions of the manuscript. C.C.A. Bernard provided reagents and edited the manuscript. R.A. Sobel performed histology and immunohistochemistry analysis. U. Schulze-Topphoff initiated the study with S.S. Zamvil, designed research, acquired and analyzed data. B.A.C. Cree analyzed data and contributed to critical revisions of the manuscript. S.S. Zamvil initiated the study, designed research, interpreted data, wrote the manuscript, and supervised the study.
STUDY FUNDING
M. Varrin-Doyer was a fellow of the US National Multiple Sclerosis Society (NMSS; FG 2067-A-1). Dr. Bernard is supported by grants from the National Health and Medical Research Council of Australia/CIRM Joint Project (APP1053621), the Victoria/CIRM Joint Project (RMI-01739), and the Department of Industry, Commonwealth of Australia (AISRF06680). U.S.-T. was a fellow of the NMSS and the Deutsche Forschungsgemeinschaft (DFG; SCHU 2587/1). S.S. Zamvil received research grant support from the NIH (RO1 AI073737 and RO1 NS063008), the NMSS (RG 4124, RG 5179, and RG 5180), the Guthy Jackson Charitable Foundation, and the Alexander M. and June L. Maisin Foundation of the Jewish Community Federation and Endowment Fund. Teva Pharmaceuticals, Ltd., provided laquinimod but did not provide financial support for this investigation.
DISCLOSURE
M. Varrin-Doyer received research support from the NMSS and Teva Pharmaceuticals. K.L. Pekarek is employed by ARMO Biosciences. C.M. Spencer reports no disclosures. C.C.A. Bernard served on the editorial board for Future Neurology, Inflammation and Regeneration, was guest editor for Inflammation and Regeneration, received research support from NHMRC of Australia, Victorian Government/CIRM, Department of Industry, Commonwealth of Australia, Eva and Les Erdi AUSiMED Fellowship in Neurologic Diseases. R.A. Sobel is editor-in-chief for Journal of Neuropathology and Experimental Neurology, associate editor for Journal of Neuroimmunology, received research support from NIH/NIAID, DOD/CDRMP, National Multiple Sclerosis Society, Progressive MS Alliance. B.A.C. Cree is an editor of Annals of Neurology, consulted for AbbVie, Biogen, EMD Serono, Novartis, Sanofi Genzyme, Shire, received research support from Acorda, Hoffmann-La Roche, MedImmune, Novartis, Receptos, Teva. U. Schulze-Topphoff is employed by Teva GmbH. S.S. Zamvil served on the data safety monitoring board for BioMS, Teva Pharmaceuticals, Inc., Eli Lilly and Co., is a member of the clinical advisory board for Myelin Repair Foundation, is deputy editor for Neurology: Neuroimmunology & Neuroinflammation, has consulted for Biogen Idec, Teva Neuroscience, EMD Serono, Genzyme, Novartis, Roche, is on the speakers bureau for Advanced Health Media, Biogen, received research support from Biogen, NIH, NMSS, Alexander M. and June L. Maisin Foundation. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Brück W, Gold R, Lund BT, et al. Therapeutic decisions in multiple sclerosis: moving beyond efficacy. JAMA Neurol 2013;70:1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Comi G, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med 2012;366:1000–1009. [DOI] [PubMed] [Google Scholar]
- 3.Vollmer TL, Sorensen PS, Selmaj K, et al. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. J Neurol 2014;261:773–783. [DOI] [PubMed] [Google Scholar]
- 4.Varrin-Doyer M, Zamvil SS, Schulze-Topphoff U. Laquinimod, an up-and-coming immunomodulatory agent for treatment of multiple sclerosis. Exp Neurol 2014;262:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulze-Topphoff U, Shetty A, Varrin-Doyer M, et al. Laquinimod, a quinoline-3-carboxamide, induces type II myeloid cells that modulate central nervous system autoimmunity. PLoS One 2012;7:e33797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jolivel V, Luessi F, Masri J, et al. Modulation of dendritic cell properties by laquinimod as a mechanism for modulating multiple sclerosis. Brain 2013;136:1048–1066. [DOI] [PubMed] [Google Scholar]
- 7.Brück W, Zamvil SS. Laquinimod, a once-daily oral drug in development for the treatment of relapsing-remitting multiple sclerosis. Expert Rev Clin Pharmacol 2012;5:245–256. [DOI] [PubMed] [Google Scholar]
- 8.Thone J, Ellrichmann G, Seubert S, et al. Modulation of autoimmune demyelination by laquinimod via induction of brain-derived neurotrophic factor. Am J Pathol 2012;180:267–274. [DOI] [PubMed] [Google Scholar]
- 9.Brunmark C, Runström A, Ohlsson L, et al. The new orally active immunoregulator laquinimod (ABR-215062) effectively inhibits development and relapses of experimental autoimmune encephalomyelitis. J Neuroimmunol 2002;130:163–172. [DOI] [PubMed] [Google Scholar]
- 10.Yang JS, Xu LY, Xiao BG, Hedlund G, Link H. Laquinimod (ABR-215062) suppresses the development of experimental autoimmune encephalomyelitis, modulates the Th1/Th2 balance and induces the Th3 cytokine TGF-beta in Lewis rats. J Neuroimmunol 2004;156:3–9. [DOI] [PubMed] [Google Scholar]
- 11.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008;358:676–688. [DOI] [PubMed] [Google Scholar]
- 12.Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011;378:1779–1787. [DOI] [PubMed] [Google Scholar]
- 13.Steinman L, Zamvil SS. Beginning of the end of two-stage theory purporting that inflammation then degeneration explains pathogenesis of progressive multiple sclerosis. Curr Opin Neurol 2016;29:340–344. [DOI] [PubMed] [Google Scholar]
- 14.von Büdingen HC, Bar-Or A, Zamvil SS. B cells in multiple sclerosis: connecting the dots. Curr Opin Immunol 2011;23:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molnarfi N, Schulze-Topphoff U, Weber MS, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med 2013;210:2921–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007;130:1089–1104. [DOI] [PubMed] [Google Scholar]
- 17.Toubi E, Nussbaum S, Staun-Ram E, et al. Laquinimod modulates B cells and their regulatory effects on T cells in multiple sclerosis. J Neuroimmunol 2012;251:45–54. [DOI] [PubMed] [Google Scholar]
- 18.Gatto D, Wood K, Caminschi I, et al. The chemotactic receptor EBI2 regulates the homeostasis, localization and immunological function of splenic dendritic cells. Nat Immunol 2013;14:446–453. [DOI] [PubMed] [Google Scholar]
- 19.Shin C, Han JA, Koh H, et al. CD8alpha(−) dendritic cells induce antigen-specific T follicular helper cells generating efficient humoral immune responses. Cell Rep 2015;11:1929–1940. [DOI] [PubMed] [Google Scholar]
- 20.Shin C, Han JA, Choi B, Cho YK, Do Y, Ryu S. Intrinsic features of the CD8alpha(−) dendritic cell subset in inducing functional T follicular helper cells. Immunol Lett 2016;172:21–28. [DOI] [PubMed] [Google Scholar]
- 21.Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol 2011;29:621–663. [DOI] [PubMed] [Google Scholar]
- 22.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 2003;197:1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Litzenburger T, Fassler R, Bauer J, et al. B lymphocytes producing demyelinating autoantibodies: development and function in gene-targeted transgenic mice. J Exp Med 1998;188:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clements CS, Reid HH, Beddoe T, et al. The crystal structure of myelin oligodendrocyte glycoprotein, a key autoantigen in multiple sclerosis. Proc Natl Acad Sci USA 2003;100:11059–11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wegner C, Stadelmann C, Pfortner R, et al. Laquinimod interferes with migratory capacity of T cells and reduces IL-17 levels, inflammatory demyelination and acute axonal damage in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol 2010;227:133–143. [DOI] [PubMed] [Google Scholar]
- 26.Shetty A, Gupta SG, Varrin-Doyer M, et al. Immunodominant T-cell epitopes of MOG reside in its transmembrane and cytoplasmic domains in EAE. Neurol Neuroimmunol Neuroinflamm 2014;1:e22. doi: 10.1212/NXI.0000000000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weber MS, Prod'homme T, Patarroyo JC, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol 2010;68:369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nurieva RI, Chung Y, Hwang D, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity 2008;29:138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linterman MA, Beaton L, Yu D, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med 2010;207:353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baumjohann D, Preite S, Reboldi A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity 2013;38:596–605. [DOI] [PubMed] [Google Scholar]
- 31.Bettelli E, Baeten D, Jager A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest 2006;116:2393–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krishnamoorthy G, Lassmann H, Wekerle H, Holz A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest 2006;116:2385–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol Neuroimmunol Neuroinflamm 2015;2:e62. doi: 10.1212/NXI.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brück W, Wegner C. Insight into the mechanism of laquinimod action. J Neurol Sci 2011;306:173–179. [DOI] [PubMed] [Google Scholar]
- 35.Brück W, Pfortner R, Pham T, et al. Reduced astrocytic NF-kappaB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathol 2012;124:411–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mishra MK, Wang J, Keough MB, et al. Laquinimod reduces neuroaxonal injury through inhibiting microglial activation. Ann Clin Transl Neurol 2014;1:409–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kramann N, Menken L, Hayardeny L, Hanisch UK, Brück W. Laquinimod prevents cuprizone-induced demyelination independent of Toll-like receptor signaling. Neurol Neuroimmunol Neuroinflamm 2016;3:e233. doi: 10.1212/NXI.0000000000000233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gudi V, Gingele S, Skripuletz T, Stangel M. Glial response during cuprizone-induced de- and remyelination in the CNS: lessons learned. Front Cell Neurosci 2014;8:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karussis D, Grigoriadis S, Polyzoidou E, Grigoriadis N, Slavin S, Abramsky O. Neuroprotection in multiple sclerosis. Clin Neurol Neurosurg 2006;108:250–254. [DOI] [PubMed] [Google Scholar]
- 40.Arnold DL. Evidence for neuroprotection and remyelination using imaging techniques. Neurology 2007;68:S83–S90; discussion S91–S96. [DOI] [PubMed] [Google Scholar]
- 41.Mohr R, Krawinkel U. Helper T-cell kinetics and investigation of antigen receptor expression on early and memory T-helper cells. Immunology 1976;31:249–255. [PMC free article] [PubMed] [Google Scholar]
- 42.Schulze-Topphoff U, Varrin-Doyer M, Pekarek K, et al. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proc Natl Acad Sci USA 2016;113:4777–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bittner S, Ruck T, Gobel K, et al. Effects of glatiramer acetate in a spontaneous model of autoimmune neuroinflammation. Am J Pathol 2014;184:2056–2065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.